

1 Introduction
Numerous asymmetric catalyzed reactions were developed with transition metal complexes using chiral phosphorus ligands [1,2]. This class of ligands is widely used because their structure can be easily modified on the phosphorus center, or in its α- or β-position, owing to numerous electrophilic or nucleophilic reactions of their P(III)- or P(IV)-precursors [2–4]. Noteworthy, over the last decade, the chemistry of phosphorus ligands has made significant breakthroughs due to the borane used as protecting group, giving stable complexes, often crystallized, and affording clean reactions [4–6]. In addition, the chirality of the phosphorus ligands can stem either from the carbon backbone or from the phosphorus center, increasing thus the number of possible stereoisomers to improve asymmetric catalysis results [7].
As the asymmetric catalysis requires the selective formation of a single enantiomer, the availability of chiral ligands in both enantiomeric forms is essential to afford the desired stereoisomer. In most cases, both enantiomers of the phosphorus ligands are obtained by chemical resolution (For resolution of BINAP, MeO-BIPHEP or SYNPHOS, see [8]), or from their chiral original backbone scaffold (For the synthesis of JOSIPHOS and MonoPHOS from chiral backbone scaffolds, see [9]). In other cases, the enantiomers are obtained by stereoselective synthesis starting from the chiral pool (For representative synthesis of DIOP and PHOX starting from the pool chiral, see [10]), or from prochiral precursors (For the asymmetric synthesis of DuPHOS, see [11]).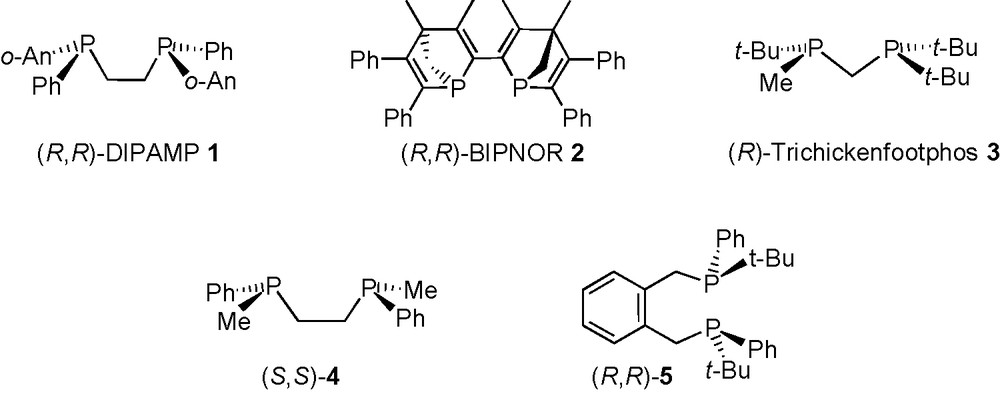
In the case of the DIPAMP 1, BIPNOR 2 and Trichickenfootphos 3, the P-chirogenic phosphines were obtained enantiomerically pure by chemical resolution or by chromatography on a chiral column, respectively [12]. However, most of the time, this class of ligands are prepared by efficient stereoselective synthesis using the phosphorus borane chemistry [4,6,13–17]. The principle of these syntheses is based on the preparation of enantiomerically enriched P-chirogenic organophosphorus borane building blocks 8–10, acting either as nucleophilic or electrophilic reagents.
In the first case, the nucleophilic reagents 8 and 9a are prepared in the presence of (−)-sparteine, by enantioselective deprotonation of the dimethylphosphine borane 6, or by dynamic thermodynamic resolution of the secondary phosphine borane 7, respectively (Scheme 1). The homocoupling of the anion 8 in the presence of CuCl2 under air, or the reaction of 9a with an electrophile, leads to the preparation of P-chirogenic ligands by C-C or P-C bond formation, such as in (S,S)-4 or (R,R)-5 (Scheme 1) [13,14]. Noteworthy, these nucleophilic methodologies have been successfully applied by T. Imamoto and coll. for the synthesis of several efficient chiral ligands in asymmetric catalysis, which are currently commercially available (MiniPHOS, BisP*, Quinox P*) [4,15].
Unfortunately both nucleophilic methodologies are restricted to bulky alkyl or methyl substituents on the phosphorus atom. Furthermore, as only (−)-sparteine is commercially avalaible, only one enantiomer is accessible. The stereoselective synthesis of the other enantiomer is still possible, but using in this case, demanding strategies [16] or the (+)-sparteine surrogate [17].
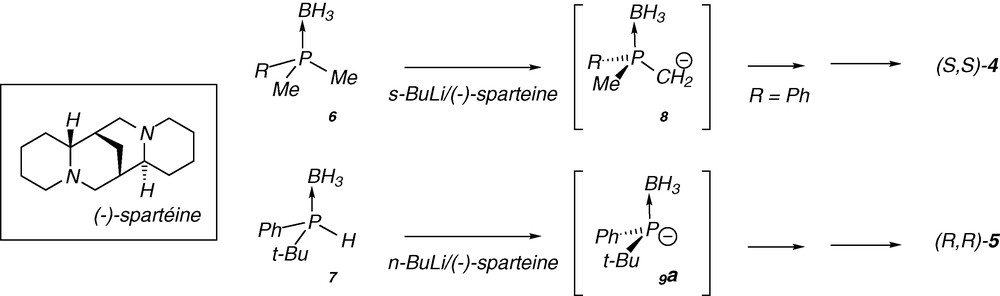
In the case of the P-chirogenic electrophilic building blocks 10, the synthesis is based on subsequent stereoselective reactions starting from the oxazaphospholidine borane complex 11, prepared from (+)-ephedrine (Scheme 2) [6(a,c),18]. After reaction with an organolithium reagent the starting complex (Sp)-11 affords the ring-opening product 12, which leads to the P-chirogenic phosphinite borane 10 by P-N bond cleavage under acidic methanolysis conditions [6(a,c),18]. The reaction of the phosphinite borane 10 with an organolithium reagent, leads to the corresponding P-chirogenic phosphines 13, after decomplexation with DABCO (Scheme 2).
As ephedrine is available in both antipodal forms (+) or (−), the stereoselective synthesis of both enantiomeric mono- or diphosphines can be achieved. In addition, we previously reported the enantiodivergent synthesis of both enantiomers starting from the same complex 11, by simply changing the order of addition of the organolithium reagents (i.e. R3Li then R2Li vs R2Li then R3Li) (Scheme 2) [6(a,c), 18]. However, this method was mainly examplified using aryl substituents on the phosphorus atom, because in the case of alkyl group, side reactions can occur by deprotonation in α-position of the phosphine borane.
We now wish to report some new efficient routes for the enantiodivergent synthesis of P-chirogenic mono- and diphosphines using the ephedrine methodology, giving easy access to both enantiomers, notably for derivatives bearing alkyl groups.
2 Results and discussion
2.1 Enantiodivergent synthesis using P-chirogenic chlorophosphine boranes as electrophilic reagents (Route A)
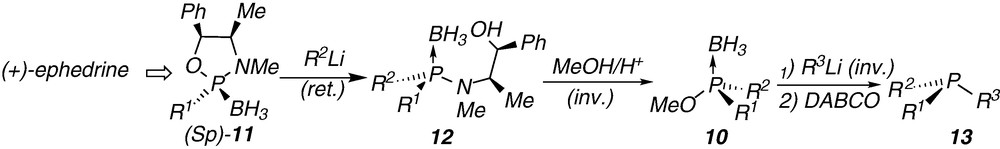
In connection with our on-going work on the asymmetric synthesis of P-chirogenic phosphorus compounds, we developed earlier the facile preparation of chlorophosphine boranes 14 as new P-chirogenic electrophilic building blocks [19]. The chlorophosphine boranes 14 are readily obtained by acidolysis of the aminophosphine boranes 12 with a toluene solution of HCl, leading to the P-N bond cleavage with inversion of configuration on the phosphorus atom (Scheme 3) [19].
The reaction of the chlorophosphine boranes 14 with organolithium reagents stereospecifically provides the corresponding P-chirogenic phosphine boranes 15, with inversion of configuration on the phosphorus atom (Scheme 3a). The route (A) was successfully applied to the enantioselective synthesis of both (R) and (S)-enantiomers of the di- or triaryl-phosphine boranes 15a-d, reported in Table 1.
Enantiodivergent synthesis of phosphine boranes 15 using chlorophosphine boranes 14.
| Entry | Aminophosphine boranes | Chlorophosphine boranes | Phosphine boranes | |||||||
| R1 | R2 | HCl (equiv) | Time (h) | R3 | Yields (%)a | e.e. (%)b | Conf. | |||
| 1 | Ph | Me | 12a | 2.1 | 1 | 14a | o-An | 80 | 90b | (S)-15a |
| 2 | Ph | o-An | 12b | 4 | 1 | 14b | Me | 90 | 98b | (R)-15a |
| 3 | Ph | o-An | 12b | 4 | 1 | 14b | Fc | 71 | 95b | (R)-15b |
| 4 | Ph | o-An | 12b | 4 | 1 | 14b | m-Xyl | 79 | 99b | (S)-15c |
| 5 | Ph | Fc | 12c | 15 | 48 | 14c | o-An | 79 | 98b | (S)-15b |
| 6 | Ph | m-Xyl | 12d | 6 | 1 | 14d | o-An | 82 | 99b | (R)-15c |
| 7 | Ph | i-Pr | 12ec | 5 | 12 | 14e | Me | 68 | 89d | (R)-15d |
| 8 | i-Pr | Ph | 12fc | 5 | 12 | 14e | Me | 86 | 96d | (S)-15d |
a Isolated yields.
b Determined by HPLC on a chiral column of the borane complex.
c Prepared from (−)-ephedrine (see Scheme 4).
d Determined by 31P NMR of the phosphine oxide derivative in presence of reagent 21.
Thus, inverting the introduction order of the substituents during the synthesis, i.e. methyl as former group then o-anisyl, or reversely, can afford either the (S)- or the (R)-PAMP borane 15a respectively, with e.e. up to 98% (entries 1,2; Table 1). Both enantiomers of the tertiary phosphine boranes 15b and 15c, were separately prepared using the same procedure starting respectively from o-anisyl-, ferrocenyl- or m-xylyl-aminophosphine boranes 12b, 12c or 12d, which are derived from the (+)-ephedrine (Table 1, entries 3–6).4 The X-ray structures of the phosphine boranes (R)-15b and (S)-15b were obtained and their ORTEP views are given in Figs. 1 and 2, with selected structural parameters.
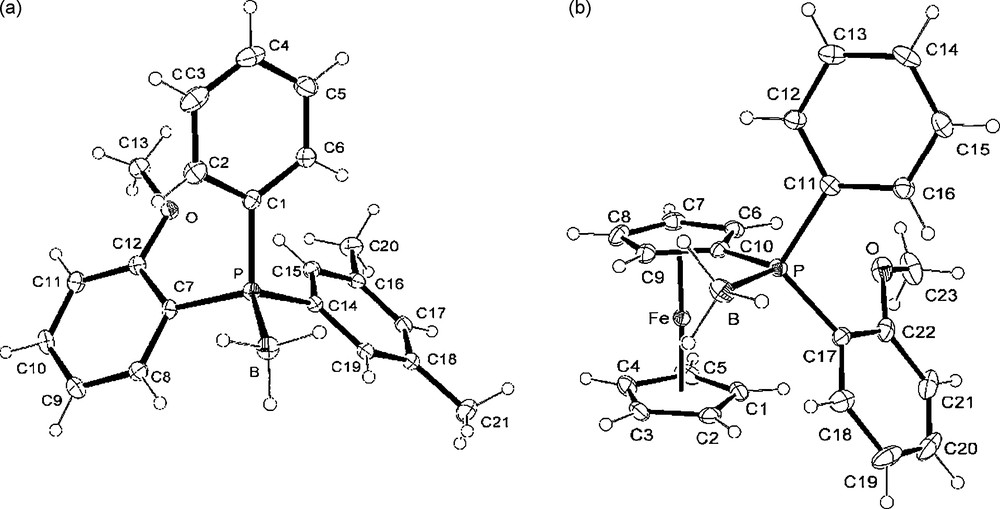
Crystal structures: (a) Phosphine borane (R)-15c. Selected bond lengths (Å), angles (°): P-B 1.917(2), P-C(1) 1.8138(18), P-C(7) 1.8170(17), P-C(14) 1.8165(16); C(1)-P-B 109.53(9); C(7)-P-B 111.37(8), C(14)-P-B 113.43(9). (b) Phosphine borane (R)-15b. Selected bond lengths (Å), angles (°): P-B 1.913(3), P-C(10) 1.793(2), P-C(11) 1.815(2), P-C(17) 1.810(2), Cp(1)-Fe 1.659(2), Cp(2)-Fe 1.645(2); C(10)-P-B 109.87(12), C(11)-P-B 107.82(11), C(17)-P-B 112.83(13), Cp(1)-Fe-Cp(2) 176.86 (12).
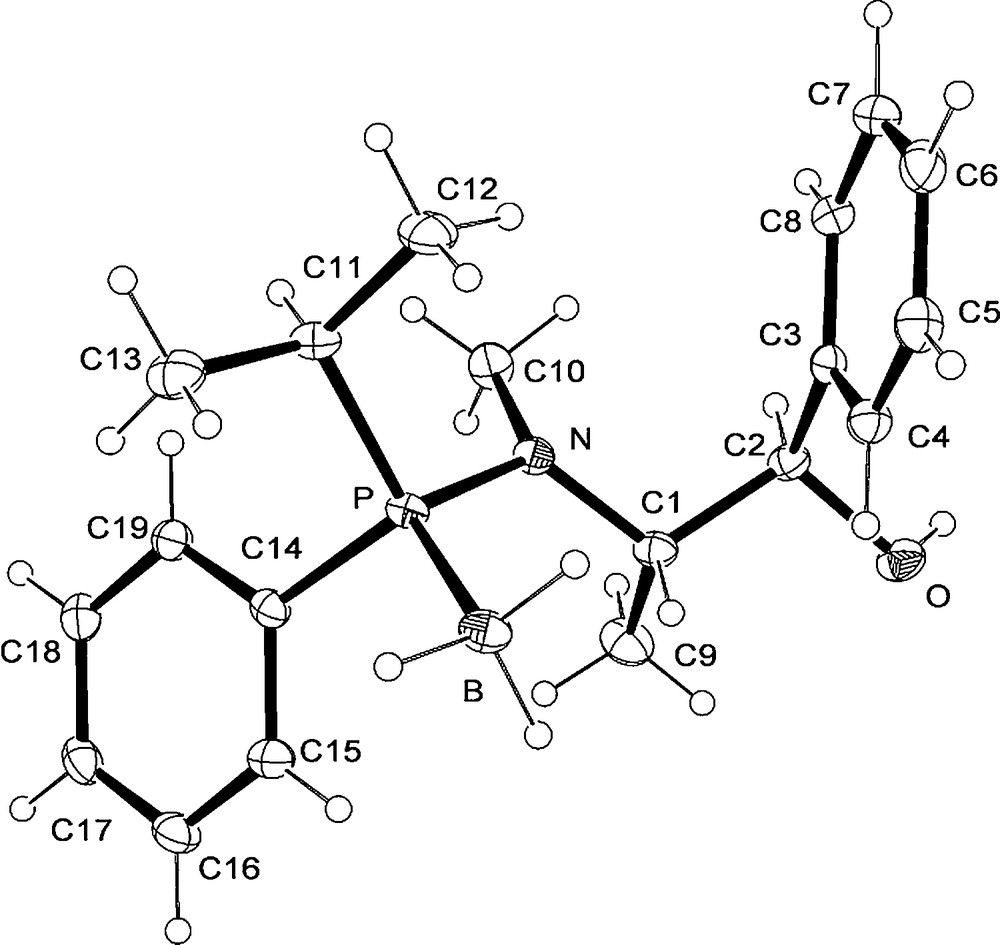
Crystal structure of aminophosphine borane 12f with selected bond lengths (Å), angles (°): P-B 1.9206(19); P-N 1.6755(13), P-C(11) 1.8285(16, P-C(14) 1.8263(16; N-P-B 114.47(8); C(11)-P-B 112.31(8), C(14)-P-B 110.93(8), C(1)-N-P 118.35(10), C(1)-N-C(10) 116.03(13), C(10)-N-P 118.84(11).
2.2 Enantiodivergent synthesis using P-chirogenic phosphide borane reagents (Route B)
Recently, the investigation of the reaction between the t-butyllithium reagent and the chlorophosphine boranes 14, led to the discovery of an unexpected new metal-halide exchange affording the corresponding P-chirogenic phosphide boranes 9 with retention of the configuration at the phosphorus atom (Scheme 3b). Following the subsequent addition of alkyl halide in excess, the corresponding phosphine boranes 15 were obtained in good to excellent yields and with e.e. up to 99% (Scheme 3b, Table 2). The route (B) was also successfully applied for the enantioselective preparation of the phosphine borane (S)-15a (PAMP.borane). The latter is obtained by acidolysis with HCl of the aminophosphine borane 12b, then metal-halide exchange with t-BuLi reagent, and finally, trapping with methyl iodide (Table 2, entry 1). Noteworthy, the phosphine borane (S)-15a, obtained in this case, is the enantiomer of that prepared following the route (A) starting from aminophosphine borane 12b (Table 1, entry 2).
Enantiodivergent synthesis of phosphine boranes 15 using chlorophosphine boranes 14 as phosphide borane 9 precursors.
| Entry | Aminophosphine boranes | Chlorophosphine boranes | Phosphine boranes | |||||||
| R1 | R2 | HCl (equiv) | Time (h) | R3X | Yields (%)a | e.e. (%) | Conf. | |||
| 1 | Ph | o-An | 12b | 4 | 1 | 14b | MeI | 75 | 92b | (S)-15a |
| 2 | Ph | i-Pr | 12ec | 5 | 12 | 14e | MeI | 73 | 96d | (S)-15d |
| 3 | i-Pr | Ph | 12fc | 5 | 12 | 14e | MeI | 71 | 96d | (R)-15d |
a Isolated yields.
b Determined by HPLC on a chiral column of the borane complex.
c Prepared from (−)-ephedrine.
d Determined by 31P NMR of the phosphine oxide derivative in presence of reagent 21.
Interestingly, route (B) offers a versatile and efficient pathway for the enantiodivergent synthesis of P-chirogenic phosphines. Thus, the i-propylaminophosphine borane 12e, which was prepared from the oxazaphospholidine complex (Rp)-11a, leads to both enantiomers of the phosphine borane 15d (Scheme 4a). On the one hand, the acidolysis of 12e with HCl, which affords the corresponding chlorophosphine-borane intermediate, provides the (S)-methylphenyl-i-propylphosphine borane 15d after a metal-halogen exchange using t-BuLi reagent, and then trapping with methyl iodide (Scheme 4a; Table 2, entry 2). On the other hand, the (R)-methylphenyl-i-propylphosphine borane 15d was obtained according to route (A), after HCl acidolysis of the aminophosphine borane 12e then reaction with MeLi (Scheme 4; Table 1, entry 7).
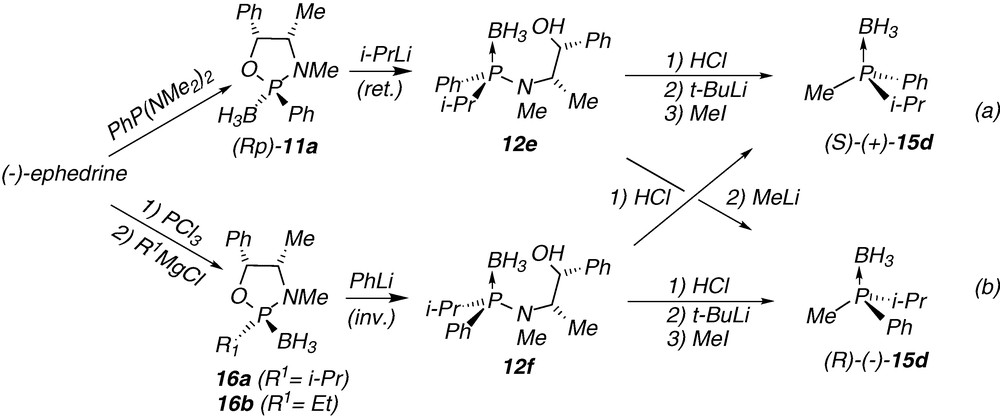
The synthesis of both enantiomers of the phosphine borane 15d was also achieved starting from the phenyl-i-propylaminophosphine borane 12f, readily obtained from the 2-i-propyloxazaphospholidine borane 16a. This new starting complex 16a was prepared in a diastereomerically pure form by subsequent reactions of PCl3 with (−)-ephedrine [21], then with i-propylmagnesium reagent and finally with BH3.DMS (Scheme 4b). The relative configuration of the complex 16a was determined by comparison with the X-Ray structure of the 2-ethyloxazaphospholidine borane 16b (R1 = Et). This latter compound was prepared by a similar procedure from (−)-ephedrine, but using ethylmagnesium chloride instead of i-propylmagnesium reagent.5 The reaction of the oxazaphospholidine borane 16a with the phenyllithium reagent affords the phenyl-i-propylaminophosphine borane 12f by P-O bond cleavage of the heterocycle (Scheme 4b). The X-ray structure of compound 12f (Fig. 2) proves the stereochemistry of ring-opening process with inversion of configuration on the phosphorus centre (Scheme 4b). After acidolysis with HCl of the epimer 12f, then metal-halide exchange with t-BuLi reagent of the chlorophosphine-borane intermediate, and finally trapping with MeI, the (R)-methylphenyl-i-propylphosphine borane 15d was obtained in 71% yield and with 97% e.e. (Scheme 4b; Table 2, entry 3).
Finally, the (S)-methylphenyl-i-propylphosphine borane 15d was also obtained from the i-propylaminophosphine borane 12f using route (A), by acidolysis with HCl followed by a reaction with MeLi (Scheme 4; Table 1, entry 8).
2.3 Enantiodivergent synthesis of the P-chirogenic 1,2-bis(methylphenylphosphino)ethane 4
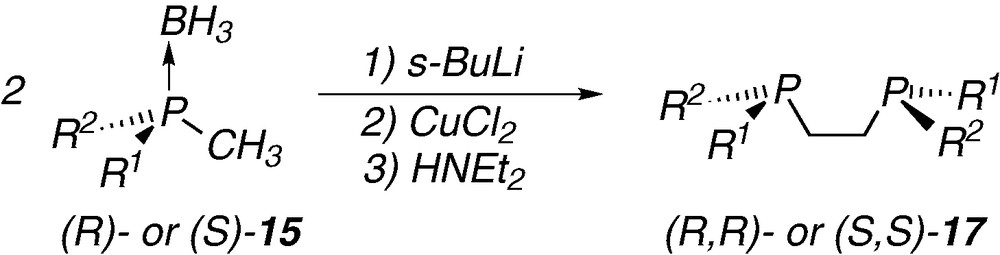
The synthesis of both enantiomers of the P-chirogenic 1,2-bis(diphosphino)ethane 17 can be achieved by a Knowles’ like coupling of the corresponding (R)- or (S)-methylphosphine borane 15, which are prepared according to one of the approaches described above (Scheme 5) [12a,18].
Preliminary results indicate that the C-C bond coupling can also be achieved with the (R)-methylphenylphosphinite borane 10a affording the corresponding (R,R)-1,2-bis(phenylphosphinito borane)ethane 18 (Scheme 6a) [21,22]. Interestingly, the reaction of two equivalents of methyl lithium reagent with the compound 18 leads to the (S,S)-diphosphine diborane 19 and then, to the (S,S)-(+)-diphosphine 4 after decomplexation of the borane protecting group (Scheme 6a, Table 3, entry 6). The other enantiomer of the diphosphine (R,R)-(−)-4 was then prepared using the same pathway, but starting from (−)-ephedrine (Scheme 6b, Table 3, entry 7).

Preparation of free mono- and diphosphines from their borane complexes.
| Entry | Borane complex | Conditions | Mono- and diphosphine | ||||
| δ31P NMR | e.e. (%) | ||||||
| 1 | (R)-15a | HNEt2 50 °C/10h | (R)-13a | +46a | −36 | 98d | |
| 2 | (S)-15b | DABCO 50 °C/10h | (S)-13b | −4.8b | −28.3 | 98d | |
| 3 | (S)-15c | DABCO 50 °C/10h | (S)-13c | −3.8b | −16.9 | 99d | |
| 4 | (S)-15d | DABCO 50 °C/10h | (S)-13d | −10b | −19.3 | 93e | |
| 5 | (R)-15d | DABCO 50 °C/10h | (R)-13d | – | – | 96e | |
| 6 | (S,S)-18 | DABCO 50 °C/10h | (S,S)-4 | +24.5b | – | 99f | |
| 7 | (R,R)-18 | DABCO 50 °C/10h | (R,R)-4 | −23.5c | −32 | 99f |
a In MeOH.
b In CHCl3.
c In CH2Cl2.
d Determined by HPLC on a chiral column of the borane complex derivative.
e Determined by 31P NMR of the phosphine oxide derivative in presence of reagent 21.
f Determined by 31P NMR in presence of chiral palladium complex 20.
The X-ray structure of the (S,S)-1,2-bis(phenylphosphinito borane)ethane 18 (Fig. 3), exhibits an unfolded conformation with the substituents disposed anti relative each other. Moreover, the (S) absolute configuration of the phosphinite borane fragments is consistent with the retention of the configuration during the C-C bond coupling of the anion resulting from 10a (Scheme 6).
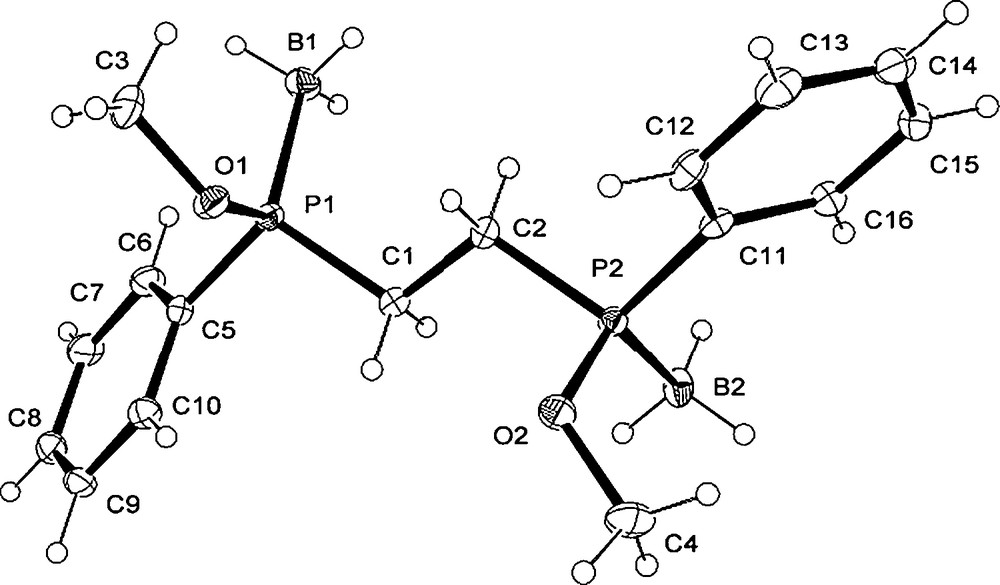
Crystal structure of the (S,S)-diphosphinite diborane 18. Selected bond lengths (Å), angles (°): (a) P(1)-C(1) 1.806(3), P(2)-C(2) 1.804(3), P(1)-C(5) 1.803(3), P(2)-C(11) 1.805(3), P(1)-O(1) 1.607(3), P(2)-O(2) 1.600(2), P(1)-B(1) 1.895(3), P(2)-B(2) 1.889(4); O(1)-P(1)-C(1) 99.90(14), O(2)-P(2)-C(2) 99.42(13), O(1)-P(1)-C(5) 106.13(13), O(2)-P(2)-C(11) 105.77(14), O(1)-P(1)-B(1) 116.66(17), O(2)-P(2)-B(2) 116.19(16).
2.4 Decomplexation of the borane complexes into P-chirogenic mono- and diphosphines
The phosphine borane complexes can directly be used without decomplexation, for the synthesis of quaternary phosphonium salts [23], (thio)phosphorylated derivatives [24], and chiral complexes or catalysts derived from transition metals [25,26b]. When the uncoordinated phosphines are required, the borane decomplexation can be achieved from a reaction with an amine [26], a strong acid [27], EtOH [28] or an olefin [24b,29], to quantitatively lead to the corresponding P(III)-compound with complete retention of configuration on the phosphorus atom. The uncoordinated P-chirogenic mono- 13 and the diphosphine 4 were obtained by heating at 50 °C either in pure diethyl amine, or in toluene in the presence of DABCO. Subsequently, the purification is achieved by fast filtration through neutral alumina (Scheme 2, Table 3). The enantiomeric excesses of the phosphines were determined either by HPLC on a chiral column after an additional complexation with BH3.DMS, or by 31P NMR in presence of the chiral palladium complex 20 (For the preparation of the chiral palladium complex 19, see [30a]; For the preparation of N,N-dimethyl-1-phenylethylamine, see [30b]; For the 31P NMR, see [30c]), or by 31 P NMR of the phosphine oxide derivatives in presence of the resolving Kagan's reagent 21 [31].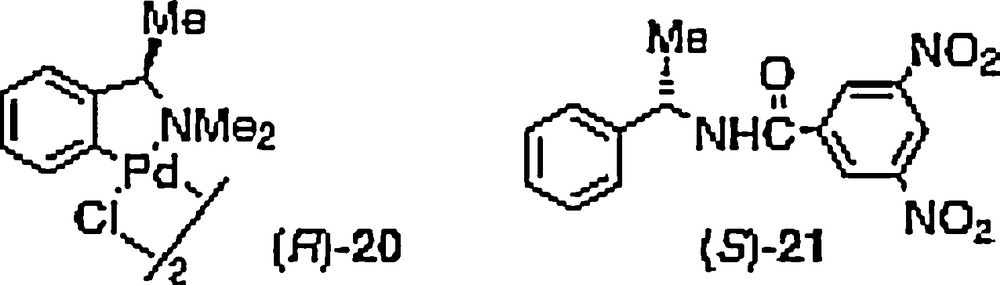
3 Conclusion
Several approaches for the enantiodivergent syntheses of P-chirogenic mono- and diphosphines were described, using the convenient ephedrine methodology and the borane complex chemistry.
Since the two enantiomers of ephedrine are available, the stereoselective synthesis of both enantiomers of mono- or diphosphine is easily performed. Furthermore, both enantiomers of a phosphine ligand can also be obtained, starting from the same oxazaphospholidine borane complex derived from (+)-ephedrine, when changing the order of addition of the organolithium reagents during the synthetic pathway.
An alternate approach is based on the P-chirogenic chlorophosphine boranes, which afford one enantiomer of a phosphine borane with inversion of configuration, by a reaction with an organolithium reagent. On the other hand, the chlorophosphine boranes also provide the corresponding P-chirogenic phosphide boranes 9 with retention of configuration on the phosphorus atom, by an unexpected metal-halogen exchange using the t-butyllithium reagent. The addition of alkyl halide to the resulting phosphide boranes then affords the corresponding phosphine boranes, but with opposite configuration.
A third approach is based on the stereoselective synthesis of the starting oxazaphospholidine borane complex derived from (−)-ephedrine. In the case where this complex is prepared using the bis(dimethylamino)phosphine, the absolute configuration of phosphorus centre is (R). When it is prepared by subsequent reactions of (−)-ephedrine with PCl3, and then with a Grignard reagent, the configuration of the phosphorus centre is (S). Consequently, depending on the method for the preparation of the starting oxazaphospholidine borane, the convenient synthesis can provide either phosphine enantiomers.
Finally, the synthesis of (R,R)- and (S,S)-1,2-bis(methylphenylphosphino)ethane 4 was also described, using both P-chirogenic diphosphinite diborane 18 enantiomers prepared from (+)- or (−)-ephedrine.
In summary, the efficiency of the ephedrine methodology is again highlighted by offering several approaches for the synthesis of both enantiomeric P-chirogenic mono- and diphosphines in the desired absolute configuration, notably bearing alkyl substituents.
4 Experimental section
All reactions were carried out under an Ar atmosphere in dried glassware. Solvents were dried and freshly distilled under an Ar atmosphere over sodium/benzophenone for THF, diethylether, toluene and benzene, CaH2 for CH2Cl2. Hexane and isopropanol for HPLC were of chromatography grade and used without further purification. Methyllithium (1.6 M in Et2O), s-butyllithium (1.4 M in cyclohexane), t-butyllithium (1.6 M in pentane), phenyllithium (1.8 M in Bu2O), isopropyllithium (0.7 M in pentane), isopropylmagnesium chloride (2 M in THF), ethylmagnesium chloride (2 M in THF), ferrocene, 2-bromoanisole, 5-bromo-m-xylene, methyl iodide, BH3.SMe2, N-methyl morpholine and 1,4-diazabicyclo[2.2.2]octane (DABCO) were purchased from Aldrich, Acros or Alfa Aesar, and used as received. (+)- and (−)-ephedrine were purchased from Aldrich and dried by azeotropic shift of toluene on rotary evaporator. Phosphorus trichloride was distilled and CuCl2 dried at 100 °C for 12 h, before use. The solution of HCl in toluene (0.2–0.4 M) was obtained by bubling HCl gas in toluene and titrated by acidimetry before use. The (2S, 4R, 5S)-(−)-3,4-dimethyl-2,5-diphenyl-1,3,2-oxazaphospholidine-2-borane 11a and its enantiomer (2R, 4S, 5R)-(+)-11a, were prepared from the appropriate (+)- or (−)-ephedrine, as previously described [19a].
The (Sp)-(+)-N-methyl-N-[(1S,2R)(1-hydroxy-2-methyl-1-phenyl-2-propyl)]aminomethylphenyl phosphine borane 12a and (Rp)-(+)-N-methyl-N-[(1S,2R)(1-hydroxy-2-methyl-1-phenyl-2-propyl)] amino-o-anisylphenylphosphine borane 12b were prepared from the (+)-ephedrine according to the published procedure [19c]. (R)-(+) and (S)-(−)-[(O-methyl)-methylphenylphosphinite]borane 10a were obtained as previously described, starting from the (+)- and (−)-ephedrine respectively [18(a,b),26c,32]. Chiral HPLC analysis were performed on SHIMADZU 10-series apparatus, using chiral columns (Chiralcel OK, Chiralcel OD, Chiralcel OD-H, Chiralpack AD, Chiralcel OJ, Lux 5 μ-cellulose-2), and with hexane/propan-2-ol mixtures as the mobile phase (Flow rate 1 mL min−1; UV detection λ = 254 nm). Thin layer chromatography (TLC) was performed on 0.25 mm E. Merck precoated silica gel plates and exposed by UV, potassium permanganate or iodine treatment. Flash chromatography was performed with the indicated solvents using silica gel 60 A, (35–70 μm; Acros) or aluminium oxide 90 standardized (Merck). All NMR spectra data were recorded on BRUKER AM 250, 300 AVANCE, 500 AVANCE DRX and 600 AVANCE II spectrometers at ambient temperature. Data are reported as s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, brs = broad singlet, brd = broad doublet, dhept = doublet of heptuplet, coupling constant(s) in Hertz. Melting points were measured on a Kofler bench melting point apparatus and are uncorrected. Optical rotations values were recorded at 20 °C on a Perkin-Elmer 341 polarimeter, using a 10 cm quartz vessel. Infrared spectra were recorded on a Bruker Vector 22 apparatus. Mass and HRMS spectra were recorded on Mass, Bruker ESI micro TOF-Q apparatus, at the Université de Bourgogne (Dijon). The major peak m/z was mentioned with the intensity as a percentage of the base peak in brackets. Elemental analyses were measured with a precision superior to 0.3% at the Microanalysis Laboratories of the Universités P. & M. Curie (Paris) and Bourgogne (EA 1108 CHNS-O FISONS Instrument). X-Ray analyses were performed at the Université de Bourgogne, and the data were collected at 115 K on a Bruker Nonius Apex II CCD system using graphite-monochromated Mo-Kα radiation. The structures were solved by direct methods (SIR92)[33] and refined with full-matrix least-squares methods based on F2 (SHELXL-97)[34] with the aid of the WINGX program [35]. All non-hydrogen atoms were refined with anisotropic thermal parameters. Hydrogen atoms were either included in theirs calculated positions or found in Fourier difference maps (CH3 and BH3). Crystallographic data and structures refinement details for 12f, 15c, 15d and 18 are summarized in Table 4.
Crystallographic data and structures refinement details for 12f, 15c, 15d and 18.
| Compounds | 12f | 15c | 15d | 18 |
| Empirical formula | C19H29BNOP | C21H24BOP | C23H24BFeOP | C16H26B2O2P2 |
| Formula weight | 329.21 | 334.18 | 414.05 | 333.93 |
| Temperature (K) | 115(2) | 115(2) | 110(2) | 115(2) |
| Crystal system | Orthorhombic | Orthorhombic | Monoclinic | Monoclinic |
| Space group | P212121 | P212121 | P21 | P21 |
| a (Å) | 8.2195(4) | 10.0720(2) | 8.5691(3) | 10.9431(5) |
| b (Å) | 14.5616(6) | 12.8584(3) | 10.4833(3) | 8.6606(6) |
| c (Å) | 16.1165(8) | 14.5612(3) | 11.6417(4) | 10.8797(8) |
| α (°) | 90 | 90 | 90 | 90 |
| β (°) | 90 | 90 | 106.7440(10) | 90.906(4) |
| γ (°) | 90 | 90 | 90 | 90 |
| Volume (Å3) | 1928.97(16) | 1885.82(7) | 1001.46(6) | 946.19(11) |
| Z | 4 | 4 | 2 | 2 |
| ρcalc. (g/cm3) | 1.134 | 1.177 | 1.373 | 1.172 |
| μ (mm−1) | 0.146 | 0.150 | 0.842 | 0.232 |
| Size (mm3) | 0.32 × 0.30 × 0.12 | 0.20 × 0.20 × 0.18 | 0.40 × 0.30 × 0.20 | 0.25 × 0.05 × 0.05 |
| F(000) | 712 | 712 | 432 | 356 |
| λ | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| sin(θ)/ λ max; Å−1 | 0.65 | 0.65 | 0.65 | 0.65 |
| Index ranges | h: −10; 10 | h: −13; 13 | h: −11; 10 | h: −12; 13 |
| k: −18; 18 | k: −16; 16 | k: −13; 13 | k: −8; 11 | |
| l: −20; 20 | l: −18; 18 | l: −15; 14 | l: −14; 14 | |
| Reflection collected | 4310 | 4307 | 6351 | 3302 |
| Rint | 0.017 | 0.023 | 0.024 | 0.026 |
| Reflection with I ≥ 2σ (I) | 4180 | 3796 | 3950 | 3132 |
| Data/restraints/parameters | 4310/0/214 | 4307/0/223 | 4472/1/248 | 3302/1/200 |
| R indices (I ≥ 2σ [I]) | R1 = 0.0341a | R1 = 0.0364a | R1 = 0.0329a | R1 = 0.0439a |
| wR2 = 0.0773b | wR2 = 0.0816b | wR2 = 0.0614b | wR2 = 0.0939b | |
| R indices (all data) | R1 = 0.0362a | R1 = 0.0464a | R1 = 0.0427a | R1 = 0.0481a |
| wR2 = 0.0792b | wR2 = 0.0855b | wR2 = 0.0644b | wR2 = 0.0981b | |
| Goodness-of-fitc on F2 | 1.082 | 1.045 | 1.046 | 1.082 |
| Absolute structure parameters | 0.04(8) | 0.04(8) | 0.000(13) | 0.00(14) |
| Δρ (e Ǻ−3) | 0.200 and −0.180 | 0.181 and −0.311 | 0.217 and −0.286 | 0.291 and −0.242 |
| CCDC deposition No. | 768 727 | 768 725 | 768 724 | 768 726 |
a R1 = Σ(||Fo|-|Fc||)/Σ|Fo|.
b where for 12f, for 15c, for 15d and for18.
c S = [Σw(Fo2-Fc2)2/(n-p)]1/2 (n = number of reflections, p = number of parameters).
CCDC 768724, 768725, 768726 and 768727 contain the supplementary crystallographic data for 15d, 15c, 18, 12f. These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif.
4.1 Synthesis of (2S,4S,5R)-(−)-2-isopropyl-3,4-dimethyl-5-phenyl-1,3,2-oxazaphospholidine borane 16a
A 250 mL Schlenk flask, equipped with a magnetic stirrer and a septum, was charged with 50 mL of toluene and 2.8 mL (25.8 mmol) of N-methylmorpholine. Addition of PCl3 (1.12 mL, 12.9 mmol) was smoky and the resulting solution was cooled to −78 °C. Then (−)-ephedrine (2.1 g, 12.9 mmol) in 10 mL of toluene was added dropwise via syringe. The reaction was allowed to reach room temperature overnight and N-methylmorpholine hydrochloride was filtered under argon. The resulting crude solution (2R,4S,5R)-2-chloro-3,4-dimethyl-5-phenyl-1,3,2-oxazaphospholidine was used further without purification. 31P NMR (CDCl3, 121.5 MHz) δ + 168.6 ([20] + 172.4).
A 250 mL Schlenk flask, equipped with a magnetic stirrer and a septum, was charged with crude solution of (2R,4S,5R)-2-chloro-3,4-dimethyl-5-phenyl-1,3,2-oxazaphospholidine in toluene. The mixture was cooled to −60 °C and 8 mL of isopropylmagnesium chloride (2 M in THF; 16.1 mmol) was added. Reaction was stirred at this temperature for 1 h and then 2 mL of BH3.BMS complex were added. After 1 h under stirring, the mixture was hydrolyzed at −60 °C, and the aqueous layer was extracted with CH2Cl2. Combined organic phases were dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash chromatography on silica gel with a mixture hexane/CH2Cl2 (15:10) as eluent to afford the oxazaphospholidine borane complex 16a as a mixture of epimers in a 20:1 ratio (1.76 g, 55% yield). Crystallisation from hexane gives 1.4 g (43%) of the major isomer 16a.
(c 1.9, CHCl3); mp = 50 °C; IR (ν cm−1): 3035, 2978, 2859, 2402, 2372, 2344, 1460, 1385, 1290, 1226, 1190, 965, 875, 836, 739; 1H NMR (CDCl3, 300.13 MHz) δ 0.0–0.90 (m, 3H), 0.59 (d, J = 6.7 Hz, 3H), 1.03 (dd, J = 15.4 and 7.2 Hz, 3H), 1.05 (dd, J = 15.4 and 7.2 Hz, 3H), 1.82 (dhept, J = 9.0 and 7.2 Hz, 1H), 2.58 (d, J = 9.4 Hz, 3H), 3.55 (dq, J = 17.1 and 6.5 Hz, 1H), 5.35 (dd, J = 5.8 and 1.6 Hz, 1H), 7.10- 7.23 (m, 5H); 31P NMR (CDCl3, 121.5 MHz) δ + 156.1 (q, J = 75.8 Hz); 13C NMR (CDCl3, 75.5 MHz) δ 12.9 (d, J = 1.8 Hz), 15.8 (d, J = 2.2 Hz), 16.2, 30.5 (d, J = 8.1 Hz), 31.7 (d, J = 29.1 Hz), 60.0 (d, J = 2.4 Hz), 84.2 (d, J = 7.4 Hz), 126.3, 126.2, 128.4, 136.5 (d, J = 5.9 Hz); HRMS (ESI) calcd for C13H23BNNaOP [M + Na]+: 274.1505; found: 274.1470.
4.2 Synthesis of the aminophosphine boranes 12
4.2.1 Preparation of organolithium reagents
4.2.1.1 Aryllithium reagents by metal-halogen exchange
In a two necked-flask equipped with a magnetic stirrer and an argon inlet, 1 equiv. of sec-butyllithium is added. The mixture is cooled to 0 °C and 1 equiv. of 1-bromoanisole (1-bromonaphthalene or bromobenzene) is slowly added with a syringe while stirring. After the formation of a white precipitate, the mixture is stirred for 1 h at 0 °C. The organolithium reagent is dissolved with a minimum of dry THF before use.
4.2.1.2 Preparation of ferrocenyllithium by deprotonation of the ferrocene
A 250 mL three-necked flask equipped with a magnetic stirrer under an argon atmosphere was charged with ferrocene (0.74 g, 4 mmol) and THF (10 mL). At 0 °C, t-BuLi (2.75 mL, 1.6 M in hexane, 4.4 mmol) was added dropwise, and the reaction mixture was stirred at 0 °C for 1 h, before use.
4.2.2 General procedure
In a 100 mL three-necked flask, equipped with a magnetic stirrer and an argon inlet, 5 mmol of the oxazaphospholidine borane complex 11a were dissolved in 5 mL of anhydrous THF. The mixture was cooled at −78 °C and 2 equiv. (10 mmol) of the organolithium reagent were slowly added. The resulting mixture was stirred and warmed to 0 °C (or RT) until the starting material had completely reacted. The reaction was monitored by TLC over silica (CH2Cl2 as eluent), and was finally hydrolyzed at 0 °C with 2 mL of water. The THF was removed under reduced pressure and the aqueous layer was extracted several times with dichloromethane. The combined organic phases were dried over MgSO4 and the solvent was removed. The residue was purified on a short column of silica gel, using a mixture of toluene/AcOEt 95:5 as eluent, to afford the aminophosphine boranes 12. The aminophosphine boranes can be recrystallized using a mixture hexane/isopropanol 7:3.
4.2.3 (Rp)-(+)-N-methyl-N-[(1S,2R)(1-hydroxy-1-phenyl-prop-2-yl]aminoferrocenylphenyl phosphine borane 12c (from (Sp)-11a)
Yield = 80%; Orange crystals; (c 1.0, CHCl3); Rf = 0.62 (toluene/EtOAc [9:1]); IR (KBr, ν cm−1): 3500 (O-H), 2372 (B-H), 1455, 1437, 1386, 1367, 1217, 1163, 1106, 1063, 1022, 998, 956, 884, 822, 763, 746, 721, 698, 646, 614; 1H NMR (CDCl3, 300.13 MHz) δ 0.20–2.00 (m, 3H), 0.91 (d, J = 6.3 Hz, 3H), 2.05 (brs, 1H), 2.38 (d, J = 8.4 Hz, 3H), 4.16–4.25 (m, 1H), 4.23–4.27 (m, 1H), 4.30 (brs, 5H), 4.51 (d, J = 11.7 Hz, 2H), 4.58–4.62 (m, 1H), 4.87 (d, J = 5.7 Hz, 1H), 7.28–7.46 (m, 10H); 31P NMR (CDCl3, 121.5 MHz) δ + 70.7 (m); 13C NMR (CDCl3, 75.0 MHz) δ 13.7, 31.3, 40.3, 58.4 (d, J = 10.6 Hz), 70.9, 71.9, 73.0, 79.6, 127.3, 128.4, 128.8, 128.9, 129.1, 131.1, 132.2 (d, J = 9.8 Hz), 143.3; Anal. calcd for C26H31BFeNOP (471.17): C 66.28, H 6.63, N 2.97; found: C 66.33, H 6.83, N 3.02.
4.2.4 (Rp)-(−)-N-methyl-N-[(1S,2R)(1-hydroxy-1-phenyl-prop-2-yl]aminophenyl-m-xylylphosphine borane 12d (from (Sp)-11a)
Yield = 70%; White crystals; (c 0.6, CHCl3); mp = 124 °C; Rf = 0.12 (toluene); IR (KBr, ν cm−1): 3549 (O-H), 3055–2796 (C–H), 2394 (B–H), 1596, 1455, 1438, 1270, 1160, 1065, 1037; 1H NMR (CDCl3, 300.13 MHz) δ 0.20–2.00 (m, 3H), 1.27 (d, J = 6.5 Hz, 3H), 1.90 (bs, 1H), 2.33 (s, 6H), 2.51 (d, J = 7.8 Hz, 3H), 4.30 (m, 1H), 4.83 (brs, 1H), 7.13 (brs, 1H), 7.17–7.24 (m, 3H), 7.28 (brs, 1H), 7.30–7.36 (m, 4H), 7.38–7.40 (m, 1H,), 7.42–7.48 (m, 3H); 31P NMR (CDCl3, 121.5 MHz) δ + 70.5 (brd, J = 82.7 Hz); 13C NMR (CDCl3, 75.0 MHz) δ 13.4, 21.4, 30.4 (d, J = 3.6 Hz), 58.1 (d, J = 9.9 Hz), 78.8 (d, J = 5.6 Hz), 126.7, 127.9, 128.2 (d, J = 10.2 Hz), 128.5, 129.9 (d, J = 10.2 Hz), 130.5 (d, J = 2.1 Hz), 131.2 (d, J = 31.5 Hz), 132.0 (d, J = 10.7 Hz), 138.0 (d, J = 10.7 Hz), 142.5; MS (EI) m/z (relative intensity) 805 (2 M+ + Na; 15), 414 (M+ + Na; 100), 392 (M+ + H; 13); HRMS (ESI) calcd for C24H31BNNaOP [M + Na+]: 392.23091; found: 392.23095; Anal. calcd for C24H31BNOP (391.302): C 73.67, H 7.99, N 3.58; found: C 73.90, H 8.02, N 3.56.
4.2.5 (Rp)-(+)-N-methyl-N-[(1R,2S)(1-hydroxy-1-phenyl-prop-2-yl]aminophenyl-i-propyl phosphine borane 12e (from (Rp)-11a)
Yield = 80%; Colorless oil; (c 0.6, CHCl3); Rf = 0.25 (CH2Cl2); IR (ν cm−1): 3510 (O-H), 2974–2874 (C-H), 2380 (B-H), 1453, 1436, 1386, 1220, 1173, 1107, 1071, 1023, 1005, 955, 914, 884, 742, 727, 698, 645, 619, 582; 1 H NMR (CDCl3, 300.13 MHz) δ 0.10–0.90 (m, 3H), 0.96 (dd, J = 17.1 and 7.2 Hz, 3H), 1.03 (d, J = 6.9 Hz, 3H), 1.09 (dd, J = 15.3 and 7.2 Hz, 3H), 2.50 (d, J = 7.2 Hz, 3H), 2.47–2.61 (m, 1H), 3.97-–4.09 (m, 1H), 4.68 (d, J = 4.8 Hz, 1H), 7.07–7.19 (m, 3H), 7.23–7.37 (m, 5H), 7.46 (m, 2H); 31P NMR (CDCl3, 121.5 MHz) δ + 76.4; 13C NMR (CDCl3, 75.0 MHz) δ 12.9 (d, J = 3.8 Hz), 17.5 (d, J = 5.3 Hz), 22.8 (d, J = 44.5 Hz), 29.9 (d, J = 3.0 Hz), 59.1 (d, J = 7.6 Hz), 79.2 (d, J = 2.3 Hz), 126.7, 128.1, 128.9, 129.0 (d, J = 2.3 Hz), 131,7 (d, J = 55.9 Hz), 131.8 (d, J = 9.1 Hz), 143.2; MS (EI) m/z (relative intensity) 352 (M+ + Na; 100), 338 (M+ - BH3 + Na; 95); HRMS (ESI) calcd for C19H29BNNaOP [M + Na]+ 352.1962; found: 352.1976.
4.2.6 (Sp)-(+)-N-methyl-N-[(1R,2S)(1-hydroxy-1-phenyl-prop-2-yl]aminophenyl-i-propyl phosphine borane 12f
A 50 mL Schlenk flask, equipped with a magnetic stirrer and a septum, was charged with 15 mL of THF and 0.75 g (3 mmol) of (2S,4S,5R)-(−)-2-isopropyl-3,4-dimethyl-5-phenyl-1,3,2-oxaza phospholidine borane 16a (major isomer). The mixture was cooled to −78 °C and 3.33 mL (6 mmol) of phenyllithium was added and stirred overnight at ambient temperature. After hydrolysis at 0 °C, the aqueous layer was extracted with CH2Cl2 and the organic phase was dried over MgSO4, filtered and concentrated in vacuo. The crude product was purified by flash chromatography using CH2Cl2 as eluent, to afford the aminophosphine borane 12f as a mixture of epimers in a 9:1 ratio (0.81 g, 82% yield). Crystallisation from hexane:isopropanol (7:3) affords 0.51 g (52%) of major isomer 12f.
(c 1.1, CHCl3); mp 130 °C; IR (ν cm−1): 3510 (OH), 2417 (BH), 2375 (BH), 2343 (BH), 1387, 1103, 1018, 999, 743, 723, 698, 585. 1H NMR (CDCl3, 300.13 MHz) δ 0.1- 0.90 (m, 3H), 0.99 (dd, J = 15.4 and 7.3 Hz, 3H), 1.03 (dd, J = 15.4 and 7.3 Hz, 3H), 1.88 (bs, 1H), 2.53 (d, J = 8.4 Hz, 3H), 2.63 (dhept, J = 16.3 and 7.1 Hz, 1H), 4.11 (dhept, J = 6.8 and 1.2 Hz, 1H), 4.78 (d, J = 5.2 Hz, 1H), 7.24- 7.52 (m, 8H), 7.70–7.76 (m, 2H); 31P NMR (CDCl3, 121.5 MHz) δ + 76.3 (brd, J = 88 Hz); 13C NMR (CDCl3, 75.0 MHz) δ 12.1, 15.6, 16.8 (d, J = 5.4 Hz), 21.5 (d, J = 47.4 Hz), 29.1 (d, J = 3.5 Hz), 58.0 (d, J = 7.0 Hz), 78.7 (d, J = 6.3 Hz), 126.4, 127.6, 128.3, 128.4, 128.5, 130.5 (d, J = 47.8 Hz), 130.9 (d, J = 2.3 Hz), 131.4 (d, J = 7.1 Hz), 142. HRMS (ESI) calc. for C19H29BNNaOP [M+ + Na] 352.19755; found 352.19823.
4.3 Preparation of the chlorophenylphosphine borane 14
4.3.1 General procedure
In a 50 mL two-necked flask equipped with a magnetic stirrer, an argon inlet and a septum was introduced 2 mmol of the aminophosphine borane 12. A solution of HCl in toluene was then added under stirring at room temperature, without previous dissolution of the compound 12 (see conditions, Table 1). After reaction (Table 1), the precipitate of ephedrine hydrochloride was filtred off on Millipore 4 μm filter, and the excess of HCl was removed by several vacuum/argon cycles. The toluene solution of chlorophosphine borane 14 obtained was immediately used without further purification in highly stereoselective synthesis. For their analyses, the chlorophosphine boranes 14 were obtained after evaporation of toluene and purification by filtration on a short column of silica gel.
4.3.2 (R)-(+)-Chloromethylphenylphosphine borane 14a
14a was prepared from the aminophosphine borane 12a derived from the (+)-ephedrine, according to the literature [19c].
4.3.3 (S)-o-Anisylchlorophenylphosphine borane 14b (from 12b derived from the (+)-ephedrine)
Yield = 99%; colorless oil; Rf = 0.80 (toluene); 1H NMR (CDCl3, 300.13 MHz) δ 0.40–2.20 (m, 3H), 3.63 (s, 3H), 6.91 (dd, J = 8.3 and 4.6 Hz, 1H), 7.11 (td, J = 7.6 and 2.6 Hz, 1H), 7.39–7.61 (m, 4H), 7.72–7.82 (m, 2H), 7.95 (ddd, J = 14.9, 7.7 and 1.6 Hz, 1H); 31P NMR (CDCl3, + 121.5 MHz) δ + 91.9 (brd, J = 51.4 Hz); 13C NMR (CDCl3, 75.0 MHz) δ 55.7, 112.0 (d, J = 4.4 Hz), 117.6 (d, J = 47.7 Hz), 121.1 (d, J = 12.3 Hz), 128.5 (d, J = 11.5 Hz), 128.7 (d, J = 50.8 Hz), 131.1 (d, J = 12.9 Hz), 132.0 (d, J = 2.6 Hz), 134.6 (d, J = 14.3 Hz), 135.5 (d, J = 1.8 Hz), 161.1 (d, J = 2.4 Hz); MS (EI) m/z (relative intensity): 263 (M-H+; 9), 250 (M+-BH3; 100), 215 (40), 183 (35), 107 (20), 91 (40), 77 (10); HRMS (ESI) calcd for C13H12ClOP [M+-BH3]: 250.0314; found: 250.0298.
4.3.4 (S)-Chloroferrocenylphenylphosphine borane 14c (from 12c derived from the (+)-ephedrine)
Yield = 80%; orange oil; Rf = 0.50 (petroleum ether/AcOEt [3:1]); 1H NMR (CDCl3, 300.13 MHz) δ 0.60–1.80 (m, 3H), 4.20 (s, 5H), 4.44–4.49 (m, 2H), 4.54–4.55 (m, 1H), 4.56–4.64 (m, 1H), 7.41–7.48 (m, 3H), 7.77–7.84 (m, 2H); 31P NMR (CDCl3, 121.5 MHz) δ + 93.3 (brs); 13C NMR (CDCl3, 75.0 MHz) δ 70.6, 71.3 (d, J = 60.0 Hz), 71.4 (d, J = 10.2 Hz), 72.7 (d, J = 9.0 Hz), 72.8 (d, J = 8.6 Hz), 73.1 (d, J = 15.3 Hz), 128.7 (d, J = 11.0 Hz), 131.2 (d, J = 12.2 Hz), 132.4 (d, J = 2.4 Hz), 132.4 (d, J = 50.7 Hz); MS (EI) m/z (relative intensity): 346 (M-Cl + O + Na+; 15), 333 (M-BH3-Cl + O + Na+; 100); HRMS (ESI) calcd for C16H17BClNaFeP [M+ + Na]: 365.0095; found: 365.0090.
4.3.5 (S)-Chlorophenyl-m-xylylphosphine borane 14d (from 12d derived from the (+)-ephedrine)
The resulting crude solution of chlorophosphine borane 14d was used without further purification. 31P NMR (CDCl3, 121.5 MHz) δ + 93.5 (brs).
4.3.6 (S)-Chlorophenyl-i-propylphosphine borane 14e (from 12e derived from the (−)-ephedrine)
Yield = 78%; colorless oil; Rf = 0.60 (petroleum ether/AcOEt [3:1]); 1H NMR (CDCl3, 300.13 MHz) δ 0.50–1.60 (m, 3H), 1.12 (dd, J = 19.1 and 7.1 Hz, 3H), 1.28 (dd, J = 17.7 and 7.0 Hz, 3H), 2.39–2.51 (m, 1H), 7.49–7.62 (m, 3H), 7.83–7.90 (m, 2H); 31P NMR (CDCl3, 121.5 MHz) δ + 113.4 (brs); 13C NMR (CDCl3, 75.0 MHz) δ 16.8, 17.2 (d, J = 3.0 Hz), 33.2 (d, J = 24.9 Hz), 129.5 (d, J = 10.6 Hz), 132.3 (d, J = 11.3 Hz), 133.3 (d, J = 2.3 Hz), 133.6 (d, J = 51 Hz); MS (EI) m/z (relative intensity): 191 (M-Cl-BH3 + O + Na+; 100).
The (R)-Chlorophenyl-i-propylphosphine borane 14e, which was prepared from 12f and the (−)-ephedrine, exhibits satisfactory analytical data in agreement with the (S)-enantiomer described before.
4.4 Preparation of phosphine boranes 15 from chlorophosphine boranes 14 (Route A)
4.4.1 General procedure
To a solution of chlorophosphine borane 14 in toluene cooled at −78 °C was added the organolithium reagent (2.5 equiv). The reaction mixture was let to warm to RT during 1 h, then hydrolysed with water (20 mL). The organic phase was removed and the aqueous layer was extracted with CH2Cl2, and the combined extracts were dried over MgSO4, then concentrated. The residue was purified by chromatography on a short column of silica gel with toluene/petroleum ether: 7/3 as eluent, to afford the phosphine boranes 15. Their recrystallisation in a mixture hexane/i-PrOH, affords the enantiomerically pure phosphine boranes 15.
4.4.2 (S)-(+)- and (R)-(−)-o-Anisylmethylphenylphosphine borane 15a
15a were prepared from 14a and 14b respectively, according to the literature [19a]. This compound exhibit satisfactory analytical data in agreement with the recent ones reported in § 4.5.2.
4.4.3 (S)-(−)-o-Anisylferrocenylphenylphosphine borane 15b (from 12c derived from the (+)-ephedrine)
Yield = 79%; orange solid; (c 1.0, CHCl3) for 98% e.e.; mp = 140 °C; Rf = 0.46 (toluene); 1H NMR (CDCl3, 300.13 MHz) δ 0.50–1.60 (m, 3H), 3.47 (s, 3H), 4.02 (s, 5H), 4.47–4.54 (m, 3H), 4.67–4.70 (m, 1H), 6.86–6.89 (m, 1H), 7.03–7.09 (m, 1H), 7.31–7.39 (m, 3H), 7.46–7.56 (m, 3H), 7.75–7.82 (m, 1H); 31P NMR (CDCl3, 121.5 MHz) δ + 14.9 (brs); 13C NMR (CDCl3, 75.0 MHz) δ 55.9, 69.1 (d, J = 70.3 Hz), 70.4, 72.0 (d, J = 8.2 Hz), 72.3 (d, J = 7.6 Hz), 74.0 (d, J = 8.4 Hz), 74.5 (d, J = 11.9 Hz), 111.3 (d, J = 63.4 Hz), 112.4 (d, J = 4.4 Hz), 120.8 (d, J = 25.9 Hz), 121.5 (d, J = 11.5 Hz), 128.5 (d, J = 10.5 Hz), 129.1 (d, J = 28.4 Hz), 130.5 (d, J = 2.4 Hz), 131.9 (d, J = 9.9 Hz), 133.2 (d, J = 62.8 Hz), 134.1 (d, J = 10.7 Hz), 136.1 (d, J = 12.3 Hz), 161.5; Anal, calcd for C23H24FeBOP (414.075): C 66.72, H 5.84; found: C 66.96, H 6.01.
The enantiomeric excess of o-anisylferrocenylphenylphosphine borane 15b was determined by HPLC analysis on a Chiralcel AD column, eluent: hexane/i-PrOH (98:2), 1 mL/min, λ = 254 nm: (R)-15b, tR = 11.6 min; (S)-15b, tR = 12.3 min.
The (R)-(+)-o-anisylferrocenylphenylphosphine borane 15b, prepared from 12b derived from the (+)-ephedrine, exhibits satisfactory analytical data in agreement with the (S)-enantiomer: Yield = 71%; (c 0.6, CHCl3) for 95% e.e.
4.4.4 (R)-(−)-o-Anisylphenyl-m-xylylphosphine borane 15c (from 12d derived from the (+)-ephedrine)
Yield = 82%; white solid; (c 1.0, CHCl3) for 99% e.e.; mp: 174–176 °C; Rf = 0.46 (toluene); 1H NMR (CDCl3, 300.13 MHz) δ 0.60–1.80 (m, 3H), 2.32 (s, 6H), 3.57 (s, 3H), 6.92–6.96 (m, 1H), 7.05–7.22 (m, 2H), 7.26–7.28 (m, 2H), 7.41–7.67 (m, 7H); 31P NMR (CDCl3, 121.5 MHz) δ + 18.1 (brs); 13C NMR (CDCl3, 75.0 MHz) δ 22.0, 55.9, 112.4 (d, J = 4.6 Hz), 117.8 (d, J = 56.5 Hz), 121.8 (d, J = 11.3 Hz), 128.9 (d, J = 10.4 Hz), 129.5 (d, J = 59.9 Hz), 130.6 (d, J = 60.3 Hz), 131.1 (d, J = 9.9 Hz), 131.2 (d, J = 2.4 Hz), 133.2 (d, J = 2.5 Hz), 133.5 (d, J = 9.8 Hz), 134.3 (d, J = 1.9 Hz), 136.7 (d, J = 11.5 Hz), 138.5 (d, J = 11.0 Hz), 162.1; HRMS (ESI) calcd for C21H24BOP: 334.1733; found: 334.1746. Anal, calcd for C21H24BOP (334.166): C 75.45, H 7.19; found: C 75.50, H 7.41.
The enantiomeric excess of o-anisylphenyl-m-xylylphosphine borane 15c was determined by HPLC analysis on a Chiralcel OK column, eluent: hexane/i-PrOH 80:20, 40 °C, 0.5 mL/min, λ = 254 nm: (R)-15c, tR = 9.00 min; (S)-15c, tR = 17.35 min.
The (S)-(+)-o-anisylphenyl-m-xylylphosphine borane 15c, prepared from 12b derived from the (+)-ephedrine, exhibits satisfactory analytical data in agreement with the (R)-enantiomer: Yield = 79%; (c 1.0, CHCl3) for 99% e.e..
4.4.5 (S)-(+)-Methylphenyl-i-propylphosphine borane 15d (from 12f derived from the (−)-ephedrine)
Yield = 86%; colorless oil; (c 1.4, CHCl3) for 96% e.e.; Rf = 0.75 (petroleum ether/ ethyl acetate [10:1]); IR (ν cm−1) 3207, 2963, 2925, 2873, 2374 (BH), 1463, 1437, 1416, 1262, 1096, 1020, 799, 553, 540, 530; 1H NMR (CDCl3, 300.13 MHz) δ 0.10–0.90 (m, 3H), 1.02 (dd, J = 15.9 and 7.2 Hz, 3H), 1.15 (dd, J = 15.6 and 7.2 Hz, 3H), 1.54 (d, J = 10.2 Hz, 3H), 2.02–2.11 (m, 1H), 7.46–7.48 (m, 3H), 7.68–7.73 (m, 2H); 31P NMR (CDCl3, 121.5 MHz) δ + 16.4 (q, JPB = 59.5 Hz); 13C NMR (CDCl3, 75.0 MHz) δ 8.7 (d, J = 38.2 Hz), 17.2, 26.7 (d, J = 36.1 Hz), 129.3 (d, J = 9.4 Hz), 129.5 (d, J = 52.4 Hz), 131.8 (d, J = 2.4 Hz), 132.6 (d, J = 8.6 Hz); MS (EI) m/z (relative intensity): 203 (M + Na+; 100); HRMS (ESI) calcd for C10H18BNaP: 203.1133; found: 203.1120.
The enantiomeric excess of the phosphine borane 15d was determined by 31P NMR (CDCl3, 121.5 MHz) of the phosphine oxide derivative prepared according reference [24b], in presence of the Kagan's reagent (S)-21 [31]: δ + 44.37 for (S)-15d and + 44.26 for (R)-15d.
The (R)-(−)-methylphenyl-i-propylphosphine borane 15d, prepared from 12e derived from the (−)-ephedrine, exhibits satisfactory analytical data in agreement with the (S)-enantiomer: Yield = 68%; 89% e.e.
4.5 Preparation of phosphine boranes 15 from phosphide boranes 9 (Route B)
4.5.1 General procedure
The solution of chlorophosphine borane 14, prepared according to the procedure described on § 4.3, was cooled at −85 °C and t-butyllithium (0.75 mmol, 3 equiv.) was added dropwise over 2 minutes. After stirring for 5 min at −85 °C, dry THF (0.5 mL) was added dropwise over 30 seconds and the reaction mixture turns deep yellow. An excess of methyl iodide (0.75 mmol, 3 equiv.) was finally added in once. The white mixture was stirred for 10 minutes at low temperature before being hydrolyzed (5 mL of water). The aqueous phase was extracted with ethyl acetate (1 × 10 mL) and with dichloromethane (2 × 10 mL), the combined organic layers were dried over MgSO4 and the solvent was removed.
4.5.2 (S)-(+)-o-Anisylmethylphenylphosphine borane 15a (from 12b derived from the (+)-ephedrine)[19a]
Yield = 75%; white crystals (hexane/dichloromethane); [α]25 = + 25.8 (c 1.3, MeOH) for 92% e.e.; mp = 76–77 °C; Rf = 0.55 (toluene); 1 H NMR (CDCl3, 300.13 MHz) δ 0.40–1.50 (m, 3H), 1.94 (d, J = 10.6 Hz, 3H), 3.67 (s, 3H), 6.88 (dd, J = 8.3 and 3.5 Hz, 1H), 7.05 (t, J = 7.4 Hz, 1H), 7.33–7.56 (m, 4H), 7.56–7.69 (m, 2H), 7.87 (ddd, J = 13.8, 7.6 and 1.6 Hz, 1H); 31P NMR (CDCl3, 121.5 MHz) δ + 9.2 (q, J = 66.1 Hz). The enantiomeric excess of o-anisylmethylphenylphosphine borane 15a was determined by HPLC analysis on a Chiralcel OK column, eluent: hexane/i-PrOH 80:20, 1 mL/min, 40 °C, λ = 210 nm: (R)-15a, tR = 11 min; (S)-enantiomer, tR = 21 min.
4.5.3 (S)-(+)-or (R)-(−)-Methylphenyl-i-propylphosphine borane 15d
15d, prepared from 12e or 12f, respectively, exhibits satisfactory analytical data in agreement with the ones described in § 4.4.5. S)-(+)-15d: yield = 73%; 96% e.e.; (R)-(−)-15d: Yield = 71%; (c 1.2, CHCl3) for 97% e.e.
4.6 Preparation of phosphines from their borane complexes
General procedure: In a 50 mL two-necked flask equipped with a magnetic stirrer and an argon inlet, 1 mmol of phosphine borane 15 and 3 equiv. of DABCO were dissolved in 2 mL of toluene. The reaction mixture was heated at 45 °C overnight. After cooling, the crude mixture was purified by chromatography on neutral alumina, using toluene as eluent.
4.6.1 (R)-(+)-o-Anisylmethylphenylphosphine 13a
Oil; [α]D + 45.7 (c 1.7, MeOH); Rf = 0.54 (toluene/hexane 8:2); IR (ν cm−1) 3068, 3001, 2964, 2906, 1584, 1574, 1474, 1462, 1295, 1268, 1241, 1179, 1073, 1043, 882, 753, 696. 1H NMR (CDCl3, 250 MHz), δ 1.55 (d, J = 4.8, 3H), 3.64 (s, 3H), 6.70–6.80 (dd, 1H), 6.81 (t, J = 9 Hz, 1H), 7.00–7.10 (m, 1H), 7.15–7.30 (m, 4H), 7.35–7.47 (m, 2H); 31P NMR (CDCl3, 101 MHz) δ −36 (s); 13C NMR (CDCl3, 63 MHz), δ 10.8 (d, J = 13.3). 55.1 (s), 110.0 (s), 120.6 (s), 127.9 (d, J = 6.7 Hz), 129.5 (s), 131.1 (s), 131.8 (d, 9.5 Hz), 139.0 (brs), 160.6 (d, J = 2.8 Hz).
4.6.2 (S)-(−)-o-Anisylferrocenylphenylphosphine 13b
Oil; [α]D −4.8 (c 1.0, CHCl3); 1H NMR (CDCl3, 300 MHz), δ 3.63 (s, 3H), 3.75–3.76 (m, 1H), 4.03 (s, 5H), 4.19–4.20 (m, 1H), 4.24–4.25 (m, 1H), 4.31–4.32 (m, 1H), 6.73–6.84 (m, 3H), 7.16–7.26 (m, 4H), 7.36–7.40 (m, 2H). 31P NMR (CDCl3,125 MHz) δ −23.3 (s).
4.6.3 (S)-(−)-o-Anisylphenyl-m-xylylphosphine 13c
Oil; [α]D −3.8 (c 0.9, CHCl3); 1H NMR (CDCl3, 300 MHz), δ 2.28 (s, 6H), 3.78 (s, 3H), 6.62–6.71 (m, 1H), 6.84–6.97 (m, 5H), 7.24–7.36 (m, 6H); 31P NMR (CDCl3, 125 MHz) δ −16.9 (s); 13C NMR (CDCl3, 75.5 MHz), δ 21.3, 55.7, 110.2 (d, J = 1.8 Hz), 121.0, 125.8 (d, J = 11.4 Hz), 128.3 (d, J = 6.9 Hz), 128.4, 130.3 (d, J = 22.8 Hz), 130.6 (d, J = 21.7 Hz), 133.7, 133.8 (d, J = 19.4 Hz), 136.1 (d, J = 9.1 Hz), 137.0 (d, J = 10.2 Hz), 137.7 (d, J = 8 Hz), 161.2 (d, J = 15.3 Hz).
4.6.4 (S)-(−)- Methylphenyl-i-propylphosphine 13d,
Oil; [α]D −10 (c 0.3, CHCl3) for 93% e.e.; 1H NMR (CDCl3, 300 MHz), δ 0.93 (dd, 3H, J = 7 and 14.8 Hz), 1.05 (dd, 3H, J = 7 and 13.7 Hz), 1.30 (d, 3H, J = 13.7 Hz), 1.78 (dhept, 1H, J = 7 and 4.3 Hz), 7.31–7.38 (m, 3H), 7.45–7.53 (m, 2H); 31P NMR (CDCl3, 125 MHz) δ −19.3 (s); 13C NMR (CDCl3, 75.5 MHz), δ 8.6 (d, J = 15.1 Hz), 18.7 (d, J = 14 Hz), 19.4 (d, J = 15.3 Hz), 28.5 (d, J = 8.2 Hz), 128.1 (d, J = 7.6 Hz), 128.6, 132.3 (d, J = 18.1 Hz), 139.1 (d, J = 15.1 Hz).
4.7 Preparation of 1,2-bis(methylphenylphosphino)ethane 4
4.7.1 Synthesis of 1,2-bis(methoxyphenylphosphino borane)ethane 18
The diphosphinite diborane 18 was prepared by coupling the α-carbanion derived from the (O-methyl)methylphenylphosphinite borane 10a, in presence of CuCl2 under air, according to the procedure described in reference [23].
In a 50 mL two-necked flask equipped with a magnetic stirrer and an argon inlet, 1 mmol of 10a was dissolved in 2 mL of dry THF at −78 °C. Then, 1 mmol of s-butyllithium was slowly added under stirring. The temperature of the mixture was kept at −78 °C for 15 min, then allowed to warm at −30 °C. After 1 hour in these conditions, dry CuCl2 (1.1 mmol) was added and the temperature was brought to 0 °C and allowed under air. After 12 hours, the mixture was hydrolyzed by HCl 10%, and extracted with CH2Cl2. The solvent was removed and the residue purified by chromatography on silica gel using hexane/CH2Cl2 (6:4) as eluent.
The (S,S)-(−)-18 was prepared as previously described starting from (−)-ephedrine [22].
The (R,R)-(+)-1,2-Bis(methoxyphenylphosphino borane)ethane 18 was prepared starting from (+)-ephedrine: yield = 80%; solid; (c 1.3, CHCl3); mp = 115 °C; Rf = 0.48 (hexane/CH2Cl2 [55:45]); 1H NMR (CDCl3, 300.13 MHz) δ 0.00–1.50 (m, 6H), 1.91–2.07 (m, 2H), 2.07–2.24 (m, 2H), 3.58 (dd, J = 6.0 and 6.0 Hz, 6H), 7.45–7.59 (m, 6H), 7.59–7.73 (m, 4H); 31P NMR (CDCl3, 121.5 MHz) δ + 118.1 (brd, J = 58.0 Hz); 13C NMR (CDCl3, 75.0 MHz) δ 23.3 (d, J = 43.0 Hz), 54.1, 128.2–132.4.
4.7.2 Synthesis of 1,2-bis(methylphenylphosphino borane)ethane 19 [13a]
In a 50 mL two-necked flask equipped with a magnetic stirrer and an argon inlet, 1 mmol of diphosphinite borane 18 was dissolved in 2 mL of dry THF at −78 °C. Then, 2 mmol of the methyl lithium reagent was added dropwise under stirring. The temperature of the mixture was kept at −78 °C for 15 min, then allowed to warm at 0 °C. The mixture was hydrolyzed and extracted with CH2Cl2. The organic phase was dried over MgSO4 and the solvent evaporated. The residue was purified by flash chromatography on silica gel using hexane/dichloromethane as eluent.
The (S,S)-(+)-19 was prepared starting from (R,R)-(+)-18 derived from (+)-ephedrine: yield = 95%; solid; mp = 167 °C (i-PrOH); Rf = 0.44 (hexane/CH2Cl2 [55:45]); (c 1, CHCl3); IR (KBr, ν cm−1): 3056, 2909 (C-H), 2378 (B-H), 1457, 1420, 1063, 739, 693; 1H NMR (CDCl3, 300.13 MHz) δ 0.00–1.40 (m, 6H), 1.53 (brd, J = 9.8 Hz, 6H), 1.80–1.88 (m, 2H), 1.96–2.04 (m, 2H), 7.39–7.51 (m, 6H), 7.54–7.64 (m, 4H); 31P NMR (CDCl3, 121.5 MHz) δ + 12.9 (brs); 13C NMR (CDCl3, 75.0 MHz) δ 10.9 (d, J = 38.0 Hz), 20.7 (d, J = 35.0 Hz), 128.9–131.7; MS (DCI, CH4) m/z (relative intensity): 287 (60), 104 (100), 91 (80), 77 (60); Anal. calcd for C16H26B2P2 (312.17): C 63.54, H 8.67; found: C 63.58, H 8.61.
The (R,R)-(−)-19 was prepared starting from (S,S)-(−)-18 derived from the (−)-ephedrine: yield = 76%; mp = 162 °C (Hexane); (c 0.3, CHCl3); 31P NMR (CDCl3, 121.5 MHz) δ + 12.2 (brd, J = 64 Hz).
4.7.3 1,2-bis(Methylphenylphosphino borane)ethane 4 [13a]
In a 50 mL two-necked flask, with a magnetic stirrer and an argon inlet, 1 mmol of diphosphine diborane 19 and 3 equiv. of DABCO were dissolved in 2 mL of toluene. The reaction mixture was heated at 45 °C overnight. After cooling, the crude mixture was purified by chromatography on neutral alumina, using toluene as eluent.
(S,S)-(+)-1,2-bis(Methylphenylphosphino)ethane 4: yield = 95%; oil; (c 0.9, CHCl3); Rf = 0.85 (hexane/toluene/ethyl acetate [20:70:10]); IR (KBr, ν cm−1): 3071, 2961 (C-H), 2899, 1485, 1433, 1096, 1069, 1027, 879, 748, 695; 1H NMR (CDCl3, 300 MHz) δ 1.19 (d, J = 2.0 Hz, 6H), 1.53 (q, J = 3 Hz, 4H), 7.17–7.51 (m, 10H); 31P NMR (CDCl3, 125 MHz) δ – 32.0; 13C NMR (CDCl3, 75.5 MHz) δ 11.3, 25.8, 128.3–131.6; MS (DCI, CH4) m/z (relative intensity): 274 (35), 259 (65), 244 (50), 227 (65), 200 (40), 185 (30), 121 (100), 91 (50), 75 (55); HRMS (ESI) calcd for C16H20P2: 274.1040; found: 274.1041. Anal. calcd for C16H20P2 (274.10): C 70.04, H 7.35; found: C 70.03, H 7.44.
(R,R)-(−)-1,2-bis(Methylphenylphosphino)ethane 4: yield = 95%; (c 0.9, CHCl3); 31P NMR (CDCl3, 125 MHz) δ – 32 (s). 1H –NMR (CDCl3, 300 MHz), δ 1.28 (d, J = 2 Hz, 6H), 1.67 (q, J = 4 Hz, 4 H), 7.30–7.35 (m, 6 H), 7.37–7.43 (m, 4 H); 13 C NMR (CDCl3, 75.5 MHz), δ (11.4, 25.9, 128.3, 128.4, 131.4, 139.6.
The enantiomeric purity was checked by 31P NMR in presence of the chiral palladium complex 20. 31P –NMR (CDCl3, 125 MHz) δ (ppm) −32 (s),
Acknowledgments
The authors gratefully acknowledge the ANR MetChirPhos, the Ministry of Research, the CNRS and the Bourgogne country council for financial supports. We thank Miss M.J. Penouilh and E. Rémond for their technical assistance, MM. A. Tessier and P. Harvey for their help in the preparation of the manuscript.
4 The o-anisylphenyl-m-xylylphosphine 12d is referred as the ChouPhos in the laboratory.
5 To be published.