

1 Introduction
Les sousphtalocyanines, molécules apparentées aux métallophtalocyanines, sont composés par trois unités iso-indoles reliées par un atome de bore. Ils possèdent un système π conjugué de 14 électrons délocalisés sur l’ensemble de la molécule. Les sousphthalocyanines de bore SubPcB ont été découverts par hasard par Meller et Ossoko [1], lorsqu’ils ont essayé de synthétiser la phtalocyanine de bore. La réaction de condensation du phtalonitrile en présence du trichlorure de bore BCl3 dans le chloronaphtalène n’a pas conduit au produit tétramérisé attendu. Les sousphtalocyanines sont donc synthétisés avec un bon rendement par la réaction de cyclotrimérisation de phtalonitriles en présence d’un dérivé trihalogéné de bore [2–4].
D’une manière générale, les halogénosousphtalocyanines SubPcBX sont obtenus par réaction entre le phtalonitrile et les halogénures de bore RBX2 (avec R = CH3(CH2)3, C6H5, Cl, F, Br et X = Cl, F, Br) dans le chloronaphtalène pendant dix minutes [1]. Les températures de réactions élevées et les longs temps de réaction conduisent à des sous-produits ou des produits secondaires qu’on ne peut pas identifier [5]. Diaz-Garcias et al. ont montré que la réaction entre le phtalonitrile et le trichlorure de bore donnait naissance à SubPcBCl et à ses dérivés mono et dichloré à la périphérie [6]. Les substitutions à la périphérie proviennent de la chloration du macrocycle par BCl3 [6]. Les trois produits subliment ensemble et sont solubles dans le chloroforme, le dichlorométhane et insolubles dans les solvants organiques apolaires comme l’hexane et dans beaucoup d’autres polaires comme le méthanol. Le mélange réactionnel contient aussi plusieurs produits de dégradation du phtalonitrile qui peuvent être éliminés par lavage au méthanol.
L’approche qui consistait à faire réagir d’autres réactifs du bore afin de varier la nature du ligand axial ne conduit pas non plus aux sousphtalocyanines de bore [7]. Cela a été prouvé par les tentatives de réactions avec les triiodures de bore, les trialkyl borates, les esters boroniques et amides boroniques [7]. Meller et Ossoko avaient déjà montré que les réactions entre les diboranes, les amines du bore et le phtalonitrile produisent des quantités importantes de la phtalocyanine base H2Pc [1]. Parmi les différents réactifs de bore (BCl3, BBr3, BF3, BPh3, BPhCl2) utilisés depuis longtemps pour la synthèse de SubPcBX (X = halogène), BCl3 est le plus communément utilisé puisqu’il offre le meilleur compromis entre le rendement de la cyclotrimérisation et la stabilité des chloroSubPcB formés [4,8]. L’inconvénient de cette méthode est que le traitement de BCl3 et sa sensibilité à l’humidité peuvent jouer sur le rendement et rendre la méthode difficilement reproductible [8].
Les SubPcBCl, en présence de la lumière, tendent à se décomposer en solution au bout de quelques semaines, voire quelques jours [9]. La faiblesse de la liaison B-Cl est probablement responsable de cette instabilité [10,11] ; la possibilité de varier la nature du ligand axial peut permettre de surmonter cette instabilité.
Un cristal de SubPcBCl a été obtenu par lente évaporation d’une solution du produit dans l’acétone [12]. Les cristaux de SubPcBCl obtenus par Meller et Ossoko sont de couleur bronze. L’étude cristallographique par rayons X a montré que les sousphtalocyanines ont une structure en forme de cône à cause des contraintes provenant des couplages entre les trois unités iso-indoles [13]. La SubPcBCl fond à une température supérieure à 300 °C et sublime à 350 °C et à une pression entre 10−6 à 10−4 Torr [1]. Ainsi, les films fins de dépôt sur surface métalliques sont préparés par sublimation sous vide à 350 °C et à une pression de 10−6 Torr.
Depuis sa découverte accidentelle par Meller et Ossoko [1], la chlorosousphtalocyanine de bore (SubPcBCl) n’a cessé d’être l’objet de nombreux travaux afin d’élucider les propriétés physicochimiques du macrocycle sousphtalocyanine. Les SubPc ont surtout été utilisées comme matériaux intermédiaires pour la synthèse de phtalocyanines (Pc) non symétriques [14–18].
Les propriétés physicochimiques de ces composés ont été modulées par l’introduction d’une variété de groupements fonctionnels à la périphérie et par le remplacement des trois sous-unités iso-indoles par trois noyaux pyrroles [19]. Quand le dérivé substitué du phtalonitrile n’est pas du groupe C2V, la SubPc obtenue est un mélange d’isomères de symétries C1 et C3 [7,20–22]. Chaque isomère est un mélange racémique de deux formes énantiomères, optiquement actives [23,24]. Très peu de composés de symétrie C3 ont été obtenus sous des formes optiquement actives [8,25].
La fonctionnalisation de la position axiale est une autre approche pour modifier les propriétés de ces composés afin d’obtenir des matériaux sur mesure [26,27].
Jusqu’au début de ce travail, à notre connaissance, les sousphtalocyanines n’ont jamais été obtenus à partir de l’acide 1,4-diboronique benzène. Dans ce travail, nous synthétisons la SubPcBCl à partir de l’acide 1,4-diboronique benzène. Le composé obtenu a été caractérisé par spectrométries de masse, IR, UV-visible et RMN du proton.
2 Partie expérimentale
Les réactifs Carlo Erba SDS, Merck, Aldrich de pureté commerciale ont été utilisés sans purification supplémentaire. Le phtalonitrile a été recristallisé dans du méthanol. Le butanol-1 et le tétrachlorure de carbone, bien que de grade anhydre, ont été séchés (mis sur tamis moléculaires).
2.1 Spectrométrie de masse
2.1.1 Conditions expérimentales
2.1.1.1 Matériel
Les analyses par spectrométrie de masse basse et haute résolution ont été effectuées avec un spectromètre de masse QStar Elite (Applied Biosystems SCIEX) équipé d’une source d’ionisation à pression atmosphérique (API) assistée pneumatiquement.
L’échantillon a été ionisé en mode électrospray positif dans les conditions suivantes : tension électrospray (ISV) : 5500 V ; tension d’orifice (OR) : 20 V ; pression du gaz de nébulisation (air) : 20 psi.
Les spectres de masse basse et haute résolution ont été obtenus avec un analyseur temps de vol (TOF).
La mesure de masse exacte (haute résolution) a été effectuée en triplicat avec un double étalonnage interne.
2.1.1.2 Méthode
« Basse résolution » : l’échantillon est dissout dans 300 μL de dichlorométhane puis dilué au 1/10 dans une solution de méthanol à 0,5 % d’acide formique.
« Haute résolution » : l’échantillon est dissout dans 300 μl de dichlorométhane puis dilué au 1/10 dans une solution de méthanol à 3 mM d’acétate d’ammonium.
Étant donnée la masse du composé ciblé, les ions choisis pour références internes sont l’ion pseudomoléculaire, [M + H]+ et l’adduit ammonium, [M + NH4]+, de deux oligomères du poly(propylène glycol) (PPG425) attendus respectivement à m/z 367.2690 et m/z 442.3374.
La solution de l’extrait est dopée avec l’étalon interne puis introduite dans la source d’ionisation par infusion à un débit de 20 μL/min.
2.2 Spectrométrie infrarouge
Le spectre IR a été enregistré à partir de pastille de KBr à 1 % sur un appareil IR Brücker IFS35 à transformée de Fourrier.
2.3 Spectrométrie UV-visible
Le spectre a été enregistré sur un spectromètre UV-LAMBDA 35 à partir des solutions dans le chloroforme et le dichlorométhane.
2.4 Spectrométrie RMN 1H
Les spectres ont été enregistrés sur spectromètre RMN de 400 MHz à partir de solution du composé dans du chloroforme deutérié CDCl3.
3 Synthèse de la chlorosousphtalocyanine de bore à partir de l’acide 1,4-diboronique benzène
Le schéma réactionnel est le suivant.
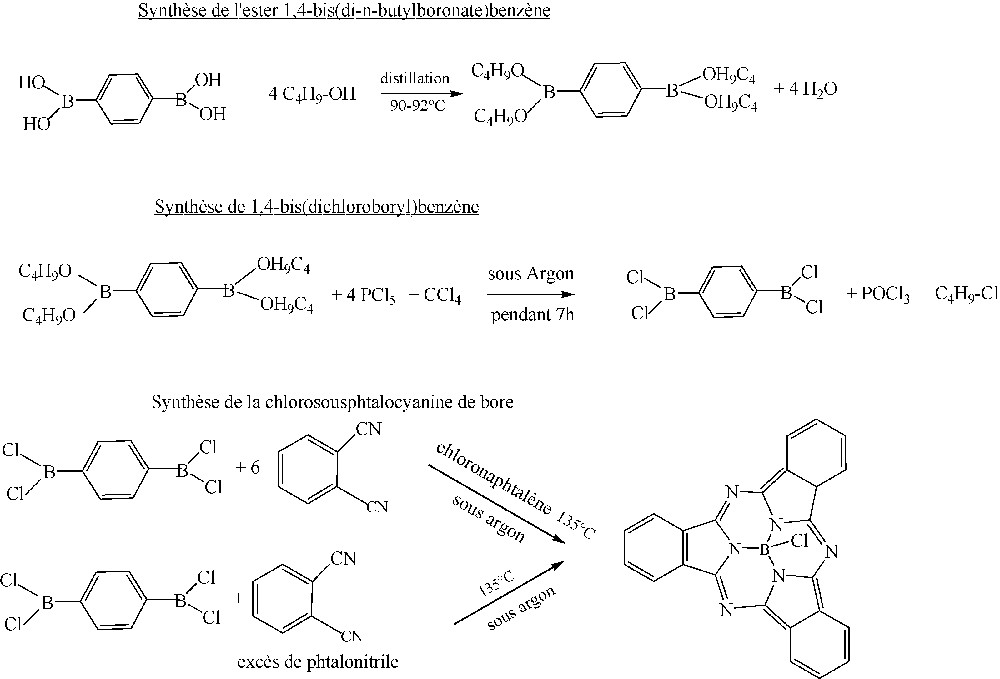
Schéma réactionnel de la synthèse.
3.1 Synthèse du 1,4-bis (di-n-butyl boronate) benzène par estérification de l’acide 1,4-diboronique benzène par le n-butanol
Nous avons adapté une méthode décrite par Nielsen et McEwen [28].
On fait réagir dans un appareil de microdistillation et dans les proportions 1:4 l’acide 1,4-bis (dihydroxy boronique) benzène (0,0121 mole) et le n-butanol (0,0725 mole) entre 90 à 92 °C. La réaction est conduite sous argon et dans des conditions rigoureusement anhydres, car l’ester obtenu s’hydrolyse rapidement avec l’eau pour redonner l’acide 1,4-bis (dihydroxy boronique) benzène et le n-butanol. Il est extrêmement sensible à l’oxygène et à l’humidité de l’air et s’hydrolyse presqu’instantanément en contact de l’humidité de l’air.
L’ester est obtenu après élimination de l’azéotrope eau-butanol (composé de 75 % d’eau et de 25 % de butanol) provenant de la réaction d’estérification à 93 °C et de l’excès de butanol.
On recueille un liquide dont la température d’ébullition est comprise entre 160 à 176 °C.
3.1.1 Synthèse du 1,4-bis (dichloroboryl) benzène
Pour cette synthèse, nous avons adapté une méthode décrite par Nielsen et McEwen [28].
Dans un schlenk contenant 10 g (0,0482 mole) de pentachlorure de phosphore PCl5, on introduit, sous argon, la solution de l’ester 1,4-bis (di-n-butyl boronate) benzène dissout dans 8,93 mL de tétrachlorométhane CCl4. Sous agitation, le mélange est porté au reflux pendant sept heures.
Après le reflux, on distille le CCl4 et les sous-produits oxychlorure de phosphore POCl3 et le chlorure de n-butyle C4H9Cl. On élimine au maximum la partie liquide en séchant sous vide pendant 24 heures. Lorsque la majeure partie du liquide a été éliminée, le mélange réactionnel beige clair devient plus foncé. Ce qui montre que la réaction de chloration se poursuivait avec l’excès de PCl5. Le produit obtenu est sublimé sous argon. Le 1,4-bis (dichloroboryl) benzène est extrêmement sensible à l’oxygène et à l’humidité de l’air et s’hydrolyse presqu’instantanément en contact de l’humidité de l’air pour donner le 1,4-bis (dihydroxy boronique) benzène.
3.2 Synthèse de la chlorosousphtalocyanine de bore
On fait réagir dans les proportions 1:6 le composé beige avec du phtalonitrile. Cette réaction peut se faire soit en présence d’un solvant organique ayant une température d’ébullition élevée, soit en utilisant un excès de phtalonitrile (le phtalonitrile fondu jouera le rôle de solvant).
Dans le réacteur contenant le produit beige [1,4-bis (dichloroboryl) benzène], on introduit 9,3 g (0,0726 mole) de phtalonitrile recristallisé dans du méthanol anhydre et 10 mL de chloronaphlalène. Sous argon, le mélange est porté au reflux à 135 °C pendant cinq heures. Après le reflux, le chloronaphtalène est éliminé ; on obtient un mélange réactionnel rouge noirâtre.
Lorsque la réaction se produit dans le phtalonitrile fondu, on obtient dans ce cas un produit solide noir.
3.3 Purification
Le produit brut obtenu est récupéré avec du méthanol. On filtre et on isole un solide marron-violacé. Le lavage au méthanol permet d’éliminer l’excès de phtalonitrile et ses produits de dégradation. Le solide est ensuite lavé à l’hexane par soxhlet pendant 24 heures afin d’éliminer les traces de phtalonitrile puis au chloroforme. On obtient un solide violet. Le SubPcBCl a été isolé par sublimation aux températures comprises entre 280 à 350 °C à une pression de 10−5 à 10−6 torrs avec un rendement de 45 %.
4 Résultats expérimentaux et discussion
4.1 Spectrométrie de masse
Les analyses par spectrométrie de masse basse et haute résolution du composé permettent de conclure que sa formule brute est C24H12N6BCl, soit une masse exacte au maximum isotopique de 430.0905, avec une erreur inférieure à 3 ppm.
4.2 Spectrométrie d’absorption infrarouge
Les spectres infrarouges des sousphtalocyanines sont essentiellement composés de déformations des élongations et de flexions des liaisons C-C et C-N. Les attributions sont faites par analogie avec ceux des Pc. La bande de déformation d’élongation de la liaison B-Cl apparaît à 953 par centimètre [1].
Nombres d’ondes en centimètre et intensité des bandes de vibrations : 1614(f), 1457(tF), 1388(e), 1325(m), 1285(f), 1232(F), 1198(m), 1133(tF), 1091(f), 1064(f), 953(F), 795(m), 758(tF), 728(f), 697(m), 633(f), 569(m), 442(f). Ces résultats sont conformes aux données de la littérature [1].
4.3 Spectrométrie UV-visible
L’allure du spectre UV-visible obtenu est identique à celle donnée dans la littérature. La bande de la transition principale (Q) apparaît à 565 nm suivi de deux épaulements à 541 nm et à 512 nm ; elle est plus intense que la bande de soret (B) qui apparaît à 306 nm.
4.4 Spectrométrie RMN 1H
Les pics apparaissent sous forme de deux multiplets aux déplacements chimiques compris entre 7,75 à 7,98 ppm et entre 8,87 à 8,92 ppm. Les données de la littérature [2,5–7,19] permettent d’attribuer ces multiplets aux protons reliés aux atomes de carbone Cγ et Cδ [5,7]. Un multiplet correspondant aux 6 H3,6 benzéniques apparaît à 8,84 à 8,81 ppm et un autre multiplet attribuable aux 6 H4,5 benzéniques sort à 7,94 à 7,91 ppm.
5 Conclusion
Le schéma réactionnel adopté nous a permis d’obtenir, pour la première fois, SuPcBCl à partir de l’acide 1,4-diboronique benzène. La nature du composé de synthèse est confirmée par les mesures spectrométriques de masse, infrarouge, UV-visible et de RMN H1.