

1 Introduction
The chemistry of the pentacoordinated phosphoranes has started from the achievement of the first synthesis of pentaorganophosphorane, pentaphenylphosphorane, by Wittig in 1949 [1]. During these studies, he had found the so-called Wittig reaction, which is known as one of the famous olefin formation methods by the reactions of phosphorus ylides and carbonyl compounds [2]. Because such pentacoordinated phosphoranes formally have 10 electrons in the valence shell, that is, break the “octet” rule, these classes of compounds have attracted much attention from the viewpoint of structural chemistry. Moreover, these compounds have been recognized as the intermediate of various organic synthetic reactions. For example, an oxaphosphetane, which is a four-membered ring compound bearing a pentacoordinated phosphorus atom, has been proved to be the intermediate of the Wittig reaction, and actually been isolated and well characterized (Scheme 1). Thus, investigations of these compounds lead to the control of the reactivity as well as the elucidation of the origin of the reactivity and selectivity and it is quite useful for the development of organic chemistry, especially organic synthetic chemistry.
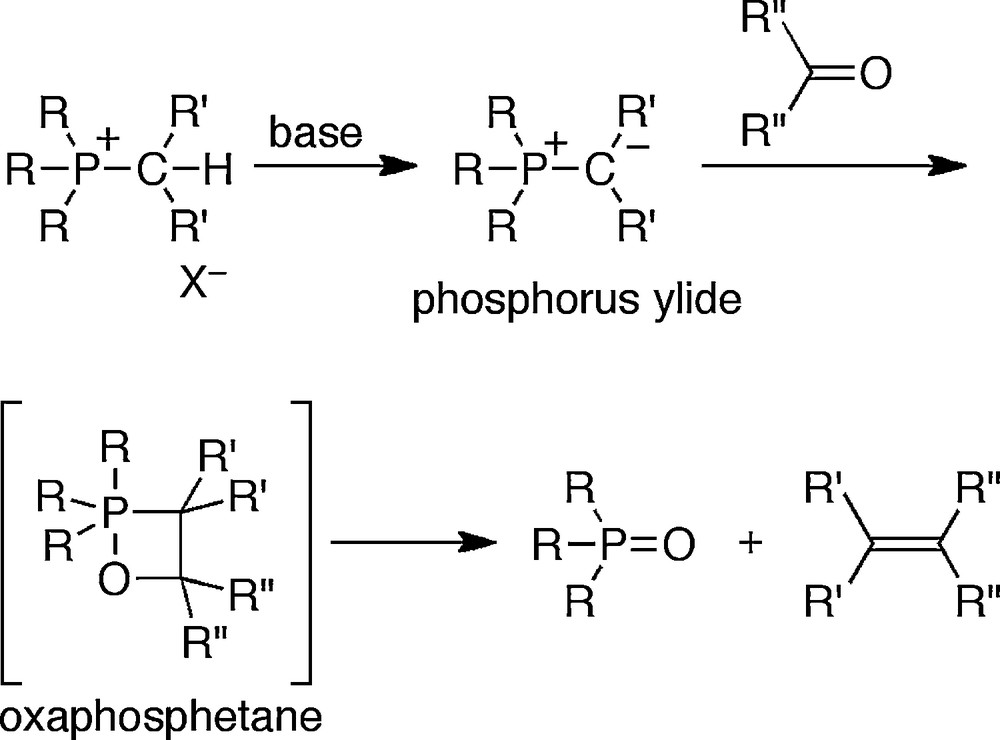
Wittig reaction.
Pentacoordinated phosphorus compounds usually take a trigonal bipyramidal (TBP) structure, in which two distinctive sites, apical and equatorial sites, exist (Fig. 1). In the TBP structure, an 1) electronegative, 2) sterically small, and 3) low π-donating ligand prefers an apical position. Such properties are called as apicophilicity and arrangement of the ligands in the pentacoordinated phosphoranes is basically decided according to the apicophilicity rule.
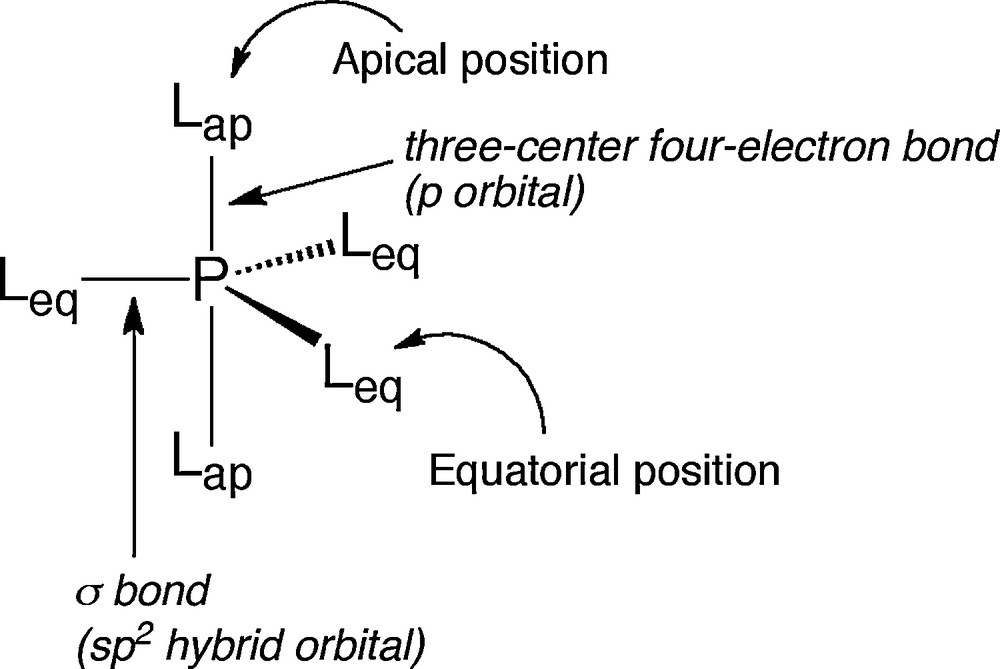
Trigonal bipyramidal structure of pentacoordinated phosphoranes.
The pentacoordinated phosphoranes, which break the apicophilicity rule, that is, phosphoranes bearing an anti-apicophilic arrangement, have been reported recently. The chemistry of such anti-apicophilic phosphoranes is also important in organic synthetic chemistry, since an anti-apicophilic isomer of oxaphosphetane is considered as the direct intermediate in the second step of the Wittig reaction; olefin formation step from oxaphosphetane [3]. This review highlights the synthesis of anti-apicophilic phosphoranes and their structures and reactivities.
2 Pentacoordinated phosphoranes
2.1 Bonding properties
Since pentacoordinated phosphoranes formally have 10 electrons in the valence shell, they show a specific bonding model. As described above, pentacoordinated phosphoranes take a TBP structure and there are two ligating sites, apical and equatorial sites. The apical bond consists of three-center four-electron bond using the p orbital of the central phosphorus atom, while the equatorial bond is a typical σ bond using sp2 hybrid orbital of the phosphorus atom (Fig. 1) [4]. This three-center four-electron bond forms three molecular orbitals as shown in Fig. 2; 1) bonding orbital, in which three atomic orbitals overlap, 2) non-bonding orbital, in which there is a node on the central atom and atomic orbitals of ligands have opposite phase, 3) anti-bonding orbital, in which all phase of atomic orbitals are arranged in parallel fashion. The HOMO of three-center four-electron bond is corresponding to the non-bonding orbital, so that negative charge is localized on the apical ligands [5].
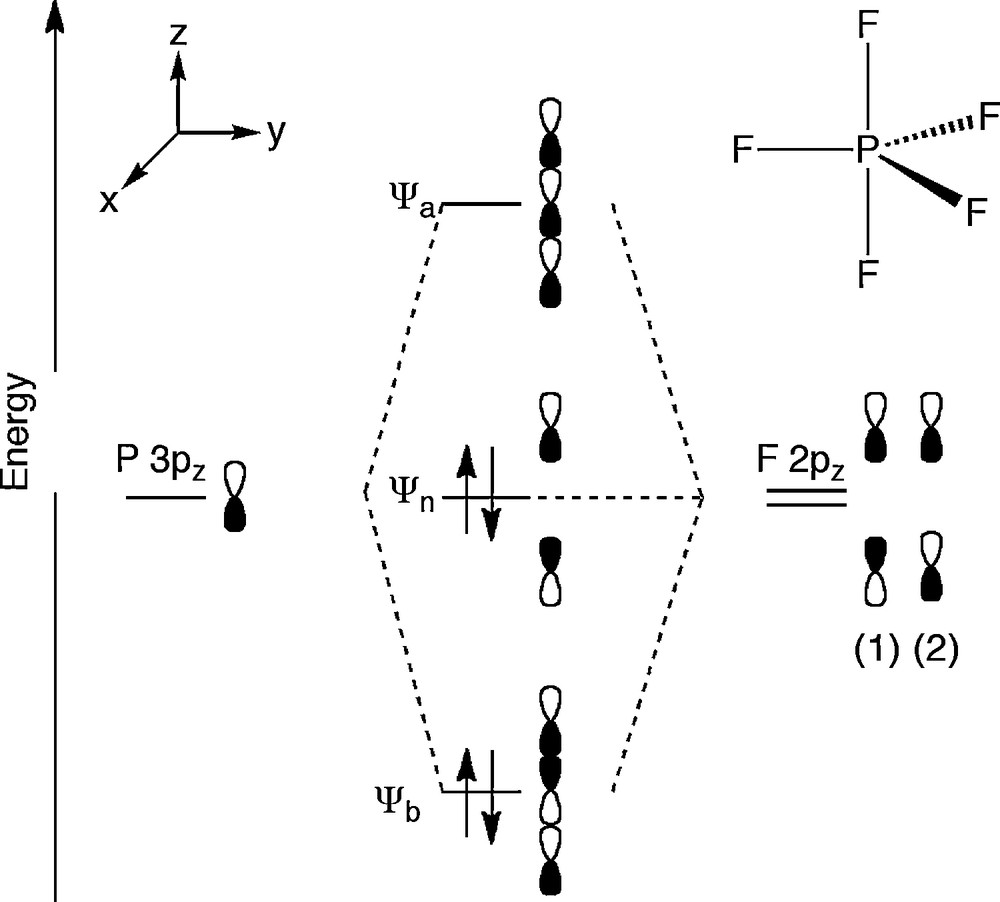
Molecular orbital of apical bonds of PF5.
2.2 Apicophilicity rule
Resulting from such bonding properties, an electronegative atom or electron-withdrawing substituent prefers the apical position to stabilize the negative charge localized at the apical ligands. Such a property, that ligands prefer the apical position, is called apicophilicity. The apicophilicity is decided by electron-withdrawing properties of ligand as well as steric hindrance and π-donating ability of ligands.
The bond angle between apical and equatorial bonds is only 90°, while that between equatorial bonds is 120°. Therefore, bulky substituents prefer the equatorial position to avoid the steric repulsion. π-donating ligands also prefer the equatorial position, because they can interact with the anti-bonding orbital (σ* orbital) of the equatorial bonds (vide infra).
The order of the apicophilicity should be decided by these features correlating with each other, so that the definition of the apicophilicity order among various ligands is quite difficult task. However, several investigations based on the experimental or theoretical examinations have been reported so far [6]. Some examples are shown in Fig. 3.
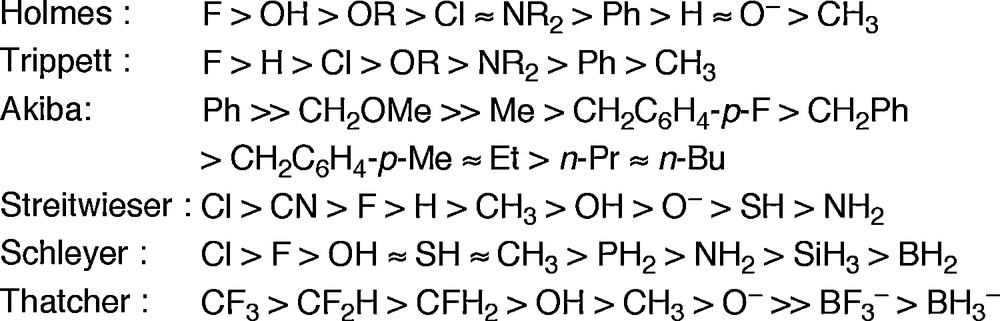
Relative apicophilicities of various substituents.
2.3 Stereoisomerizations
It is generally known that pentacoordinated phosphoranes rapidly undergo intramolecular positional isomerization without bond cleavage. A very rapid nondissociative intramolecular site exchange is usually explained by the Berry pseudorotation (BPR) mechanism [7]. The motion is illustrated in Scheme 2 where one of the equatorial ligands is taken as a pivot (3) and the originally apical bond (1, 2) undergo an angular bending from 180 to 120°, while two equatorial ligands (4, 5) bend from 120 to 180° in a plane including the pivot ligand. On this pathway, a C4v square pyramidal (SP) structure is the transition state.
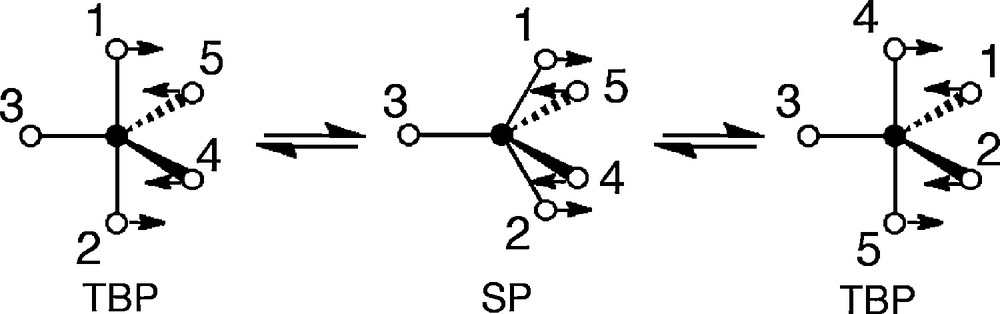
Berry pseudorotation mechanism.
In 1970, Ramirez and Ugi proposed the turnstile rotation (TR) as an alternative mechanism for the site exchange [8,9]. The motion of this process is shown in Scheme 3. Here one apical and one equatorial ligand are taken as a pair, and the other three ligands as a trio, and the two local skeletal symmetry axes of C2 of the pair and C3 of the trio are superimposed by “tilt”. Internal rotation of 60° around this axis gives a new TBP geometry.

Turnstile rotation mechanism.
The final results of BPR and TR are identical. It has been found that the BPR mechanism is more favored than the TR mechanism but the latter is not ruled out. The calculated barrier for BPR of PH5 is only 2 kcal/mol (with a C4v structure corresponding to the transition state), while that for TR is 8.2 kcal/mol. It should be noted that the energy difference is not so large, so the TR mechanism could be preferred to the BPR mechanism depending upon the substituent pattern around the phosphorus atom.
Thus, the energy barrier for the stereoisomerization based on the BPR mechanism is generally low and the stereoisomerization occurs at room temperature. Therefore, the ligands on the pentacoordinated phosphoranes are arranged to give the thermodynamically most stable isomer, according to the apicophilicity rule.
3 Synthesis of anti-apicophilic phosphoranes
As described in the previous section, pentacoordinated phosphorus compounds undergo rapid stereoisomerization through BPR mechanism or TR mechanism, so that suitable molecular design is required for the synthesis and isolation of the pentacoordinated phosphoranes with an anti-apicophilic arrangement.
3.1 Combination of two bidentate ligands
Various bidentate ligands based on a five-membered ring structure have been widely used for the stabilization of pentacoordinated compounds. The so-called Martin ligand (Fig. 4) is a well-known ligand, which has achieved the synthesis of various pentacoordinated compounds, because it is based on a five-membered ring structure and electronic effect is also incorporated into it.
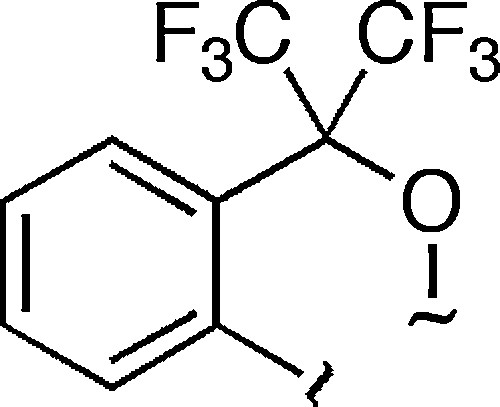
Martin ligand.
Martin et al. reported the synthesis of spirobihydrophos-phorane 1 bearing two Martin ligands [10]. On the other hand, Akiba et al. have reported the anti-apicophilic stereoisomer of 1, in which one of the two Martin ligands are directed to the opposite direction [11]. Starting from 1, the reaction with 2 equivalents of n-butyllithium led to the introduction of butyl group on the phosphorus atom and cleavage of one of the five-membered rings giving hydrophosphorane 2a in 90% yield with the formation of by-product 3a (Scheme 4).

Reaction of hydrophosphorane 1 with butyllithium.
Thermal reaction of 2a in refluxing toluene afforded 3a, quantitatively. When 2a was treated with 2 equivalents of pyridine in tetrahydrofuran at 60 °C, however, the formation of a new compound 4a was confirmed by 31P NMR (Scheme 5).

Synthesis of an anti-apicophilic phosphorane.
X-ray crystallographic analysis of 4a revealed that this compound was a stereoisomer of 3a with trigonal bipyramidal structure. The stereoisomerization of 4a was also investigated and revealed that 4a was irreversibly converted to 3a and its activation enthalpy was estimated as 22 kcal/mol. Usually, the activation energy of BPR is low and BPR proceeds rapidly at room temperature; the intermediate for the stereoisomerization from 4a to 3a should be very unstable. Thus, pseudorotation was kinetically suppressed and it led to the isolation of anti-apicophilic phosphoranes. It was considered that 4a kinetically formed, since the transition state for the cyclization reaction from 2a to 4a was close to the structure of 4a (Scheme 5). Then, Akiba and Yamamoto have developed the oxidative cyclization instead of thermal cyclization reaction [12]. Treatment of 1 or 2a with nucleophiles such as methyllithium, n-butyllithium, and t-butyllithium afforded the dianionic species 5a–c, and oxidation of these intermediates by iodine gave the desired anti-apicophilic phosphoranes 4a–c (Scheme 6).

Synthesis of anti-apicophilic phosphoranes by oxidative cyclization.
Oxidation with iodine gave the best result compared to that with 30% hydrogen peroxide or m-chloroperbenzoic acid. The reaction with various aryllithiums was also investigated and succeeded in the introduction of various aromatic substituents generating anti-apicophilic arylphos-phoranes 4e–m [13a,b]. The yield of the anti-apicophilic isomer seems to be more dependent on the steric bulkiness of the aryl substituent than the electronic character. Indeed, ortho di-substituted aryl ligand led to the exclusive formation of the anti-apicophilic isomer. Similar reactions were reported for the synthesis of amino derivatives. The reaction of in-situ generated chlorophosphorane with primary amines afforded the anti-apicophilic aminophos-phoranes 4n–q (Scheme 7) [13c]. In these cases, the positive effect of steric bulkiness of the amino group on the yields of anti-apicophilic isomer was also observed.

Synthesis of anti-apicophilic aminophosphoranes.
Moreover, Akiba et al. applied this methodology to the synthesis of the anti-apicophilic isomer of the oxaphosphetane [14]. Hydrophosphorane 6 was allowed to react with n-butyllithium to generate dianion and subsequent oxidation by iodine at 0 °C afforded the anti-apicophilic isomer 7, in which the oxygen atom of oxaphosphetane ring was located at the equatorial position against the apicophilicity rule, as a 1:1 mixture with O-apical isomer 8 (Scheme 8).

Synthesis of an anti-apicophilic phosphrane bearing an oxaphosphetane ring.
The formation ratio of 7 could not be improved by changing the reaction conditions. However, anti-apicophilic 7 was isolated by recrystallization (isolated yield 18%) and its structure was determined by X-ray crystallographic analysis. In the Wittig reaction, which gave an olefin by the reaction of the phosphorus ylide with carbonyl compounds, a pentacoordinated phosphorus compound bearing an oxaphosphetane ring has been generally recognized as an intermediate. On the basis of the general belief that PC bond dissociation is more advanced than that of PO in the transition state, the C-apical isomer has been assumed by many to be the intermediate that gives the olefinic product and the isolation of 7 is the first experimental demonstration of the existence of the C-apical isomer.
The authors also reported the nitrogen analogue of 9 prior to Akiba's report [15]. They have succeeded in observing anti-apicophilic C-apical isomer 10 as a mixture with 9. There is equilibrium between 9 and 10 in solution, and its equilibrium ratio was estimated as 6:1 in toluene-d8 (Scheme 9).
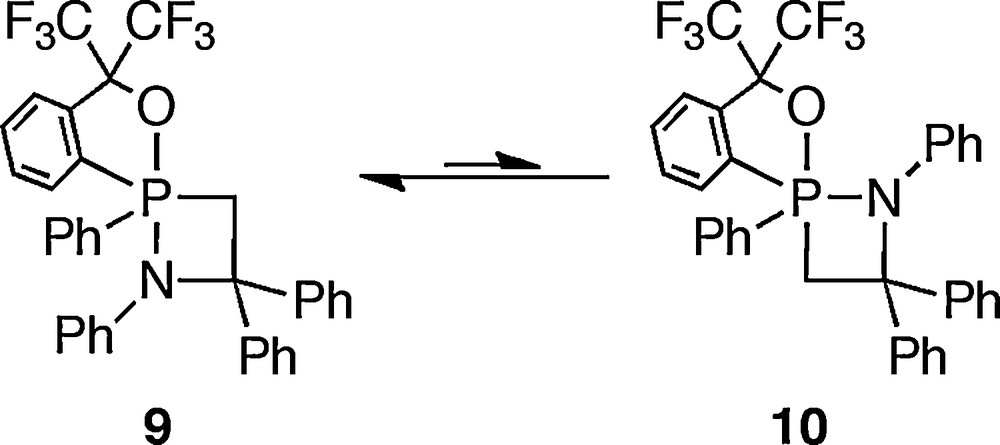
Equilibrium between stereoisomers of an azaphosphetidine.
Holmes et al. have succeeded in synthesizing of anti-apicophilic phosphoranes by using different strategy [16]. While the bidentate ligand based on a five-membered ring usually occupies one apical and one equatorial site, such a tendency is weakened as the ring size is shrunk. In the case of eight-membered ring structure, the occupation of two equatorial sites can be favored. Especially, when steric bulkiness was attached to the bidentate ligand which forms an eight-membered ring skeleton, the occupation of two equatorial sites becomes more stable than that of one apical and one equatorial site. Holmes et al. employed such an eight-membered ring bidentate ligand with the combination of a five-membered ring ligand to fix the ligand arrangement around the phosphorus atom. The synthesis of anti-apicophilic phosphoranes 11a,b was achieved by the introduction of an electropositive phenyl or ethyl group at the remaining apical position. The desired products were synthesized by the reaction of the corresponding trivalent phosphorus compound with o-chloranil (Scheme 10). When the steric bulkiness of the eight-membered ring bidentate ligand was reduced, the usual apicophilic arrangement, in which the bidentate ligand occupied one apical and one equatorial sites, is dominant. Kumara Swamy et al. reported the synthesis of t-butyl derivative bearing same molecular structure [17]. They also reported the methyl derivative and revealed that the stability of the anti-apicophilic arrangement is higher in the t-butyl derivative 11c than in the methyl derivative 11d, which exists in equilibrium with the apicophilic isomer.
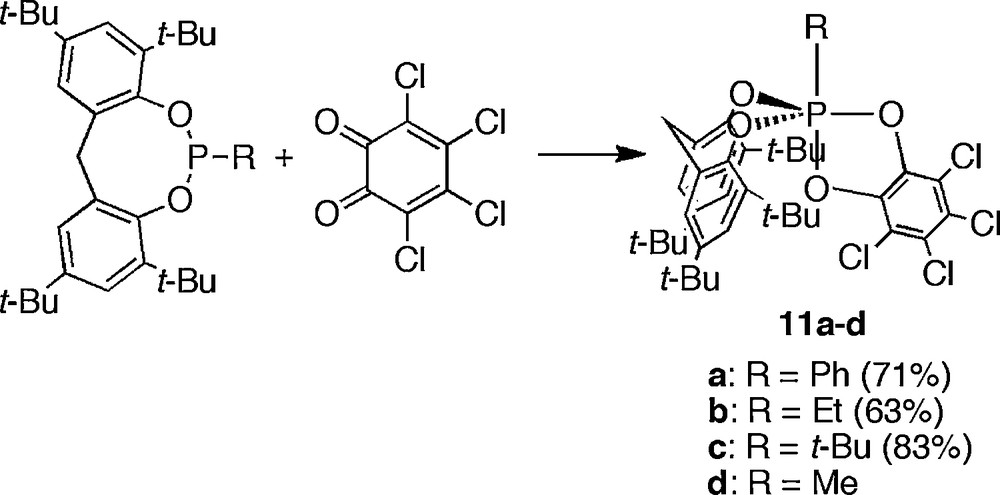
Synthesis of anti-apicophilic phosphoranes bearing an 8-membered ring structure.
3.2 Syntheses of anti-apicophilic phosphoranes bearing a tridentate ligand
du Mont et al. have reported that the reaction of diphosphane 12 with 3 equivalents of hexafluoroacetone afforded compound 13 (Scheme 11), which is corresponding to the three hexafluoroacetone adduct of formally dissociated species of 12 [18].

Reaction of diphosphane 12 with hexafluoroacetone.
The tridentate ligand prevents the BPR isomerization and it leads to the isolation of the anti-apicophilic compound. Röschenthaler succeeded in synthesizing phosphoranes 14a–c by the reaction of the phenol derivative bearing two trifluoroacetyl groups at the ortho position with dialkyl(isocyanato)phosphite (Scheme 12) [19].
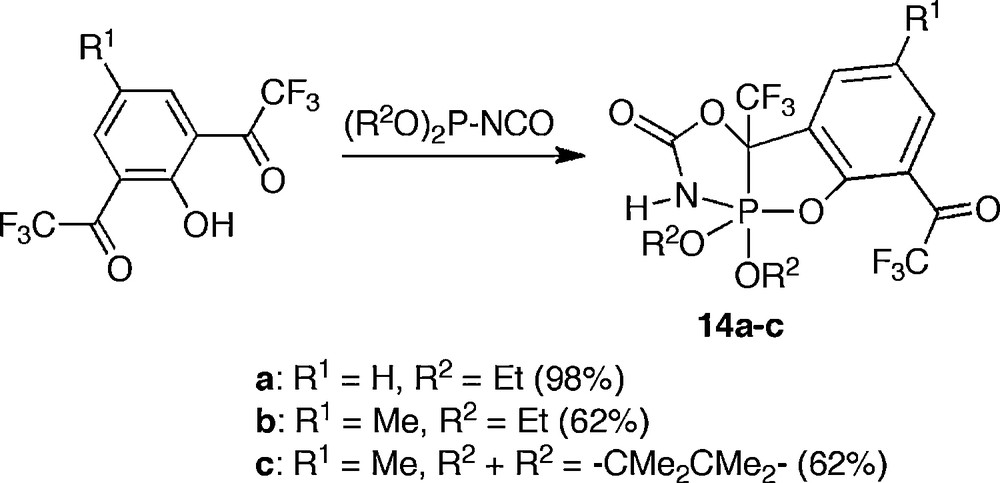
Synthesis of anti-apicophilic phosphoranes 14a-c.
Pseudorotation was suppressed, also in the cases of 14a–c, by the introduction of the tridentate ligand. The carbon atom is located at the bridgehead position of the tridentate ligand, so that 14a–c have an anti-apicophilic arrangement. Since this apical carbon atom has an electron withdrawing trifluoromethyl group, however, electronic perturbation of trifluoromethyl group cannot be neglected.
3.3 Syntheses of anti-apicophilic phosphoranes bearing a tetradentate ligand
As described above, suppression of stereoisomerization based on BPR mechanism is an important point for the synthesis of anti-apicophilic phosphoranes. The introduction of a tetradentate ligand into the phosphorus atom can completely suppress the stereoisomerization, leading to the isolation of anti-apicophilic compounds. Atranes are a class of compounds, in which the central atom takes a pentacoordinated structure, suffering from the intramolecular coordination from the nitrogen atom of a ligand, triethanolamine. Many anti-apicophilic compounds are reported in the atrane derivatives.
Milbrath and Verkade reported the reactions of 1-phospha-5-aza-2,8,9-trioxabicyclo[3.3.3]undecane (prophosphatrane) 15 with various electrophiles, giving phosphatrane derivatives bearing an anti-apicophilic arrangement. The first phosphatrane 16a was reported in 1976, and it was eventually obtained by the reaction of 15 with Meerwein reagent, because contaminated acid protonated the phosphorus atom (Scheme 13) [20].
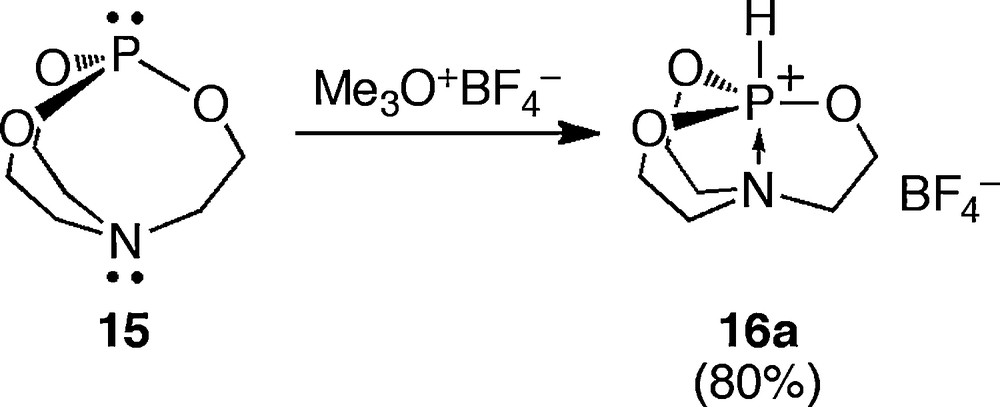
Synthesis of a phosphatrane 16a.
In addition, various alkyl halides, triphenylmethylcarbenium tetraphenylborate, borane THF complex and tungsten(CO)5 acetonitrile complex reacted with 15 to give the corresponding phosphatranes 16b–e, respectively (Fig. 5).
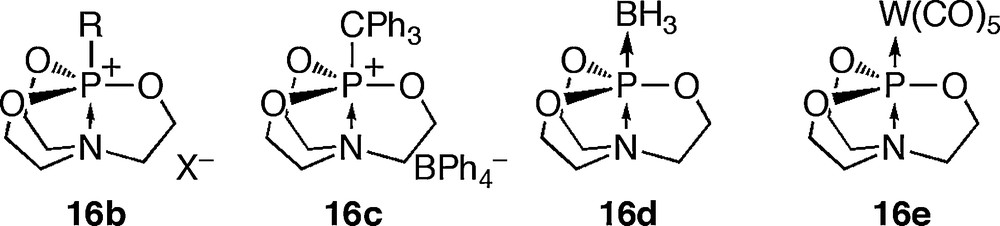
Phosphatrane derivatives 16b-e.
Although the structures of 16d and 16e are not yet fully determined, the more electropositive boron atom or transition metal is reported to introduce on the phosphorus atom.
On the other hand, the authors reported 5-carbaphosphatrane, a 5-carbon analogue of a phosphatrane, in which the nitrogen atom at the 5-position was replaced by an electropositive carbon atom. In 5-carbaphosphatrane, all equatorial positions are occupied with electronegative oxygen atoms and remaining apical positions with electropositive carbon and hydrogen atoms, that is, the structure around the phosphorus atom can be regarded as a completely anti-apicophilic arrangement. 1-Hydro-5-carbaphosphatrane 18 was prepared by the demethylation of the precursor 17 with boron tribromide (Scheme 14) [21].

Synthesis of 5-carbaphosphatrane 18.
When iodotrimethylsilane was employed as a demethylating reagent and this reaction was carried out in a sealed tube, 1-methyl-5-carbaphosphatrane 19 was obtained instead of 18 (Scheme 15).
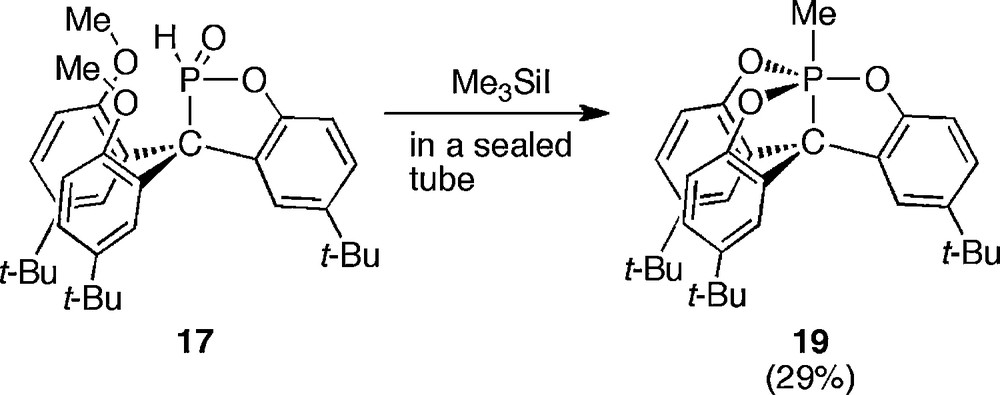
Synthesis of 1-methyl-5-carbaphosphatrane 19.
Starting from 18, the reaction with alkyllithiums or aryllithiums and subsequent thermal or oxidative cyclization afforded the corresponding 1-alkyl- or 1-aryl-5-carbaphosphatranes 20a–d (Scheme 16) [22].

Synthesis of 1-alkyl- and 1-aryl-5-carbaphosphatranes 20a-d. a yields were estimated by 1H NMR.
The authors also reported the ring expanded analogue of 5-carbaphosphatrane, 6-carbaphosphatrane [23]. Its synthesis started from the corresponding phosphonium salt 21, that is, its reduction by lithium tri(t-butoxy)aluminum hydride afforded 1-hydro-6-carbaphosphatrane 22 (Scheme 17).

Synthesis of 6-carbaphosphatrane 22.
Phosphatrane 22 existed in the equilibrium with its trivalent tautomer 23 in solution and both isomers 22 and 23 were isolated by solvent dependent recrystallization and their structures were determined by X-ray crystallographic analysis. This is the first example of isolation and structural determination of both isomers at the same time.
4 Structure
4.1 Trigonal bipyramidal structure
Pentacoordinated phosphoranes usually take a trigonal bipyramidal structure and occupation of the apical positions by an electronegative atom stabilizes the three-center four-electron bond as described in section 1.1. The structure of anti-apicophilic compounds summarized in this review is very interesting from the viewpoint of the relationship between anti-apicophilic arrangement and the geometry around the phosphorus atom. In this section, the structures of anti-apicophilic compounds will be discussed.
For the spirophosphorane 4a bearing two Martin ligands, crystallographic analysis was performed as well as on its isomer 3a with the usual apicophilic arrangement and both structures can be compared [11]. While 3a has an ideal trigonal bipyramidal structure around the phosphorus atom, the apical bond angle of 4a was 170.6° and slightly deviated from the ideal value for the trigonal bipyramidal structure (180°). However, the geometry around the phosphorus atom of 4a maintained the trigonal bipyramidal structure. Similarly, anti-apicophilic oxaphosphetane 7 also has a trigonal bipyramidal structure around the phosphorus atom [14].
Spirophosphoranes 11a–c, carbaphosphatranes 18–20, 22, and phosphoranes 14a–c reported by Röschenthaler also take an ideal trigonal bipyramidal structure [16,17,19,21–23].
On the other hand, the geometry around the phosphorus atom of 13 is strongly distorted from a trigonal bipyramidal structure, and the apical bond angle is 163°, largely deviating from its ideal value [18]. It is probably because the ligand contained in 13 is a tridentate ligand based on a fused structure of four-membered and five-membered rings and the distortion is too large to stabilize a trigonal bipyramidal structure.
Therefore, it is suggested that anti-apicophilic compounds basically take a trigonal bipyramidal structure, although employment of multidentate ligands, which can stabilize a trigonal bipyramidal structure, should be taken in the consideration. However, anti-apicophilic geometry is not considered to be unstable for breaking a trigonal bipyramidal structure.
4.2 Bond length
The apical bond in a trigonal bipyramidal structure consists of three-center four-electron bond and is accepted to be longer than a usual covalent bond. Indeed, the apical PC bond length of 4 is 1.860–1.885 Å and about 3% longer than the equatorial PC bond length (1.806–1.812 Å) [11–13] Such a tendency was also observed in oxaphosphetane 7. The apical PC bond of 7 (1.914 Å) was 5% longer than the equatorial PC bond of its isomer 8 with the usual apicophilic arrangement [14], reflecting the nature of three-center four-electron bond.
For spirophosphoranes 11a–c, their apical PC bond length is 1.838–1.879 Å, and proved to be elongated compared to the sum of covalent radii of phosphorus and carbon atoms (1.83 Å) [16,17]. For 1-t-butyl-5-carbaphosphatrane 20b, although the bond length of the PC bond incorporated in the tetradentate ligand cannot be discussed precisely, the PC bond length with the t-butyl group was estimated as 1.877 Å and elongation of the bond length was observed [22].
5 Spectral properties
5.1 31P NMR chemical shift
The chemical shift of 31P NMR provides important information about the electronic state of the phosphorus atom. Pentacoordinated phosphoranes usually give their signals in the region 0 to −100 ppm. Since the anti-apicophilic phosphoranes reviewed in this paper also show their chemical shifts in this region, the anti-apicophilic arrangement is considered to maintain the electronic properties as pentacoordinated phosphoranes. 31P NMR chemical shifts are strongly affected by the surrounding environment, that is, a kind of ligand and stereochemical structure and it is difficult to interpret them comprehensively. However, Kajiyama et al. and the authors have succeeded in the observing both isomers of spirophosphoranes bearing the Martin ligand in apicophilic and anti-apicophilic arrangement, so comparison of the chemical shifts of both isomers enables one to discuss the electronic perturbation on the phosphorus atom caused by anti-apicophilic arrangement.
The chemical shifts of spirophosphoranes reported by Akiba and Yamamoto are summarized in Table 1 [13,24].
31P NMR chemical shifts of pentacoordinated phosphoranes 3 and 4.
| 31P NMR chemical shift in Et2O (δ) | |||
| R | 3 | 4 | |
| a | n-butyl | −18.8 | −3.5 |
| b | methyl | −22.6 | −6.3 |
| c | t-butyl | −9.8 | 7.7 |
| d | benzyla | −8.0 | −22.0 |
| e | phenyl | −31.9 | −8.6 |
| f | 4-t-butylphenyl | −32.5 | −8.5 |
| g | p-anisyl | −33.1 | −8.3 |
| h | 4-(dimethylamino)phenyl | −33.8 | −9.2 |
| i | 2-methylphenyl | −26.6 | −5.6, −8.8 |
| j | 2,6-dimethylphenyl | −26.4 | −4.7 |
| k | 2,4,6-trimethylphenyl | −26.2 | −3.9 |
| l | 2,4,6-triethylphenyl | −25.8 | −3.1 |
| m | 2,4,6-triisopropylphenyl | −25.7 | −2.7 |
a Measured in CDCl3.
In all cases, the isomers 4 with an anti-apicophilic arrangement gave their signals in a downfield shifted region compared to apicophilic isomers 3. Such trend was also observed in the case of oxaphosphetane 7 (δP −6.3 ppm) and its isomer 8 (δP −10.9 ppm), although the difference was small [14]. Akiba and Yamamoto guessed that this phenomena may derive from the σ accepting property of the equatorial oxygen in the anti-apicophilic isomer.
However, azaphosphetidine reported by the authors showed the opposite property. While apicophilic isomer 9 showed its signal at −29.9 ppm, the signal of anti-apicophilic isomer 10 was observed about 20 ppm upfield (δP −50.9 ppm) [15]. The reason for the difference between oxaphosphetane and azaphosphetidine is unclear, but it is probably derived from the difference between oxygen and nitrogen atoms at the equatorial position, because these two compounds have an almost identical structure.
5-Carbaphosphatranes showed the 31P NMR signals at the downfield shifted region compared to those of usual pentacoordinated phosphoranes, since the chemical shift of 18 was 2.7 ppm and those of 19 and 20 were observed around 20 ppm, respectively [21,22]. Electronegative oxygen atoms occupied all equatorial positions and the electron density of the phosphorus center was lowered due to the σ accepting property of the oxygen atom as is the case of spirophosphoranes bearing two Martin ligands. Although three oxygen atoms of 11a–c are also located at the equatorial positions, they show their signals at the usual region for the pentacoordinated phosphorus compounds (−32.29 ppm for 11a, −23.14 ppm for 11b, −22.6 ppm for 11c) [16,17].
5.2 Coupling constants
It is well known that the one bond coupling constants (1J) are dominated by the s character of the corresponding bond. The 1J value becomes large as the s character of the bond increases. In the pentacoordinated phosphoranes, the apical bond consists of three-center four-electron bond using the p orbital of phosphorus atom, whereas the equatorial bond forms by using the sp2 hybrid orbital of phosphorus atom. Therefore, the coupling constant of apical bond is generally known to be small than that of the equatorial bond.
The one bond-coupling constant (1JPC) of the apical bond of 4a was estimated as 16 Hz, and the 1JPC value of the equatorial bond was 160 Hz, much larger than that of the apical bond [11]. The similar trend was also observed in the case of oxaphosphetanes 7 and 8. The 1JPC value of the PC bond in the oxaphosphetane ring of 7, which is the apical bond, was 29.4 Hz, whereas that of 8, corresponding to the equatorial bond, was 106.7 Hz, reflecting the bonding nature of pentacoordinated phosphoranes [14]. The apical coupling constant of 13 was 10 Hz, which is the normal value for the coupling constant of the apical bond [18].
On the other hand, spirophosphoranes 11a–c and carbaphosphatranes 18–20 and 22 showed a quite different property for the apical coupling constant (Table 2) [16,17,21–23]. For carbaphosphatranes, 1JPH values were 852 Hz (18) and 882 Hz (22), which is almost three times as much as the usual apical coupling constant for the PH bond. For carbaphosphatranes, the 1JPC for the bond between phosphorus atom and the trityl carbon atom of the tetradentate ligand was 125 to 137 Hz and the 1JPC for the opposite apical bond of alkyl and aryl derivatives 20a–d was over 200 Hz, which is extraordinarily large for the apical coupling constant of PC bond. Spirophosphorane 11c also showed a large apical coupling constant as being 192 Hz [17]. A common feature for these two classes of compounds is that both compounds have three oxygen atoms at the equatorial positions and such a trend was also observed when one of three oxygen atoms is replaced by nitrogen atom. The apical coupling constants of 14a,b was 125 Hz [19]. These results indicated that the extraordinarily large apical coupling constants should be derived from the perfectly anti-apicophilic arrangement, in which all equatorial positions are occupied with electronegative atoms such as oxygen and nitrogen atom. However, the details are still unclear and further investigation is expected.
Apical 1JPC values of anti-apicophilic phosphoranes.
| Compounds | 1JPC |
| 11c | 192 |
| 14a | 124.1 |
| 14b | 124.2 |
| 18 | 125 |
| 19 | 215, 133 |
| 20a | 207, 135 |
| 20b | 208, 137 |
| 20c | 131a |
| 20d | 248, 124 |
a 1JPC value between phosphorus atom and phenyl group was not determined.
6 Reactivity
Oxaphosphetane is an intermediate consequence of the Wittig reaction and its thermolysis is attracting much attention. Heating a solid sample of 7 at the melting point temperature (ca. 120 °C) for 5 min gave only its isomer 8 as a consequence of pseudorotation. Only after prolonged heating of 8 at the melting point (140 °C), could quantitative olefin formation be observed (Scheme 18) [14].

Thermolysis of anti-apicophilic oxaphosphetane 7.
It is apparent that the bond strength of the apical PC bond of the oxaphosphetane ring for 7 makes the bond cleavage process (Wittig reaction) much higher in energy than stereomutation.
Yamamoto and Akiba have also investigated the reactivity of anti-apicophilic phosphoranes 4a. They predicted that the nucleophilic attack on the phosphorus atom will be encouraged due to the energetically low σ*PO orbital formed in the equatorial plane and an α anion will be stabilized at the same time [24]. Indeed, 4a showed higher reactivity for the addition of fluoride or alkyllithiums than 3a (Schemes 19 and 20).

Reactions with tetrabutylammonium fluoride.
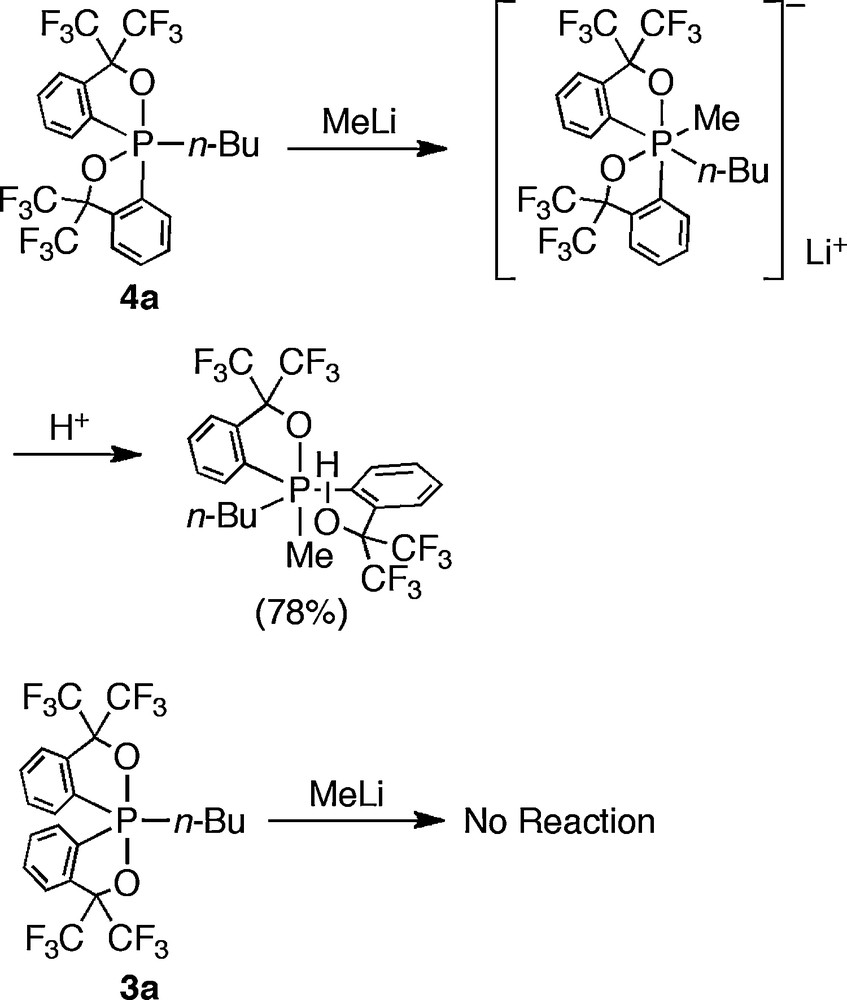
Reactions with methyllithium.
The α proton of 4d showed higher acidity than that of 3d (Scheme 21) and this can be explained by the stability of the corresponding conjugate anions.

Deprotonation of α-proton of pentacoordinated phosphoranes.
The reactions of generated anions with electrophiles showed clear differences derived from the conformation. In the reaction of anion (3d–Li) generated from usual conformer (3d) with benzoyl chloride or isobutyl chloroformate, the corresponding adducts were obtained (Scheme 22). However, in the case of the anion of 4d (4d–Li), unexpected phosphoranes were formed via rearrangement of the acylated products followed by pseudorotation. This rearrangement was induced by the enhanced electrophilicity of the σ*PO orbital (Scheme 23).
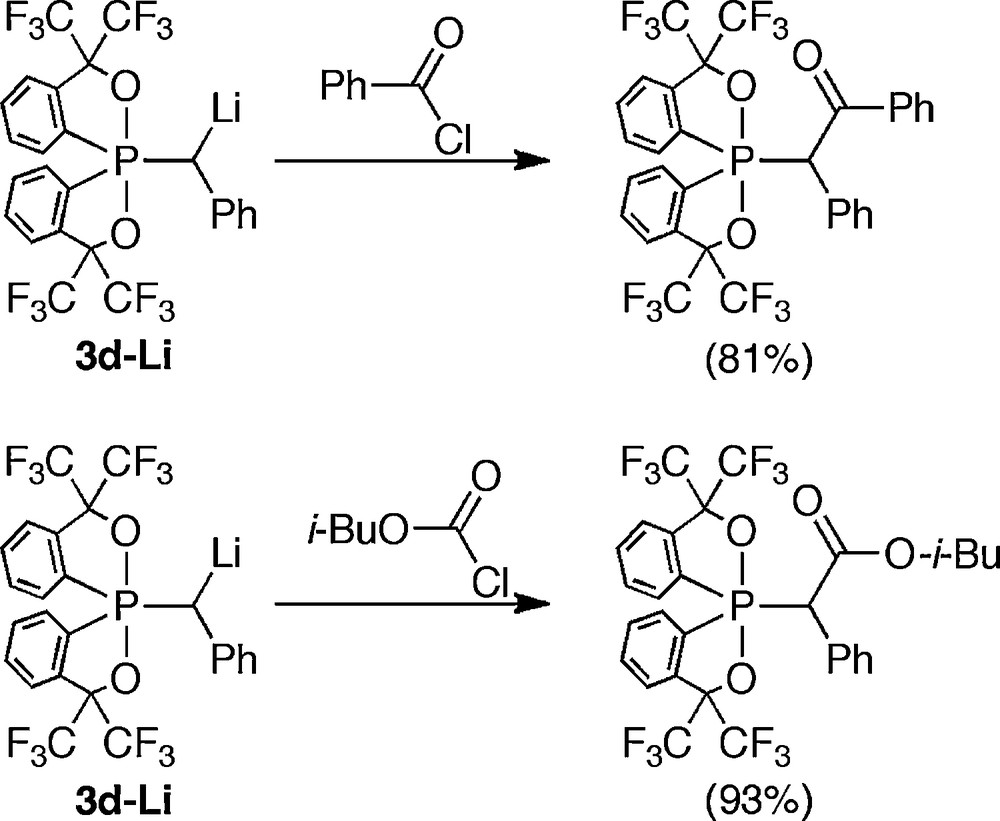
Acylation of α-anion of phosphorane 3d.
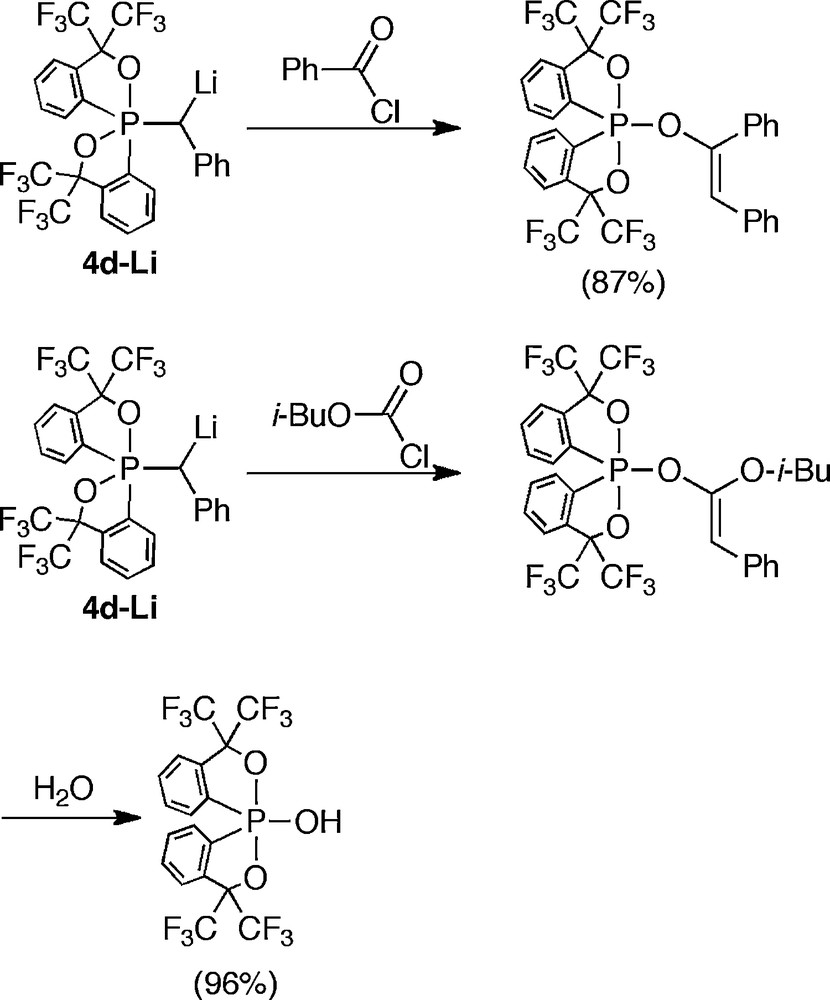
Reaction of α-anion of anti-apicophilic phosphorane.
Furthermore, the reaction of this anion and benzaldehyde gave the corresponding adduct, which forms hexacoordinated phosphate 24 bearing oxaphosphetane ring by treatment with base. The hexacoordinated oxaphosphetane 24 was isolated as a stable crystal (Fig. 6) and thermolysis of 24 lead to the quantitative formation of the corresponding stilbene, indicating that 24 can be regarded as an intermediate of the Wittig-type reaction using pentacoordinated phosphorus stabilized carbanion.

Hexacoordinated oxaphosphetane.
The enhancement of electrophilicity of anti-apicophilic phosphoranes was also recognized in 5-carbaphosphatrane 18. In the addition reaction of n-butyllithium to spirophosphorane 1, n-butyllithium worked as a base and deprotonation on the phosphorus atom occurred prior to the nucleophilic addition [11]. On the other hand, deprotonation did not occur and only nucleophilic addition proceeded in the case of 18 (Scheme 16) [22]. Thus, the electrophilicity of 18 was considered to be enhanced, because oxygen atoms were located at the equatorial positions and the acidity of the hydrogen atom on the phosphorus atom should decrease due to the fixation at the apical position.
Among the phosphatranes reported by Tang and Verkade, the reaction intermediates of the important organic synthetic reactions have been reported. Triaryl isocyanurate 28 is a trimer of aryl isocyanate and useful as an activator for the continuous anionic polymerization and postpolymerization of ɛ-caprolactam to nylon-6. Tang and Verkade have reported that proazaphosphatrane 25 worked as an efficient catalyst for the trimerization of aryl isocyanate and they found the formation of azaphosphatranes 26 and 27 bearing an anti-apicophilic arrangement during the reaction [25] (Scheme 24).

Reaction pathway for the trimerization of ArNCO catalyzed by 25.
7 Conclusion
In this review, syntheses of anti-apicophilic phosphoranes and their structures, properties and reactivities are summarized. Although these compounds are considered to be thermodynamically unstable, the syntheses of these compounds were achieved by contriving synthetic methods and molecular design. They maintained a trigonal bipyramidal structure; nevertheless, their conformation is thermodynamically disfavored. The apical coupling constant of the perfectly anti-apicophilic phosphoranes were extraordinarily large. The main reason of these phenomena is still unclear, but it can be derived from electronegative atoms located at the equatorial positions.
Characteristic reactivities of anti-apicophilic phosphoranes were also found, such as a high electrophilicity and the formation of a stable a α anion. Such reactivities originated from the high electron-accepting properties of σ*PO bond in the equatorial plane.