

1 Introduction
Since the 1960s, solid phase supported organic synthesis has proven to be one of the best systems for synthesis of libraries of compounds in a short period of time [1]. Phase discrepancy is allowing drastic simplification in purification and, despite the addition of large excesses of reagents usually used to drive reactions to completion, only filtration and washes are often necessary to obtain products with good purities. However, some limitations remain to this appealing technology, including low loading capacity and poor kinetics. Indeed, the heterogeneous nature of mixture is slowing down reactions due to poor diffusion of reagents and/or difficulties in accessing the reaction site on the resin. Moreover, monitoring reactions is somehow tricky and can require special analysis techniques such as HR MAS NMR. As an answer to this drawback, some systems have been developed in solution phase. If perfluorous systems are efficient in multiphasic reactions [2–4], other approaches using dendrimeric alcohols [5], soluble polystyrene [6–8], or polyethylene glycols were successful as well. The last, for example have been used for synthesizing peptides [9], oligosaccharides [10], or oligonucleotides [11]. Despite their success, multimeric soluble systems still have drawbacks limiting their applicability. Indeed, low loading capacity (PEG 5000 has a 0.2 mmol.g−1 loading capacity, for example) along with high water solubility prevent large scale use of PEG as support in synthesis. Furthermore, reagents and side products often co-precipitate during the purification step leading to impure products after cleavage.
An alternative to these systems has arisen recently from the field of ionic liquids. Liquid below 100 °C, ionic liquids consist only of ions conferring them a unique structure and characteristics otherwise unattainable [12]. Besides their extremely low vapor pressure, inflammability and thermal stability, their physical and chemical properties can be finely tuned by adjusting a volo the nature of the cation and the anion. Indeed, by grafting a functional group to an ionic core, one can administrate special chemical properties to an onium salt which can then serve as a reagent or reactant. These so-called Task Specific Ionic Liquids (TSILs) [13] possess all ionic liquid advantages and can be used as templates for supported organic synthesis on onium salts in solution [14,15]. Besides offering a nice alternative to solid phase supported synthesis, advantages of these soluble supports are numerous: reactions are carried out under homogeneous phase, recovering liquid phase kinetics; a classical work-up would consist only in filtration and washing several times to remove excess reagent and side products; solubility properties of reagent can be adjusted by changing the counterion; reactions are easily monitored by simple 1H NMR experiments; these supports have high loading capacity (4.87 mmol.g−1 for [HO(CH2)3NMe3][BF4] for example). Moreover, if they are not liquid at room temperature or slightly above, due, for example, to functionalization which increases their melting point, they can be used as solutions in molecular solvents or ionic liquids. These binary task specific ionic liquids have already been successfully used for the synthesis of numerous species [13,16]. Some representative examples include peptides synthesis [17], radical reactions [18], multicomponent reactions [19–21], biochemical transformations [22–27] and transition metal catalyzed reactions [28–30]. The last are particularly interesting in the sense that ionic liquids have already been shown to be good solvents for Heck reactions [31–34] for multiple reasons [35–41]. Indeed, the ionic nature of the solvent naturally favors dissolution and capture of catalyst in the ionic liquid phase which can be recycled with or without supporting a ligand [42,43]. It also accelerates considerably kinetic rates by in situ formation of NHC-carbene ligands when using imidazolium salts [44].
In this article, we would like to report the use of task specific onium salts as soluble supports in Heck reactions. Supporting either olefins or aryl halides has been envisioned and used with a comparable success. Moreover, by investigating low catalyst loading, we discovered how [Pd(OAc)2] by reaction with [tmba][PF6] is forming palladium nanoparticles leading to TON up to 106 and TOF up to 2 × 105 h−1.
2 Results and discussion
2.1 Supports synthesis
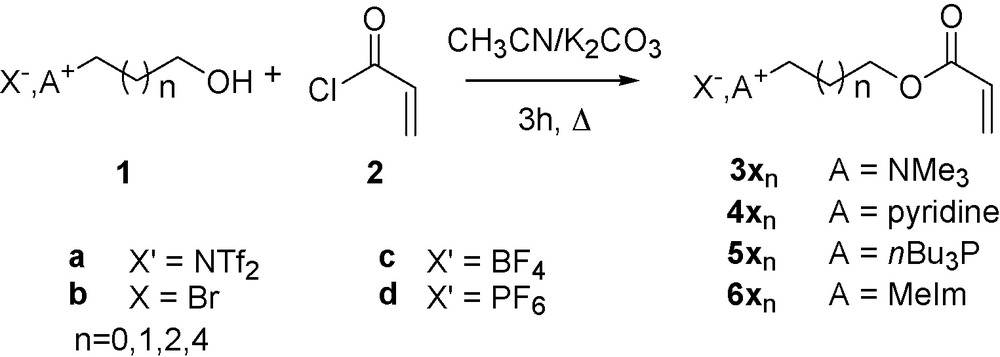
First, onium salt supported acrylates were obtained via coupling between acryloyl chloride 2 and ω-hydroxylated onium salts 1. Interestingly, triethylamine, classically used in this type of reaction was efficient but led to purification difficulties of salts 3–6 which were bypassed by using potassium carbonate instead followed by a simple filtration of mineral salts after reaction. Following this procedure, 10 different supported acrylates 3 were obtained in good yields (Table 1) with various cations such as imidazolium (Entry 1, 6a1), pyridinium (Entry 2, 4a1), tributylphosphonium (Entry 3, 5a1) or trimethylammonium (Entry 4, 3a1). Chain spacing length can also be modified from two to six carbons without notably affecting yields (Entry 4–7). Similarly to triflimide, other anions can be used in this reaction, such as bromide (Entry 8, 3b1), tetrafluoroborate (Entry 9, 3c1) or hexaflurophosphate (Entry 10, 3d1). In all cases, acrylates were obtained under pure form with no remaining starting alcohol.
Supported acrylates prepared following Scheme 1 synthesis.
| Entry | A+ | n | X− | Product | Yielda (%) |
| 1 | Imidazolium | 1 | NTf2− | 6a1 | 85 |
| 2 | Pyridinium | 1 | NTf2− | 4a1 | 80 |
| 3 | Tri(n-butyl)phosphonium | 1 | NTf2 | 5a1 | 77 |
| 4 | Trimethylammonium | 1 | NTf2− | 3a1 | 98 |
| 5 | Trimethylammonium | 0 | NTf2− | 3a0 | 90 |
| 6 | Trimethylammonium | 2 | NTf2− | 3a2 | 77 |
| 7 | Trimethylammonium | 4 | NTf2− | 3a4 | 78 |
| 8 | Trimethylammonium | 1 | Br− | 3b1 | 98 |
| 9 | Trimethylammonium | 1 | BF4− | 3c1 | 93 |
| 10 | Trimethylammonium | 1 | PF6− | 3d1 | 90 |
a Isolated yield after purification.
Eventually, one could envisage supporting the Heck reaction via the iodoarenes instead of the acrylates. Therefore, we decided to synthesize supported iodobenzene with the same sort of strategy. Benzoylation of N,N-dimethyl-1-aminobutan-4-ol 7 with 4-iodo benzoyl chloride under the same reaction conditions as previously described afforded N,N-dimethyl-1-aminobutan-4-yl-4’-iodobenzoate 9 in 88% yield. Quaternisation of tertiary amine 9 led to supported iodobenzene 10xn bearing a counteranion depending on the reagent used for methylation (triflate, iodide, methylsulfate). More diversity could be introduced by a simple ion metathesis in water leading to triflimide, hexafluorophosphate and tetrafluoroborate analogs (Method A). After anion metathesis, halide content turned out to be negligible as the silver nitrate test was negative (Schemes 1 and 2, Table 2).
Preparation of onium salt supported acrylates.
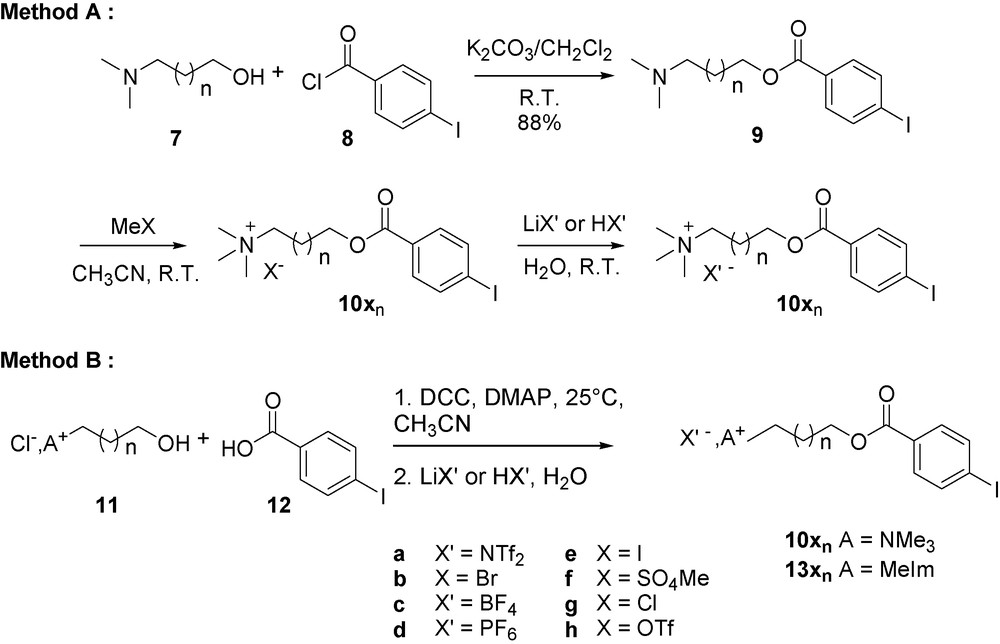
Preparation of onium salt supported aryl iodides.
Supported aryl iodides prepared following Scheme 2 procedure.
| Entry | Product | Method | Reagent | Yielda (%) |
| 1 | 10e2 | A | MeI | 99 |
| 2 | 10h2 | A | MeOTf | 96 |
| 3 | 10f2 | A | Me2SO4 | 94 |
| 4 | 10a1 | B | LiNTf2 | 95 |
| 5 | 10d2 | A | MeI then LiNTf2 | 90 |
| 6 | 10c0 | B | HBF4 | 90 |
| 7 | 10c1 | A | MeI then HBF4 | 86 |
| 8 | 10c2 | B | HBF4 | 83 |
| 9 | 10c4 | A | MeI then HBF4 | 87 |
| 10 | 10d2 | A | MeI then HPF6 | 96 |
| 11 | 10g0 | B | - | 93 |
| 12 | 10g1 | B | - | 95 |
| 13 | 13b0 | B | -b | 92 |
| 14 | 13c0 | B | HBF4 | 85 |
a Isolated yield from 7 or 11.
b Bromide was used instead of 11.
2.2 Conditions optimization
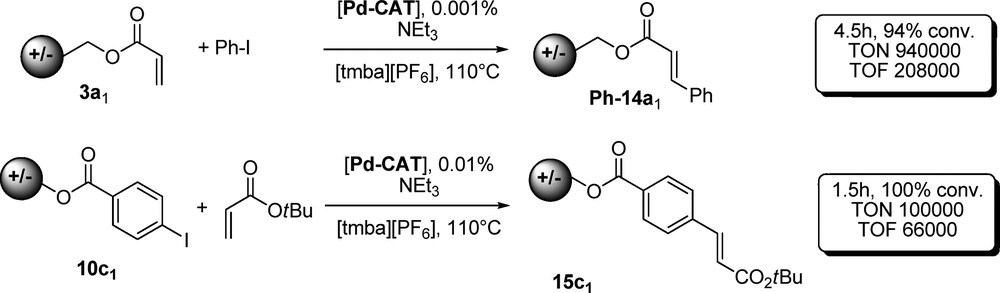
Indeed, the Heck reaction has already been largely studied in ionic liquids. This coupling reaction is particularly well suited to ionic liquids physicochemical properties as it is favored by using polar solvents or in the presence of ammonium salts [45] which stabilize Pd complexes [46]. Preliminary experiments were conducted in both ionic liquid and molecular solvents. In the presence of 1% of [Pd(OAc)2] as a catalyst and potassium carbonate as a base, supported olefin 3a1 reacts efficiently at 80 °C with iodobenzene to afford cinnamate Ph-14a1. Quantitative conversions were achieved in [tmba][NTf2] and [emim][NTf2] after 2 h at 110 °C, but only 70% were observed in acetonitrile. Furthermore E/Z selectivity was highly superior in ionic liquids as only the (E)-isomer is observed where 12% of the (Z)-compound was obtained in CH3CN. Notably, supported acrylate is much more reactive than the corresponding methyl ester as unsupported acrylate underwent reaction with a poor 26% conversion under the same conditions in acetonitrile (Scheme 3).
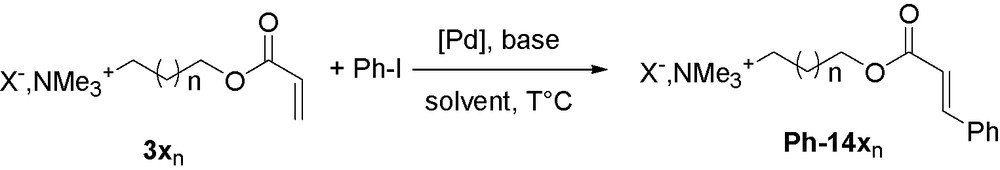
Mizoroki-Heck cross-coupling with supported acrylates.
Like traditionally observed in the case of the Heck reaction, increasing temperature is improving kinetics, and either organic or mineral bases can alternatively be used although Et3N was found to accelerate reactions. An attempt to diminish palladium concentration turned out to be particularly interesting. Indeed, for practical reasons, when using less than 100 ppm of Pd, simply weighting palladium acetate on a precision scale is not sufficiently satisfactory enough for loading accuracy. We decided to dilute palladium acetate in an ionic matrix whose melting point is above room temperature [tmba][PF6] (m.p. = 156 °C). By simply heating above this temperature a mixture of 0.1% [Pd(OAc)2] in [tmba][PF6] under inert atmosphere, a grey powder Pd-CAT was obtained after cooling which can be used directly to catalyze Heck reactions. This powder Pd-CAT is composed of palladium nanoparticles (4 nm) probably responsible for activity and larger aggregates (∼10 μm) (Fig. 1).
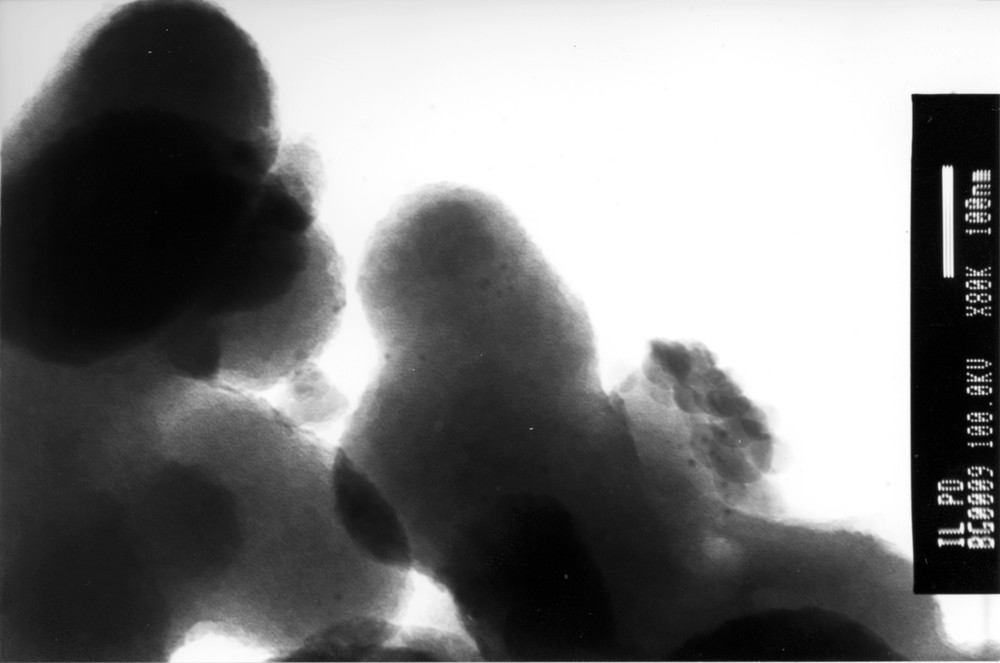
TEM picture of Pd-CAT.
Our best result was obtained using [tmba][PF6] (m.p. = 156 °C) as a solvent: at 110 °C, using only 1 ppm of Pd-CAT, 94% conversion was obtained after 270 min corresponding to a TON of 9.4 × 105 and a TOF of 2 × 105 h−1. These results are particularly significant for ligandless palladium catalysis as only few examples [47] are available in the literature with catalytic loading reaching below 10 ppm, even using a specially designed ligand (Scheme 4).
Mizoroki-Heck cross-coupling using Pd-CAT.
Similarly, supported iodobenzene was tested in Heck reaction with acrylates. To our delight, the reaction is equally efficient and cinnamic esters are isolated in good yields after only 10 min at 110 °C using 0.1% Pd-CAT. Catalyst loading diminution was also investigated in this case, and using 10 ppm of Pd-CAT in the same conditions as previously described, quantitative conversion was achieved after a short 90-min period. However, attempts to use only 1 ppm of palladium turned out to be less successful as only 35% conversion was reached after 12 h where 99% were attained using 3a1 under the very same conditions. Transesterification of the crude cinnamic ester Ph-14a1 in boiling methanol in the presence of traces of HCl gave quantitatively the methyl trans cinnamic ester after removal of excess of MeOH under vacuum and extraction with ether in an excellent state of purity > 99%, as indicated by GC/MS analysis. By using a two-carbon chain onium salt support, the hydrolysis of cinamate esters occurred during work-up. For linkers bearing more than four carbons, the final transesterification was too slow for practical applications. Three carbons turned out to be the best compromise between reactivity and stability.
Concerning use of supported iodobenzene, the reaction was found to be much more efficient in ammonium salt than in imidazolium based ionic liquids, probably due to inhibition of the catalyst by formation of Pd-NHC complexes [48]. However, this effect was only detectable using less than 100 ppm of palladium, slowing down the reaction enough to accurately perform kinetic experiments. Interestingly, support counter anion showed great influence on kinetics. Using 100 ppm of [Pd(OAc)2] in [tmba][NTf2], the reaction was quantitative after 1 h with 10c2 (BF4) where lower conversion was observed for the same time with another anion (PF6, 10d2, 93%; NTf2, 10a2, 93%; OTf, 10h2, 82%; MeSO4, 10f2, 41%; I, 10e2, 28%). Ester 15c1 could be transesterified under the conditions described above to the corresponding methyl cinnamic diester in very good yield. After transesterification, the remaining Binary Task Specific Ionic Liquids could be recycled several times without loss of matter beside other than mechanical loss during solid transfer. Interestingly, if the ppm level of palladium could not be reused, no side reactions were observed even after several cycles.
2.3 Synthesis of a library of cinnamic esters
In order to validate our methodology for parallel synthesis, we used this method to prepare a small library of substituted acrylates. Hence, a mixture of nine substituted iodobenzenes has been coupled with the supported acrylate 3a1. Noteworthy, all conversions were quantitative after 3 h in [tmba][NTf2]. Supported cinnamates were obtained with good purity as no (Z)-isomer could be detected by 1H NMR (< 1%) (Table 3).
3 Conclusion
We showed here that the onium salt supported organic synthesis (OSSOS) strategy is highly versatile and can also be used in combination with palladium catalyzed reaction. Noteworthy, we found that a simple catalyst, prepared from a mixture of a high melting point onium salt and palladium acetate displays exceptional activity as a ligandless catalyst [49]. Moreover, this material, mostly composed of nanoparticles is a non-hygroscopic, grey powder, stable at room temperature under inert atmosphere.
4 Experimental
4.1 General methods
All glassware was oven-dried prior to use and reactions were run under an inert atmosphere of argon using standard Schlenk techniques. Yields reported were from isolated products and determined to be pure by GC (DB-5 column) or TLC (pre-coated plates of silica gel 60 F254, stained with a potassium permanganate solution), and NMR spectroscopy. Solvents were carefully dried and purified following known procedures prior to use [50]. All other standard chemicals were purchased from Fisher, Alfa Aesar or Aldrich Chemical Co. and used without further purification. Reactions were monitored by gas chromatography (GC-MS) (GC system: HP 6890 series, Mass selective detector HP 5973) using a capillary column DB-5MS. Column chromatography purifications were performed on silica gel Si 60 (40–63 μm, 230–400 mesh, Merck). Melting points were determined on an electrothermal IA9300 digital melting point instrument. NMR spectra were recorded on a Bruker ARX 200 (1H: 200.13 MHz, 13C: 50.32 MHz) or AC 300 P (1H: 300.13 MHz), 1H chemical shifts (δ) are given with apparent multiplicities in ppm relative to TMS as external standard, J values in Hz, 13C chemical shifts are given relative to the central signal of CDCl3 at 77.0 ppm. IR spectra were obtained on a Bio-Rad FTS 175C FT-IR spectrophotometer. High resolution mass spectra measurements were performed at the Centre regional de mesures physiques de l’Ouest (CRMPO, University of Rennes 1) using a Varian MAT 311 with BE geometry (reversed Nier-Johnson) and EI athod or Micromass ZABSpec TOF with EBE OA TOF geometry with LSIMS ionization (liquid secondary ion mass spectrometry) at 8 kV with Cs+ gun in m-nitrobenzyl alcohol (mNBA).
4.2 General procedure for supported acrylate synthesis (Procedure I)
To a solution of hydroxyl derivatized onium salt and K2CO3 (5 eq) in acetonitrile (0.1 M) was added 3 eq. of acylating agent. After 2 h at 80 °C, solvents and unreacted acyl chloride were removed under vacuum. Product was extracted with 3 × 10 mL.mmol−1 CH2Cl2. Organic layers were combined and concentrated under reduced pressure to afford analytically pure onium salt supported acrylate.
4.3 General procedure for anion metathesis with HPF6 (Procedure II)
To a solution of onium halide (0.1 mmol) in 15 mL of water was added 0.15 mmol of an aqueous HPF6 solution (60%). After 2 h at r.t., a solid has formed. Filtration and washes (3 × 10 mL Et2O) afforded analytically pure onium hexafluorophosphate.
4.4 General procedure for anion metathesis with LiNTf2 (Procedure III)
To a solution of onium halide (0.1 mmol) in 10 mL of water was added 0.11 mmol of an aqueous LiNTf2 solution (10 mL). After 2 h at r.t., the aqueous phase was extracted with 3 × 15 mL of CH2Cl2. Organic fractions were combined and concentrated under reduced pressure. To remove remaining traces of water, the oil was dried under vacuum (0.5 mmHg) at 60 °C for 2 h.
4.5 General procedure for anion metathesis with HBF4 (Procedure IV)
To a solution of onium halide (0.1 mmol) in 15 mL of water was added 0.15 mmol of an aqueous HBF4 solution (50%). After 12 h at r.t., water was removed under reduced pressure affording a white solid. The residue was filtered and washed with 2 × 15 mL Et2O affording analytically pure onium tetrafluoroborate.
4.6 General procedure for Heck coupling with supported acrylate (Procedure V)
A mixture of 3a1 (1.5 mmol), iodobenzene (839 μL, 7.5 mmol, 5 eq), [Pd(OAc)2] (2.43 mg, 0.015 mmol, 1%), and K2CO3 (331 mg, 2.25 mmol, 1.5 eq) in 2 mL of [tmba][NTf2] or (DMF) was heated at 80 °C. Reaction was monitored by 1H NMR. After completion of the reaction, remaining reactants were eliminated with washes using alternatively water and Et2O.
4.7 General procedure for Heck coupling with supported iodobenzoic acids (Procedure VI)
A mixture of 10a2 (1.5 mmol), olefin (7.5 mmol, 5 eq), [Pd(OAc)2] (2.43 mg, 0.015 mmol, 1%), and K2CO3 (331 mg, 2.25 mmol, 1.5 eq) in 2 mL of [tmba][NTf2] (or DMF) was heated at 80 °C. Reaction was monitored by 1H NMR; at the end of the reaction, remaining reactants were eliminated with washes using alternatively water and Et2O.
4.8 Onium salt supported products
4.8.1 [AcrEtTMA][NTf2] 3a0 (2-acryolyloxyethyl)trimethylammonium bistrifluoromethanesulfonamide
Following general procedure I using (2-hydroxyethyl)trimethylammonium bistrifluoromethanesulfonamide [HOEtTMA][NTf2] and acryloyl chloride, [AcrEtTMA][NTf2] was obtained in 90% yield as a pink oil. 1H NMR (200 MHz, acetone-d6): δ = 3.50(s, 9H), 4.03–4.40 (m, 2H), 4.73–4.85 (m, 2H), 6.05 (dd, 1H, J1 = 1.9 Hz, J2 = 10.5 Hz), 6.25 (dd, 1H, J1 = 10.5 Hz, J2 = 17.3 Hz), 6.45 (dd, 1H, J1 = 1.9 Hz, J2 = 17.3 Hz). 13C (50 MHz, acetone-d6): δ = 54.0 (t, J = 4.2 Hz), 58.2, 65.2, 121.0 (q, JCF = 374 Hz), 128.8, 132.2, 165.5. LRMS(APCI): [C+] (C8 H16NO2) 158.3.
4.8.2 [AcrPrTMA][Br] 3b1 (3-acryolyloxypropyl)trimethylammonium bromide
Following general procedure I using (3-hydropropyl)trimethylammonium bromide [HOPrTMA][Br] and acryloyl chloride, [AcrPrTMA][Br] was obtained in 98% yield as a colorless oil. 1H NMR (200 MHz, acetone-d6): δ = 2.15–2.20 (m, 2H), 3.15 (s, 9H), 3.48–3.52 (m, 2H), 4.18 (t, 2H, J = 6.0 Hz), 5.75 (dd, 1H, J1 = 1.9 Hz, J2 = 10.5 Hz), 6.15 (dd, 1H, J1 = 10.5 Hz, J2 = 17.3 Hz), 6.15 (dd, 1H, J1 = 1.9 Hz, J2 = 17.3 Hz). 13C (50 MHz, acetone-d6): δ = 21.7, 52.2 (t, J = 4.2 Hz), 60.4 (t, J = 3.0 Hz), 62.6, 127.4, 130.6, 165.0. HRMS(FAB): [C+] (C9H18NO2). Calcd: 172.1338, found: 172.1356.
4.8.3 [AcrPrTMA][BF4] 3c1 (3-acryolyloxypropyl)trimethylammonium tetrafluoroborate
Following general procedure I using (3-hydropropyl)trimethylammonium tetrafluoroborate [HOPrTMA][BF4] and acryloyl chloride, [AcrPrTMA][BF4] was obtained in 93% yield as a white solid. 1H NMR (200 MHz, acetone-d6): δ = 2.28–3.31 (m, 2H), 3.32 (s, 9H), 3.06–3.15 (m, 2H), 4.52 (t, 2H, J = 6.6 Hz), 5.80 (dd, 1H, J1 = 1.9 Hz, J2 = 10.0 Hz), 6.05 (dd, 1H, J1 = 18.3 Hz, J2 = 10.0 Hz), 6.15 (dd, 1H, J1 = 1.9 Hz, J2 = 18.3 Hz). 13C (50 MHz, acetone-d6): δ = 22.8; 53.3, 61.5, 63.8, 128.5, 131.7, 167.3. LRMS(APCI): [2C+, BF4] 431.
4.8.4 [AcrPrTMA][NTf2] 3a1 (3-acryolyloxypropyl)trimethylammonium bistrifluoromethanesulfonamide
Following general procedure I using (3-hydropropyl)trimethylammonium bistrifluoromethanesulfonamide [HOPrTMA][NTf2] and acryloyl chloride, [AcrPrTMA][NTf2] was obtained in 90% yield as a pale yellow oil. 1H NMR (200 MHz, acetone-d6): δ = 2.22–2.25 (m, 2H), 3.25 (s, 9H), 3.60–3.75 (m, 2H), 4.15 (t, 2H, J = 6.0 Hz), 5.80 (dd, 1H, J1 = 1.9 Hz, J2 = 10.7 Hz), 6.05 (dd, 1H, J1 = 17.2 Hz, J2 = 10.7 Hz), 6.15 (dd, 1H, J1 = 1.9 Hz, J2 = 17.2 Hz). 13C (50 MHz, acetone-d6): δ = 29.2, 54.2 (t, JC-N = 4.0 Hz), 65.2, 65.2, 121.0 (q, JCF = 374 Hz), 129.4, 132.1, 165.6. 19F (282 MHz, acetone-d6): −79.8. HRMS(FAB): [C+] (C9H18NO2). Calcd: 172.1338, found: 172.1346.
4.8.5 [AcrPrTMA][PF6] 3d1 (3-acryolyloxypropyl)trimethylammonium hexafluorophosphate
Following general procedure I using (3-hydropropyl)trimethylammonium hexafluorophosphate [HOPrTMA][PF6] and acryloyl chloride, [AcrPrTMA][PF6] was obtained in 90% yield as a white solid. 1H NMR (200 MHz, acetone-d6): δ = 2.28–3.31 (m, 2H), 3.32 (s, 9H), 3.06–3.15 (m, 2H), 4.52 (t, 2H, J = 6.0 Hz), 5.80 (dd, 1H, J1 = 1.9 Hz, J2 = 10.2 Hz), 6.05 (dd, 1H, J1 = 17.1 Hz, J2 = 10.2 Hz), 6.15 (dd, 1H, J1 = 1.9 Hz, J2 = 17.2 Hz). 13C (50 MHz, acetone-d6): δ = 22.4, 52.7 (t, J = 3.9 Hz), 61.0, 63.8 (t, J = 3.2 Hz), 128.1, 131.2, 165.7. HRMS(FAB): [2C+, PF6] (C18H36N2O4PF6). Calcd: 489.2317, found: 489.2319.
4.8.6 [AcrBuTMA][NTf2] 3a2 (6-acryolyloxybutyl)trimethylammonium bistrifluoromethanesulfonamide
Following general procedure I using (4-hydroxybutyl)trimethylammonium bistrifluoromethanesulfonamide [HOBuTMA][NTf2] and acryloyl chloride, [AcrBuTMA][NTf2] 3a2 was obtained in 77% yield as a pale yellow oil. 1H NMR (200 MHz, acetone-d6): δ = 1.70–1.90 (m, 2H), 2.00–2.20 (m, 2H), 3.40 (s, 9H), 3.58–3.72 (m, 2H), 4.25 (t, 2H, J = 6.0 Hz), 5.80 (dd, 1H, J1 = 1.9 Hz, J2 = 10.7 Hz), 6.05 (dd, 1H, J1 = 17.2 Hz, J2 = 10.7 Hz), 6.15 (dd, 1H, J1 = 1.9 Hz, J2 = 17.2 Hz). 13C (50 MHz, acetone-d6): δ = 20.9, 26.4, 54.0, 64.5, 67.4, 122.2 (q, JCF = 374 Hz), 129.7, 131.8, 167.0. HRMS(FAB): [C+] (C10H20NO2). Calcd: 186.1494, found: 186.1506.
4.8.7 [AcrHexTMA][NTf2] 3a4 (6-acryolyloxyhexyl)trimethylammonium bistrifluoromethanesulfonamide
Following general procedure I using (6-hydrohexyl)trimethylammonium bistrifluoromethanesulfonamide [HOHexTMA][NTf2] and acryloyl chloride, [AcrHexTMA][NTf2] 3a4 was obtained in 78% yield as a pale yellow oil. 1H NMR (200 MHz, acetone-d6): δ = 1.40–1.60 (m, 4H), 1.65–1.85 (m, 2H), 1.90–2.10 (m, 2H), 3.32 (s, 9H), 3.06–3.15 (m, 2H), 4.52 (t, 2H, J = 6.4 Hz), 5.80 (dd, 1H, J1 = 2.1 Hz, J2 = 10.1 Hz), 6.05 (dd, 1H, J1 = 17.2 Hz, J2 = 10.1 Hz), 6.15 (dd, 1H, J1 = 2.1 Hz, J2 = 17.2 Hz). 13C (50 MHz, acetone-d6): δ = 23.4, 26.1, 26.5, 26.5, 53.6, 64.8, 67.4, 121.0, 129.6, 131.1, 166.6.
4.8.8 [AcrPrTBP][NTf2] 5a1 (3-acryolyloxypropyl)tributylphosphonium bistrifluoromethanesulfonamide
Following general procedure I using (3-hydropropyl)tributylphosphonium bistrifluoromethanesulfonamide [HOPrTBP][NTf2] and acryloyl chloride, [AcrPrTBP][NTf2] was obtained in 77% yield as a pale yellow oil. 1H NMR (200 MHz, acetone-d6): δ = 1.00 (t, 9H, J = 7.2 Hz), 1.45–1.85 (m, 12H), 2.10–2.28 (m, 2H), 2.48–2.70 (m, 8H), 4.30 (t, 2H, J = 6.3 Hz), 5.58 (dd, 1H, J1 = 10.2 Hz, J2 = 1.9 Hz), 6.18 (dd, 1H, J1 = 17.2 Hz, J2 = 10.2 Hz), 6.40 (dd, 1H, J1 = 17.2 Hz, J2 = 1.9 Hz). 13C (50 MHz, acetone-d6): δ = 13.1, 17.8, 18.8, 21.1, 23.4, 23.5, 23.9, 63.8, 121.1 (q, JCF = 374 Hz), 128.6, 131.1, 165.7.
4.8.9 [AcrPrPyr][NTf2] 4a1 (3-acryolyloxypropyl)pyridinium bistrifluoromethanesulfonamide
Following general procedure I using (3-hydropropyl)pyridinium bistrifluoromethanesulfonamide [HOPrPyr][NTf2] and acryloyl chloride, [AcrPrPyr][NTf2] was obtained in 80% yield as a pale yellow oil. 1H NMR (200 MHz, acetone-d6): δ = 2.43–2.72 (m, 2H), 4.15 (t, 2H, J = 5.8 Hz), 5.08 (t, 2H, J = 6.8 Hz), 5.90(dd, 1H, J1 = 10.3 Hz, J2 = 2.0 Hz), 6.18 (dd, 1H, J1 = 17.0 Hz, J2 = 10.3 Hz), 6.40 (dd, 1H, J1 = 17.0 Hz, J2 = 2.0 Hz), 8.10 (t, 2H, J = 7.0 Hz), 8.52 (m, 1H), 8.98 (m, 2H). 13C (50 MHz, acetone-d6): δ = 30.4, 60.0, 61.4, 121.0 (q, JCF = 374 Hz), 128.7, 129.0, 131.4, 145.5, 146.5, 165.8. HRMS(FAB): [C+] (C11H14NO2). Calcd: 192.1025, found: 192.1026.
4.8.10 [AcrPrMIm][NTf2] 6a1 (3-acryolyloxypropyl)methylimidazolium bistrifluoromethanesulfonamide
Following general procedure I using (3-hydropropyl) methylimidazolium bistrifluoromethanesulfonamide [HOPrMIm][NTf2] and acryloyl chloride, [AcrPrMIm][NTf2] was obtained in 85% yield as a pale yellow oil. 1H NMR (200 MHz, acetone-d6): δ = 2.30–2.50 (m, 2H), 4.15 (s, 3H), 4.28 (t, 2H, J = 6.0 Hz), 4.63 (t, 2H, J = 6.9 Hz), 5.95 (dd, 1H, J1 = 1.9 Hz, J2 = 10.7 Hz), 6.18 (dd, 1H, J1 = 17.2 Hz, J2 = 10.7 Hz), 6.40 (dd, 1H, J1 = 1.9 Hz, J2 = 17.2 Hz), 7.80 (t, 1H, J = 1.7 Hz), 7.95 (t, 1H, J = 1.7 Hz), 9.2 (s, 1H). 13C (50 MHz, acetone-d6): δ = 29.3, 36.2, 47.4, 61.4, 121.0 (q, JCF = 374 Hz), 123.0, 124.3, 128.4, 131.3, 136.9, 166.1. HRMS(FAB): [C+] (C10H15N2O2). Calcd: 195.1134, found: 192.1132.
4.8.11 4-Dimethylaminobutyl 4-iodobenzoate 92
To a solution of 4-hydroxybutyldimethylamine (2.8 g, 23.9 mmol) and K2CO3 (6.6 g) in 10 mL of CH2Cl2 were added at 0 °C 4-iodobenzoyl chloride (7.0 g, 26.3 mmol). The reaction mixture was allowed to warm up to room temperature. After 3 h at r.t., the mixture was filtered and concentrated under reduced pressure to afford 7.3 g of a colorless oil in analytically pure form (88% yield). 1H NMR (200 MHz, CDCl3): δ = 1.78–1.90 (m, 4H), 2.50(s, 6H), 2.65 (t, 2H, J = 5.3 Hz), 4.35 (t, 2H, J = 6.0 Hz), 7.70–7.85 (m, 4H). 13C (50 MHz, CDCl3): δ = 20.2, 24.9, 43.5, 57.3, 63.5, 99.0, 128.4, 129.4, 136.2, 164.3.
4.8.12 [IBzBuTMA][OTf] 10h2 {4-(4-iodobenzoyloxy)butyl}trimethylammonium trifluoromethanesulfonate
To a solution of 92 (4.0 g, 11 mmol) in acetonitrile (10 mL) was added at 0 °C methyltriflate (1.1 mL, 13.5 mmol). After 15 min at 0 °C, the white solid is filtered and washed with 3 × 10 mL of Et2O to afford 4.83 g of a white solid (96% yield). m.p. 176–178 °C. 1H NMR (200 MHz, CD3CN): δ = 1.80–2.10 (m, 4H), 3.06 (s, 9H), 3.35–3.45 (m, 2H), 4.40 (t, 2H, J = 6.1 Hz), 7.85 (d, 2H, J = 8.6 Hz), 8 (d, 2H, J1 = 8.6 Hz). 13C (50 MHz, CD3CN): δ = 19.2, 24.7, 52.5 (t, J = 4.0 Hz), 63.7, 65.6 (t, J = 3.2 Hz), 99.9, 121.3 (q, JC-F = 321 Hz), 129.6, 130.6, 137.6, 165.4. LRMS(FAB): (2C+, OTf−)+ (C29H42N2O7F3I2S) 873.1.
4.8.13 [IBzBuTMA][MeSO4] 10f2 {4-(4-iodobenzoyloxy)butyl}trimethylammonium trifluoromethanesulfate
To a solution of 92 (4.0 g, 11 mmol) in acetonitrile (10 mL) was added at 0 °C dimethylsulfate (1.24 mL, 13.2 mmol). After 15 min at 0 °C, the white solid was filtered and washed with 3 × 10 mL of Et2O to afford 4.99 g of a white solid (96% yield). m.p. 190–192 °C. 1H NMR (200 MHz, MeOD): δ = 1.80–2.10 (m, 4H), 3.20 (s, 9H), 3.38–3.50 (m, 2H), 3.70 (s, 3H), 4.40 (t, 2H, J = 6.0 Hz), 7.78 (dd, 2H, J1 = 2.0 Hz, J2 = 6.8 Hz), 7.90 (dd, 2H, J1 = 2.0 Hz, J2 = 6.8 Hz). 13C (50 MHz, MeOD): δ = 21.3, 27.0, 54.3 (t, J = 3.9 Hz), 56.0, 66.1, 67.8, 102.4, 131.1, 132.5, 139.7, 168.5. HRMS(FAB): (C+) (C14H21NO2I). Calcd: 362.0617, found: 362.0618.
4.8.14 [IBzBuTMA][I] 10f2 {4-(4-iodobenzoyloxy)butyl}trimethylammonium iodide
To a solution of 92 (4.0 g, 11 mmol) in acetonitrile (10 mL) was added at 0 °C iodomethane (820 μL mL, 13.2 mmol). After 15 min at 0 °C, the white solid was filtered and washed with 3 × 10 mL of Et2O to afford 5.32 g of a white solid (99% yield). m.p. 248–250 °C. 1H NMR (200 MHz, acetone-d6): δ = 1.90–2.05(m, 4H), 3.20 (s, 9H), 3.42–3.55 (m, 2H), 4.45 (t, 2H, J = 6.1 Hz), 7.80 (d, 2H, J = 8.7 Hz), 7.95 (d, 2H, J1 = 8.7 Hz). 13C (50 MHz, acetone-d6): δ = 19.9, 25.5, 53.4 (t, JC-N = 4.1 Hz), 65.1, 66.4, 101.6, 131.2, 131.4, 138.4, 166.8. HRMS(FAB): (2C+, I−) (C14H21NO2I2) 851.1.
4.8.15 [IBzBuTMA][PF6] 10d2 {4-(4-iodobenzoyloxy)butyl}trimethylammonium hexafluorophosphate
Following procedure II using 10f2, [IBzBuTMA][PF6] was obtained as a white solid in 96% yield. m.p. 204–206 °C. 1H NMR (200 MHz, acetone-d6): δ = 2.05–2.20 (m, 2H), 2.28–2.47 (m, 2H), 3.6 (s, 9H), 3.80–3.97 (m, 2H), 4.6 (t, 2H, J = 6.3 Hz), 8 (d, 2H, J = 8.6 Hz), 8.15 (d, 2H, J = 8.6 Hz). 13C (50 MHz, acetone-d6): δ = 21.0, 26.6, 54.1 (t, J = 4.0 Hz), 65.3, 67.4 (t, J = 3.2 Hz), 101.6, 131.2, 132.3, 139.2, 166.7. HRMS(FAB): (2C+, PF6−)+ (C28H42N2O4F6I2P). Calcd: 869.0876, found: 869.0879.
4.8.16 [IBzBuTMA][NTf2] 10a2 {4-(4-iodobenzoyloxy)butyl}trimethylammonium bistrifluoromethylsulfonamide
Following procedure III using 10f2, [IBzBuTMA][NTf2] was obtained as a white solid in 90% yield. m.p. 48–50 °C. 1H NMR (200 MHz, acetone-d6): δ = 1.82–2.00 (m, 2H), 2.10–2.22 (m, 2H), 3.38 (s, 9H), 3.60–3.72 (m, 2H), 4.40 (t, 2H, J = 6.2 Hz), 7.80 (d, 2H, J = 8.5 Hz), 7.95 (d, 2H, J = 8.5 Hz). 13C (50 MHz, acetone-d6): δ = 26.7, 29.1, 54.1 (t, JC-N = 4.1 Hz), 65.3, 67.4, 101.6, 121.2 (q, JC-F = 321 Hz), 131.2, 132.3, 139.2, 166.7. HRMS(FAB): (2C+, NTf2−)+ (C30H42N3O8F6I2S2). Calcd: 1004.0407, found: 1004.0427.
4.8.17 [IBzBuTMA][Cl] 10g2 {4-(4-iodobenzoyloxy)butyl}trimethylammonium chloride
A solution of [HOBuTMA][Cl], DCC (1.5 eq), 4-iodobenzoic acid (1.5 eq) and DMAP (0.2 eq) in acetonitrile (0.1 M) was allowed to react 4 h at room temperature. After concentration under reduced pressure, 4-iodobenzoic acid crushed out upon water addition (10 mL). After filtration and concentration under vacuum, product was recrystallized in acetone, and used directly in anion metathesis reactions without further purification.
4.8.18 [IBzBuTMA][BF4] 10c2 {4-(4-iodobenzoyloxy)butyl}trimethylammonium tetrafluoroborate
Following procedure IV using 10g2, [IBzBuTMA][BF4] was obtained as a white solid in 86% yield. m.p. 184–186 °C. 1H NMR (200 MHz, acetone-d6): δ = 1.90–2.00 (m, 2H), 2.15–2.28 (m, 2H), 3.40 (s, 9H), 3.65–3.80 (m, 2H), 4.42(t, 2H, J = 6.3 Hz), 7.85 (d, 2H, J = 8.6 Hz), 8.15 (d, 2H, J = 8.6 Hz). 13C (50 MHz, acetone-d6): δ = 21.0, 26.6, 54.0 (t, J = 4.0 Hz), 65.3, 67.4, 101.5, 131.2, 132.3, 139.2, 166.7. HRMS(FAB): (2C+, BF4−)+ (C28H42N2O4F4I2B). Calcd: 811.1263, found: 811.1259.
4.8.19 [IBzPrTMA][Cl] 10g1 {3-(4-iodobenzoyloxy)propyl}trimethylammonium chloride
A solution of [HOPrTMA][Cl], DCC (1.5 eq), 4-iodobenzoic acid (1.5 eq) and DMAP (0.2 eq) in acetonitrile (0.1 M) was allowed to react 4 h at room temperature. After concentration under reduced pressure, 4-iodobenzoic acid crushed out upon water addition (10 mL). After filtration and concentration under vacuum, product was recrystallized in acetone, and used directly in anion metathesis reactions without further purification. (95% yield) m.p. 218–220 °C. 1H NMR (200 MHz, D2O): δ = 2.12–2.35 (m, 2H), 3.12 (s, 9H), 3.38–3.52 (m, 2H), 4.32 (t, 2H, J = 5.9 Hz), 7.65 (dd, 2H, J1 = 1.8 Hz, J2 = 6.6 Hz), 7.83 (dd, 2H, J1 = 1.8 Hz, J2 = 6.7 Hz). 13C (50 MHz, D2O): δ = 22.6, 53.4 (t, JC-N = 3.9 Hz), 62.8, 64.2, 102.0, 128.8, 131.2, 138.2, 167.8. LRMS(APCI): (C+) (C13H19INO2) 348.3.
4.8.20 [IBzPrTMA][NTf2] 10a1 {3-(4-iodobenzoyloxy)propyl}trimethylammonium bistrilfuoromethylsulfonamide
Following procedure III using 10g1, [IBzBuTMA][NTf2] was obtained as a white solid in 95% yield. m.p. 48–50 °C. 1H NMR (200 MHz, acetone-d6): δ = 2.40–2.60 (m, 2H), 3.47 (s, 9H), 3.80–4.00 (m, 2H), 4.53 (t, 2H, J = 5.9 Hz), 7.83 (d, 2H, J = 8.3 Hz), 7.97 (d, 2H, J = 8.3 Hz). 13C (50 MHz, acetone-d6): δ = 23.1, 53.4 (t, J = 3.8 Hz), 62.2, 64.5, 100.9, 120.5 (q, JCF = 321 Hz), 130.0, 131.5, 138.3, 165.7. LRMS(APCI): (C+) (C13H19INO2) 348.3.
4.8.21 [IBzPrTMA][BF4] 10c1 {3-(4-iodobenzoyloxy)propyl}trimethylammonium tetrafluoroborate
Following procedure IV using 10g1, [IBzBuTMA][BF4] was obtained as a white solid in 83% yield. m.p. 190–192 °C. 1H NMR (200 MHz, acetone-d6): δ = 2.38–2.63 (m, 2H), 3.45(s, 9H), 3.75–3.93 (m, 2H), 4.52 (t, 2H, J = 5.9 Hz), 7.85 (dd, 2H, J1 = 1.9 Hz, J2 = 6.6 Hz), 7.98 (dd, 2H, J1 = 1.9 Hz, J2 = 6.7 Hz). 13C (50 MHz, acetone-d6): δ = 23.0, 53.2 (t, J = 4.0 Hz), 62.3, 64.2, 100.8, 130.0, 131.6, 138.3, 165.8. LRMS(APCI): (C+) (C13H19INO2) 348.3.
4.8.22 [IBzEtTMA][Cl] 10g0 {2-(4-iodobenzoyloxy)ethyl}trimethylammonium chloride
A solution of [HOEtTMA][Cl], DCC (1.5 eq), 4-iodobenzoic acid (1.5 eq) and DMAP (0.2 eq) in acetonitrile (0.1 M) was allowed to react 4 h at room temperature. After concentration under reduced pressure, 4-iodobenzoic acid crushed out upon water addition (10 mL). After filtration and concentration under vacuum, product was recrystallized in acetone, and used directly in anion metathesis reactions without further purification (93% yield).
4.8.23 [IBzEtTMA][BF4] 10c0 {2-(4-iodobenzoyloxy)ethyl}trimethylammonium tetrafluoroborate
Following procedure IV using 10g0, [IBzEtTMA][BF4] was obtained as a white solid in 90% yield. m.p. 200–202 °C. 1H NMR (300 MHz, CD3CN): δ = 3.18 (s, 9H), 3.68–3.80 (m, 2H), 4.50–4.73 (m, 2H), 7.78 (dd, 2H, J1 = 1.9 Hz, J2 = 6.7 Hz), 7.90 (dd, 2H, J1 = 1.8 Hz, J2 = 6.6 Hz). 13C (75 MHz, CD3CN): δ = 53.4 (t, J = 3.8 Hz), 58.3, 64.4 (t, J = 3.8 Hz), 100.6, 128.6, 130.7, 137.8, 164.7. LRMS(APCI): (C+) (C12H17INO2) 334.0.
4.8.24 [IBzHexTMA][Cl] 10g4 {6-(4-iodobenzoyloxy)hexyl}trimethylammonium chloride
A solution of [HOEtTMA][Cl], DCC (1.5 eq), 4-iodobenzoic acid (1.5 eq) and DMAP (0.2 eq) in acetonitrile (0.1 M) was allowed to react 4 h at room temperature. After concentration under reduced pressure, 4-iodobenzoic acid crushed out upon water addition (10 mL). After filtration and concentration under vacuum, product was recrystallized in acetone, and used directly in anion metathesis reactions without further purification.
4.8.25 [IBzHexTMA][BF4] 10c4 {6-(4-iodobenzoyloxy)hexyl}trimethylammonium tetrafluoroborate
Following procedure IV using 10g4, [IBzHexTMA][BF4] was obtained as a white solid in 87% yield. m.p. 170–172 °C. 1H NMR (300 MHz, CD3CN): δ = 1.30–1.62 (m, 4H), 1.70–2.02 (m, 4H), 3.30 (s, 9H), 3.45–3.60 (m, 2H), 4.30 (t, 2H, J = 6.5 Hz), 7.80 (dd, 2H, J1 = 1.7 Hz, J2 = 6.7 Hz), 7.90 (dd, 2H, J1 = 1.7 Hz, J2 = 6.7 Hz). 13C (75 MHz, CD3CN): δ = 23.4, 26.2, 26.5, 53.5 (t, J = 3.8 Hz), 65.6, 67.3, 101.0, 128.6, 131.0, 131.8, 138.8, 166.4. LRMS(APCI): (C+) (C16H25INO2) 390.0.
4.8.26 [IBzEtMIm][Br] 13b0 {3-(4-iodobenzoyloxy)ethyl}methylimidazolium bromide
A solution of [HOEtMIm][Br], DCC (1.5 eq), 4-iodobenzoic acid (1.5 eq) and DMAP (0.2 eq) in acetonitrile (0.1 M) was allowed to react 4 h at room temperature. After concentration under reduced pressure, 4-iodobenzoic acid crushed out upon water addition (10 mL). After filtration and concentration under vacuum, product was recrystallized in acetone, and used directly in anion metathesis reactions without further purification. (92% yield).
4.8.27 [IBzEtMIm][BF4] 13c0 {3-(4-iodobenzoyloxy)ethyl}methylimidazolium tetrafluoroborate
Following procedure IV using 10b0, [IBzEtMIm][BF4] was obtained as a white solid in 90% yield. m.p. 146–148 °C. 1H NMR (300 MHz, acetone-d6): δ = 4.10 (s, 3H), 4.73–4.82 (m, 2H), 4.85–4.92 (m, 2H), 7.75–7.85 (m, 3H), 7.92–8.00 (m, 3H), 9.27 (s, 1H). 13C (75 MHz, acetone-d6): δ = 36.7, 49.5, 64.2, 101.6, 124.0, 124.9, 130.0, 132.1, 138.2, 138.9, 166.0. LRMS(APCI): (C+) (C13H14IN2O2) 357.0.
4.8.28 [CinPrTMA][NTf2] Ph-14a1 {3-(3-phenylprop-2-enoylox)propyl}trimethylammonium bistrifluoromethanesulfonamide
Following general procedure V using 3a1 and iodobenzene in DMF, [CinPrTMA][NTf2] was obtained in 86% yield as a purple solid. m.p. 172–174 °C 1H NMR (300 MHz, CD3CN): δ = 2.18–2.35 (m, 2H), 3.15 (s, 9H), 3.43–3.58 (m, 2H), 4.34 (t, 2H, J = 5.9 Hz), 6.55 (d, 1H, J = 16.1 Hz), 7.41–7.53 (m, 3H), 7.65–7.88 (m, 3H). 13C (75 MHz, CD3CN): δ = 22.9, 53.6, 61.3, 64.2, 118.1, 128.7, 129.4, 131.0, 134.7, 145.4, 167.0. LRMS(APCI): [C+] (C15H22NO2) 248.2.
4.8.29 [tBuOCOCHCHBzPrTMA][NTf2] 15c1 {3-(2-tert-butoxycarbonylvinyl)benzoyloxypropyl}trimethylammonium bistrifluoromethanesulfonamide
Following general procedure VI using 10c1 and tertbutylacrylate in DMF, [tBuOCOCHCHBzPrTMA][NTf2] was obtained in 86% yield as a brown solid. m.p. 170–172 °C 1H NMR (300 MHz, CD3CN): δ = 1.53 (s, 9H), 2.18–2.35 (m, 2H), 3.15 (s, 9H), 3.43–3.58 (m, 2H), 4.44 (t, 2H, J = 5.8 Hz), 6.55 (d, 1H, J = 16.1 Hz), 7.58–7.78 (m, 3H), 8.09 (d, 2H, J = 8.4 Hz). 13C (75 MHz, CD3CN): δ = 22.0, 26.9, 52.6 (t, J = 4.0 Hz), 61.4, 63.4 (t, J = 3.3 Hz), 80.6, 122.3, 127.8, 129.6, 130.4, 138.8, 141.7, 165.6, 165.6. LRMS(APCI): [C+] (C20H30NO4) 348.3.
4.8.30 Methyl cinamate [103-26-4]
A 1 M HCl solution of supported cinnamate (0.1 M) was heated to reflux under argon atmosphere. After 3 h, methanol was removed under reduced pressure. The product was extracted from the remaining oil by adding five times 4 mL/mmol of Et2O. After concentration of the ether fraction under reduced pressure, methyl cinnamate was obtained in 97% yield with > 99% purity (GC/MS).