

1 Introduction
An efficient organic synthesis is characterized by a small number of steps to reach the target molecule, a high selectivity and good atom economy, readily available starting materials and low costs for reagents and for performing the reaction. Green chemistry prefers those routes that additionally provide waste reduction, non-toxic reagents and solvents, high safety standards and efficient use of resources and energy (see Chapter 3.) Electrolysis offers C–C bond formations and functional group interconversions (FGIs) for all compounds that are electroactive or can react with an electrogenerated reagent. This holds for a large variety of substances as with increasing potential of the electrode each compound becomes electroactive and as most of the electrogenerated reagents are sufficiently reactive. To which extent organic electrosynthesis (ES) meets the requirements of green chemistry will be examined in sections 4–9; earlier contributions to this topic are quoted in [1]. In Chapter 2. a short introduction to the reactions and the practice of organic ES will be given.
2 Introduction to reactions and practice of organic electrosynthesis
Electrochemical synthesis uses the combination of an electron transfer at an electrode with a chemical reaction. The electron transfer converts the substrate to a reactive intermediate (ion radical, radical, anion and cation) or generates a reagent (electrophile, nucleophile, acid and base). As the electron is transferred at a potential that is specific for the electroactive group, the electron transfer is potential selective, which can be employed in a potential controlled electrolysis. Most often thermal activation is not required, which decreases the energy costs and permits one to increase the selectivity of the chemical follow-up reaction. Furthermore the succeeding chemical reaction can be influenced by changing the concentration of the intermediates via the current density.
The reactive intermediates can form C–C bonds by coupling, by electrophilic, nucleophilic and radical addition. Most important is the change of the substrate reactivity by the electron transfer. A nucleophile can be converted to an electrophile at the anode and an electrophile to a nucleophile at the cathode. This in situ redox-umpolung [2] is a unique reaction of ES and that allows one to reduce the number of steps and avoid the widespread use of auxiliaries.
For all functional group interconversions (FGIs) that involve an oxidation or reduction, the electrode has advantages for economic and ecological reasons. The electron in electrolysis is one of the cheapest reagents in chemistry; it furthermore produces no waste in contrast with chemical oxidants or reductants. In addition, the electrode offers FGIs that are different to chemical FGIs and this way it opens new strategies in synthesis (Scheme 1).
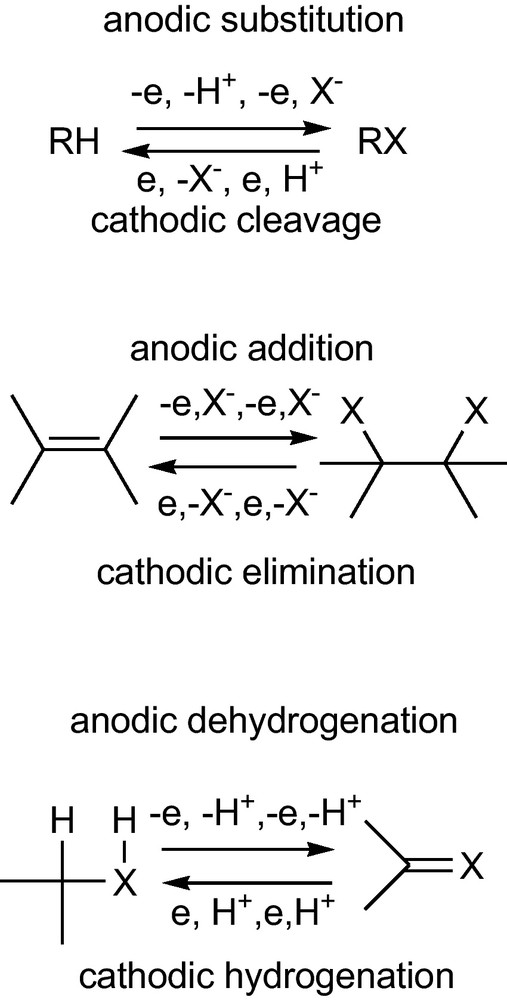
Electrochemical functional group interconversions (FGIs).
Anodic substitution (Scheme 1) allows the replacement of a hydrogen atom for a nucleophile, which in non-electrochemical reactions is not possible in one step. At the cathode, the reverse reaction can happen; the cathodic cleavage replaces a nucleophilic leaving group X by a proton, which is at least a two-step reaction in a non-electrochemical conversion.
In an anodic addition, two nucleophiles can be added to a double bond in one step. The reverse reaction at the cathode is the cathodic elimination, which generates a double bond by removal of two vicinal nucleophiles. In the non-electrochemical counterparts, an electrophile and a nucleophile are added or eliminated.
Non-electrochemical hydrogenation of double bonds is achieved catalytically with molecular hydrogen, or in the case of electron-deficient double bonds, by addition of a hydride ion. Cathodic hydrogenation can be achieved by a proton and an electron, which may lead to chemoselectivities and stereoselectivities alternative to those found in non-electrochemical hydrogenation.
In order to get an indication on the ease of an electrochemical conversion one estimates the oxidation- or reduction-potential of the substrate either from literature data or by taking a current/voltage curve (see below). In general, a substrate is the more easily reduced or oxidized the more extended is its π-system. Reduction is made easier, when the substrate contains empty orbitals and/or electron attracting substituents that stabilize the negative charge. Oxidations are facilitated vice versa by electron donating n-orbitals and/or electron donating substituents that stabilize the created electron hole.
For the electrolysis, one needs an electrolysis cell, electrodes and a solvent/supporting electrolyte (E) (Fig. 1a). The electrolysis cell contains a working electrode (WE) to convert the substrate to a reactive intermediate. A Luggin-capillary (L) allows one to measure the potential at the WE versus the reference electrode (RE) with a high resistance voltmeter (V). The set-up permits one to determine current/voltage curves by measuring the current density (mA cm−2, current per electrode surface) with an amperemeter (A) while changing the potential at the WE with an adjustable direct-current source (S). This way information about the oxidation or reduction potential of the substrate is obtained. If one keeps a constant potential at the WE versus the RE, one can achieve a potential selective conversion of the substrate (controlled potential electrolysis). The cell voltage between the working and the counter electrode (CE) is provided from an adjustable direct-current source (S); a maximal power of 1 A and 50 V is sufficient. As anode material one can use, in acidic and basic medium, a thin platinum foil on a Teflon-support, a graphite plate or rod, a glassy carbon plate or lead dioxide deposited on a graphite plate. Recently boron doped diamond electrodes (BDDEs) have become commercially available; they are corrosion resistant at high potentials and have a high oxygen and hydrogen overvoltage [1f]. In basic medium a nickel or steel electrode can be applied. As cathode material platinum, graphite, glassy carbon, steel, magnesium, zinc, aluminium or the BDDE can be used; they are employed as foils, plates or rods.

a) Schematic presentation of a divided cell: WE: working electrode, CE: counter electrode, RE: purchasable reference electrode, L: Luggin capillary (glass tube with d = 3 mm and bent tip with d = 0.2 mm; upper end connected via a G4-frit with reference electrode; the tip is about 1 mm distant from the surface of the WE; the capillary is filled with electrolyte), V: high resistance voltmeter, A: amperemeter, S: adjustable direct-current source, E: electrolyte, D: diaphragm; b) Undivided cell: double walled beaker type cell [100 ml] (1), platinum foil electrode [3 × 7 cm] on a Teflon-support (2), glassy carbon electrode (3) with Luggin capillary (4), platinum net electrode (5), Teflon-stopper (6).
The amount of organic solvent in the electrolyte has been drastically reduced with a new cathode that was applied in the cathodic addition of ethyl 2-bromoalkanoates to benzaldehydes. The cathode consisted of compressed graphite powder that was impregnated with a mixture of the substrates and 0.02 ml of a 0.1 M TBABF4 solution in methanol [3].
In some cases, the working electrode and the counter electrode have to be separated by a diaphragm (ion-exchanger membrane, porous glass or ceramic disc) to avoid the conversion of substrate or product at the counter electrode. Frequently diaphragms, which make the electrolysis more difficult, are replaced in cathodic reductions by sacrificial counter anodes that are oxidized to form metal cations. An elegant way to avoid a diaphragm is a paired-electrolysis. Here both electrodes are used to synthesize products. For examples of paired-electrolyses see [1b] and for those in a technical scale see Chapter 9. Both the diaphragm and the supporting electrolyte could be avoided in a microflow reactor. In a paired-electrolysis, benzyl chlorides were reduced to toluenes in up to 87% yield at the cathode and simultaneously 1-phenylethanols were oxidized to acetophenones in up to 61% yield in a 30 mmol scale. The reaction was performed in a microflow reactor in acetonitrile without a supporting electrolyte in a single pass of substrate using a parallel laminar flow [4].
The selectivity of reactive electrogenerated intermediates could be remarkably increased by using a micromixer. With N-acyl iminium ions as initiators for the cationic polymerization of butyl vinyl ether the Mw/Mn-ratio improved from > 2 (batch reactor) to 1.14 using the micromixer. For 1,3,5-trimethoxybenzene, the selectivity for mono- to dialkylation increased from 32% (batch reactor) to 92% (micromixer) [5].
For preparative-scale electrolysis, the author has used in many cases an undivided beaker type cell (Fig. 1b). Platinum electrodes made from thin platinum foils were attached to a Teflon-support. The electrodes were fixed via steel rods as current feeders in holes of a Teflon stopper.
The electrolyte consists of a solvent and a supporting electrolyte (sse). The supporting electrolyte has to be soluble in the solvent and has to provide a good conductivity, which is achieved in less polar solvents with large lipophilic ions. Solvent and supporting electrolyte should be electrochemically and chemically stable, be either protic or aprotic as desired, good or poor nucleophiles or electrophiles, easy to purify and to recycle.
Electrolytes play a key role in defining the environment surrounding an electrode. A lipophilic electrolyte, e.g. tetraethylammonium tosylate and THF will favour nonpolar species relative to polar ones, whilst lithium perchlorate in methanol will do the reverse [6].
In the search for good reaction conditions, one should begin with electrodes and sse's that are inert at the reduction/oxidation potential of the substrate. A good choice is a platinum or graphite electrode and a 0.1 M Bu4NBF4 solution in acetonitrile, where the anodic limit is 2.7 V (vs. Ag/AgCl) and the cathodic limit −2.6 V (vs. Ag/AgCl). After being familiar with the reaction conditions possibly greener solvent/supporting electrolytes with narrower limits can be applied. As solvents for oxidations can be used: methanol, water, acetonitrile, and for reductions can be applied: methanol, water, tetrahydrofuran or dimethylformamide. As supporting electrolytes tetraethylammonium tosylate, tetrabutylammonium tetrafluoroborate and triethylmethylammonium methyl sulfate are useful; in polar organic solvents, they afford a good conductivity and exhibit a wide potential window. For cathode processes in water or methanol alkali halides can be used as supporting electrolytes; for anode processes in these solvents potassium fluoride, sodium benzenesulfonate, potassium nitrate or sodium methanesulfonate are applicable. More information on equipment, electrodes, solvent/supporting electrolytes or on the selection of favourable electrolysis conditions are found in textbooks [7].
In the Chapters 4.–9., examples of C–C bond formation and FGI at the anode and cathode are given to illustrate how organic ES accords with the principles of green chemistry (Chapter 3.). Representative reactions have been chosen; they are organized via type of reaction and substrate. In many cases, the electrochemical conversion is compared with the conditions of the non-electrochemical synthesis leading to the same or a similar molecule1. Before these individual comparisons are done, the general agreement of organic ES with the principles of green chemistry is summarized in Chapter 3.
3 Organic electrosynthesis and the principles of green chemistry
Organic ES agrees considerably with the principles of green chemistry [8].
3.1 Prevention of waste
In ES, waste is reduced or prevented as the reagent: electron is not bound to a chemical compound and is therefore inherently pollution free.
3.2 Atom economy
Each molecule can become electroactive at a certain potential. As the electron is transferred to or from electroactive groups in the molecule no additional groups have to be attached (see also 3.8.).
3.3 Hazards
In ES, hazards are decreased due to the replacement of frequently toxic oxidants or reductants by electricity.
3.4 Safer chemicals
In ES, conditions are chosen mostly in such a way that the source of the electron, the electrode does not corrode. The cathode material: mercury, formerly applied for suppressing unwanted proton reductions, is today, whenever possible, replaced by non-toxic materials, e.g. graphite.
3.5 Safe solvents
ES in the laboratory employs in many cases safe solvents; in technical processes water or mixtures of water and alcohols are often used in flow reactors that run continuously for months or even years. Conductivity is achieved with 0.05 M to 0.5 M solutions of an inert supporting electrolyte in the solvent. Ionic liquids [9] with an inherent good conductivity as R3N·nHF and R4NF·nHF are successfully used for partial fluorinations [10]. In addition, ionic liquids are also attractive because of their low volatility and their wide potential window at the anode and cathode. For that reason, applications in small-scale synthesis are emerging; examples are carboxylations, conversions with electrogenerated bases (EGBs), reductive elimination, pinacolization, polymer modifications or reactions with organometallics [1(g),11] and further reactions shown in following chapters. The use of ionic liquids in larger-scale electrolyses should be cautiously considered. Most conversions in ionic liquids are described for a mg-scale, some of the liquids are ecological risky and fairly expensive; furthermore they are characterised by low diffusion coefficients, which lead to low current densities that require large and expensive electrode areas. Sometimes products, especially polar ones, are hard to extract, which lowers the yields and makes the reuse of the liquid difficult.
The solvent/supporting electrolyte (sse) commonly used in organic ES is in most cases cheap, flexible to use, easy to recycle and allows a simple product isolation because the solvent can be easily removed and recycled by distillation.
3.6 Energy efficiency
In ES, the processes are energy efficient as the reactions are conducted mostly at ambient pressure and temperature and cheap electricity is used as redox-reagent and driving force for the reaction.
3.7 Renewable feedstock
In ES electrochemical conversions can be applied to all substrates that either bear electroactive groups (electrophores) or can react with electrogenerated intermediates. This also holds for renewable feedstock as carbohydrates, fatty acids, amino acids or hydroxy acids, plant oils, cellulose, lignin [1(d),12].
3.8 Unnecessary derivatization
In ES activation through derivatives becomes unnecessary because the substrate is selectively activated by electron transfer to or from the inherent electrophore. The possibility to change the polarity of a substrate, e.g. from an electrophile to a nucleophile, by electron transfer (redox-umpolung [2]) saves reaction steps as the otherwise not possible reaction with an electrophilic reagent becomes possible. Electrochemistry allows one to conduct radical reactions easily and on large scale. As polar groups are tolerated in most radical conversions, protecting groups become less necessary.
3.9 Catalysis
The electrode can collect or provide electrons in catalytic oxidations or reductions and this way can regenerate the active form of chemical catalysts or enzymes [13].
3.10 Degradation
Electrolysis with boron-doped diamond electrodes (BDDE) allows the total degradation of toxic organic material [1(f),14].
3.11 Analytical methodology
The analytical methodology is based to an appreciable extent on electrochemical sensing [1c]. The electrical signal can be used as feedback to control the reaction conditions in electrolysis (potential, current density, current consumption) in order to prevent the formation of polluting products.
3.12 Safer chemistry
The large repertoire of reactions in ES allows one to circumvent hazardous compounds or to produce and use them in small amounts for in situ reactions.
Recently N-isobutyl-(2E,6Z)-dodecadienamide was synthesized in four steps exclusively by non-electrochemical reactions (route 1) and by electrochemical reactions (route 2). For three steps the organic ES led to higher or equal yields compared with the non-electrochemical conversion. In one step, a Wittig reaction, the organic ES failed, because in the electrogeneration of the base the phosphonium salt was cleaved but not deprotonated [15].
4 C–C bond formation at the anode
4.1 Aromatic compounds
Aromatic compounds can be coupled intermolecularly or intramolecularly at the anode. It has been shown that the first step is the oxidation of the aromatic compound 1 to the cation radical 2. Radical coupling of 2 with subsequent deprotonation leads to biaryls 4 (Scheme 2, path a)). Deprotonation of 2 and loss of an electron produces the benzyl cation 3, which can react in an electrophilic aromatic substitution with 1 to diphenylmethanes 5 (Scheme 2, path b)). Path a) is favoured by a low charge density on an unsubstituted carbon atom of 2, whilst a low charge density on a substituted carbon atom aids path b) [16a].

Intermolecular coupling of aryl compounds 1 to biaryls 4 and/or diphenylmethanes 5.
One example for path a) and one for path b) is shown for 1,3,5-trimethylbenzene and 1,2,4,5-tetramethylbenzene, respectively in Scheme 3(a) and (b). For the scope of the reaction and details on the mechanism, see [16(a)].

Anodic coupling of 1,3,5-trimethylbenzene and 1,2,4,5-tetramethylbenzene1 [16].
Reaction conditions, selectivities and mechanistic pathways for the intermolecular anodic coupling of phenols, arylethers and arylamines are compiled in [17]. The yields in phenol coupling vary from very low to excellent depending on the structure of the phenol and the choice of the reaction conditions. Phenol oxidations often suffer from further oxidation of the products. This overoxidation lowers the yield and can lead to an insulating deposit on the electrode (passivation). Recently the boron doped diamond (BDD) anode [1f] and hexafluoroisopropanol as solvent additive have been applied to phenol oxidation. Passivation and unwanted side products could be decreased that way, because due to the high oxygen overpotential of the BDD anode deposits and electroactive side products are degraded slightly faster than the product. Thereby the phenols 6–12 could be coupled in good to moderate yields (Scheme 4) [18]. In addition phenol 11 can be coupled in 29% yield in acetonitrile/LiClO4 at a glassy carbon anode [19].

Coupling of phenols 6–12 at the boron doped diamond (BDD) anode (arrow indicates coupling position)1 [18].
The cross coupling of 4-methylguajacol (A) with eight different arylethers (B) in a ratio A: B = 1: 10 affords at the BDD anode unsymmetrical biaryls in a one step reaction without using activating auxiliaries (Scheme 5). Depending on current consumption, current density and structure of the arylether cross coupling products A–B are formed in selectivities of A–B: B–B ranging from 1.5: 1 to > 50: 1 with yields of 16 to 47%. The substrate is electrolyzed neat with only a small amount of hexafluoroisopropanol as additive, which can be recycled.
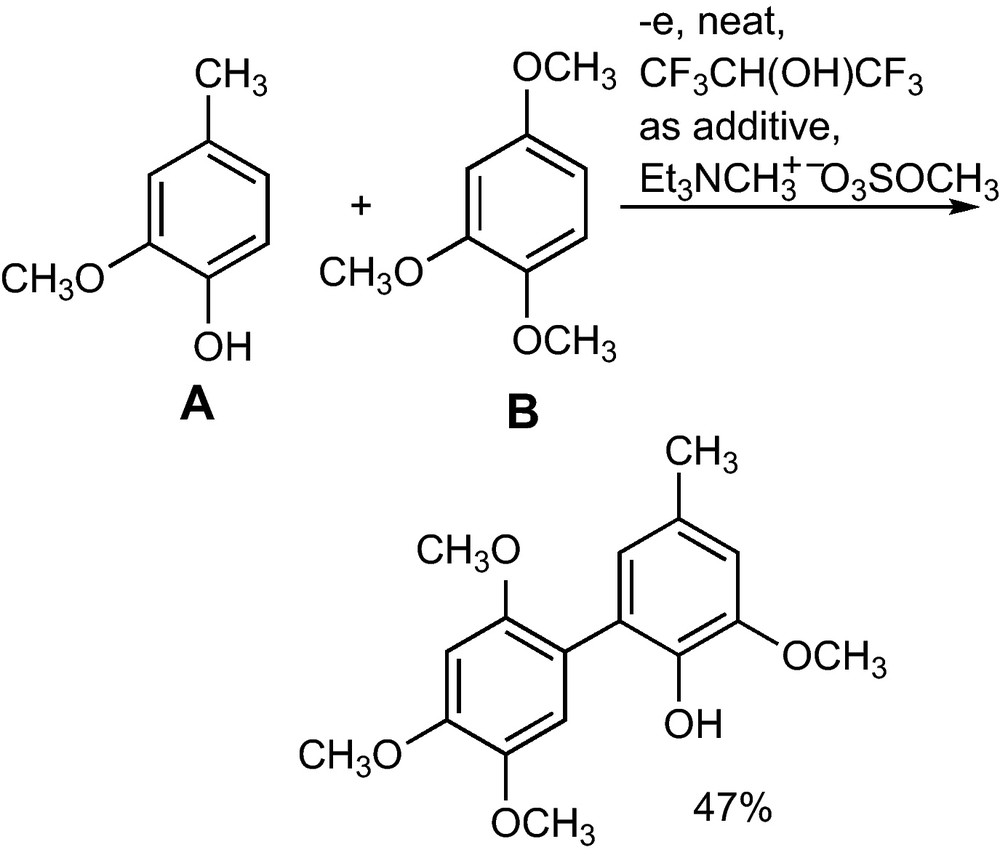
One-step anodic cross coupling of 4-methylguajacol (A) with 1,2,4-trimethoxybenzene (B) without activating auxiliaries1 [20].
Polycyclic aromatic compounds can be obtained by intramolecular coupling via a cation radical that undergoes an electrophilic aromatic substitution or a radical coupling. This way, by combination with an electron transfer (redox-umpolung) [2], two groups of similar polarity can be connected in one step. An example leading to a 9,10-dihydrophenanthrene is shown in Scheme 6 [21]. The intramolecular coupling shown in Scheme 6 is one of many examples compiled in [22]. These cyclizations provide a facile access to core structures of natural products for example of the morphine skeleton [23] or of isoquinolines [24].

Anodic intramolecular coupling of arylethers (DCM = dichloromethane, TFA = trifluoroacetic acid)1 [21].
4.2 Olefins
Olefins with substituents at one vinylic carbon atom that stabilize an electron deficient cation radical as the phenyl, vinyl, amino or alkoxy group dimerize anodically at the vinylic carbon atom that carries no stabilizing substituent. Intermediates are probably dimers of the cation radical. These 1,4-dications either undergo solvolysis to generate 1,4-dimethoxy-dimers and/or deprotonation to form 1,3-dienes.
Enolethers couple to acetals of 1,4-dicarbonyl compounds that hydrolyze to form 1,4-dicarbonyl compounds (Scheme 7), which are valuable intermediates for the preparation of five membered heterocycles [25].

Intermolecular anodic coupling of an enolether1 [25].
Aryl olefins dimerize depending on the substitution and the reaction conditions to form either 1,4-dimethoxy-1,4-diphenylbutanes or to produce 1,4-diphenyl-1,3-butadienes by methanolysis or deprotonation of the intermediate dimer dication, respectively [26]. 2-Phenyl-1-propene leads exclusively to the E,E-diene (Scheme 8).

Anodic coupling of 2-phenyl-1-propene1 [26].
Enaminoketones or -esters couple to symmetrical pyrroles (Scheme 9) [27]. Here the 1,4-dication forms the pyrrole by intramolecular aminolysis, deprotonation and elimination of benzylamine.
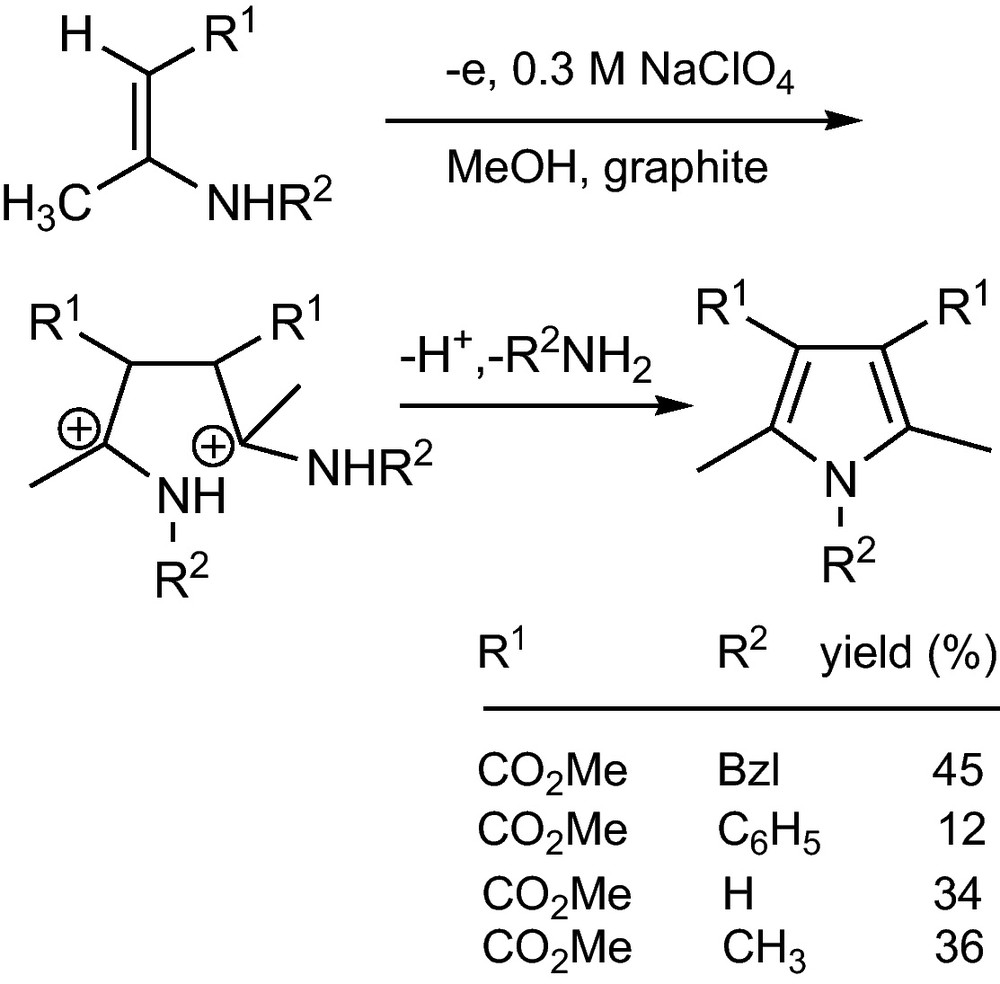
Anodic coupling of enaminoesters1 [27].
Anodically initiated [2 + 2]-cycloadditions between aryl enolethers and alkenes have been achieved in a lithium perchlorate/nitromethane electrolyte. Probably the enol ether cation radical adds to the alkene, which is followed by an electron transfer from the enolether to the ring closed cation radical, whereby a new enolether cation radical for the next cycle is generated (Scheme 10).

Electrocatalytic cycloaddition of enolethers [28].
Intramolecular coupling allows the regioselective and stereoselective C–C bond formation between two equal or two different electrophores. The connection of an enolether with an allylsilane permits the smooth formation of quaternary carbon atoms. In some cases, the reactions can be run without loss of selectivity and yield in a simple beaker type cell with a carbon rod as anode and a 6 V lantern battery as power supply (Scheme 11) [29].

Intramolecular coupling of different electrophores [29].
The intramolecular anodic coupling between two different enolethers is the key step in the synthesis of alliacol A [2(c)].
Intramolecular anodic olefin additions can be used to generate new carbon-oxygen bonds and to synthesize furanose and pyranose C-glycosides [6]. Similarly new carbon-nitrogen bonds can be created to afford substituted pyrrolidine and piperidine rings (Scheme 12) [30].
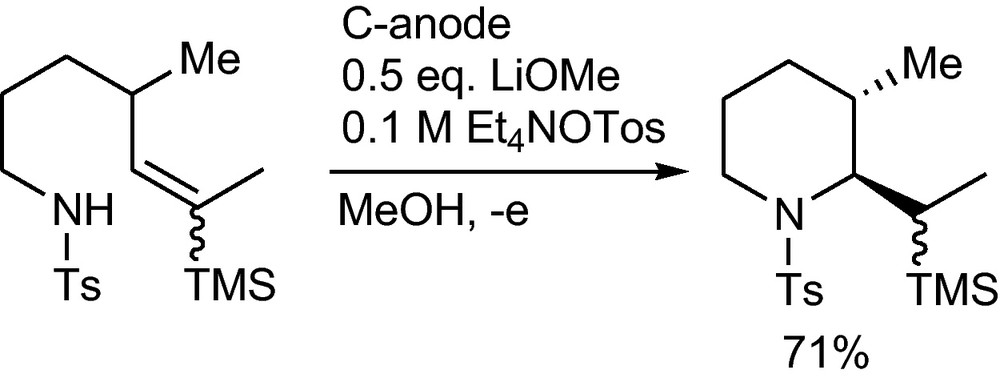
Anodic carbon-nitrogen bond formation to afford substituted pyrrolidine and piperidine rings [30].
While optimizing these cyclizations mechanistic principles emerged, these were: a more polarized cation radical favours C–C bond formation, whilst a less polarized cation radical supports C-heteroatom formation. These principles were an excellent guide for choosing the donor groups at the double bond that improved the wanted selectivity for either C–C or C–heteroatom bond formation [31].
4.3 Carboxylic acids
C–C bond formation via radicals can be accomplished by way of radical coupling or radical addition. A versatile and experimentally simple to handle source for radicals is the Kolbe-electrolysis of carboxylic acids [32]. An undivided beaker type cell with a platinum anode and a steel or platinum cathode is sufficient to do the anodic decarboxylation of carboxylates to radicals (Fig. 1b). One applies a high constant current density in an undivided cell from a battery or power source, no potential control is necessary. The acid is dissolved in methanol and neutralized to about 5%; consumed carboxylate is continuously regenerated from the acid by reduction of the protons at the cathode. High yields of symmetrical dimers (homocoupling) are obtained using these simple experimental conditions. The acid should be either unsubstituted at C-2 or have an electron attracting group at C-2 to suppress the competing non-Kolbe electrolysis. This way homomuscone or the C-disaccharide decitol, derived from glucose have been obtained (Schemes 13 and 14) [1(d),33,34]. When a low current density is applied and the acid is substituted with an electron donating group at C-2, the intermediate radical is further oxidized to a carbocation that undergoes substitution or elimination (non-Kolbe electrolysis, see Chapter 6.1.). Unsymmetrical coupling products are obtained in the coelectrolysis of two different acids (heterocoupling). Mostly the cheaper acid is taken in excess, to favour a maximal incorporation of the more costly acid into the product. This way C-glycosides (Scheme 15) [35] and pheromones (Scheme 16) [36] have been prepared. Further examples of pheromone syntheses via heterocoupling of carboxylic acids are shown in [32].
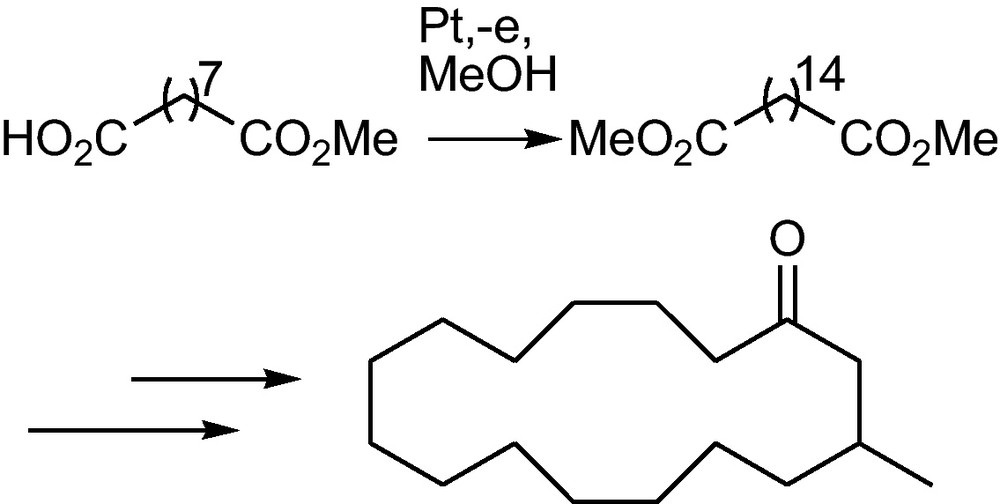
Homomuscone via homocoupling of methyl azelate1 [33].

Anodic coupling of 2-desoxy-gluconic acid to 5,6-didesoxydecitol octaacetate 1[1d].

Heterocoupling of fatty acids with carbohydrate carboxylic acids1 [35].

Lepidoptera polyene pheromone by heterocoupling of a carboxylic acid with linolenic acid1 [36].
The addition of anodically generated radicals to olefins can lead to an additive dimer, when the primary adduct dimerizes; an additive monomer is formed, when the primary adduct couples with the Kolbe-radical. The ratio of additive dimer to additive monomer can be influenced by the current density and the olefin concentration. In the electrolysis of methyl malonates in the presence of styrene, the intermediate benzyl radical couples to an additive dimer (Scheme 17) [37].
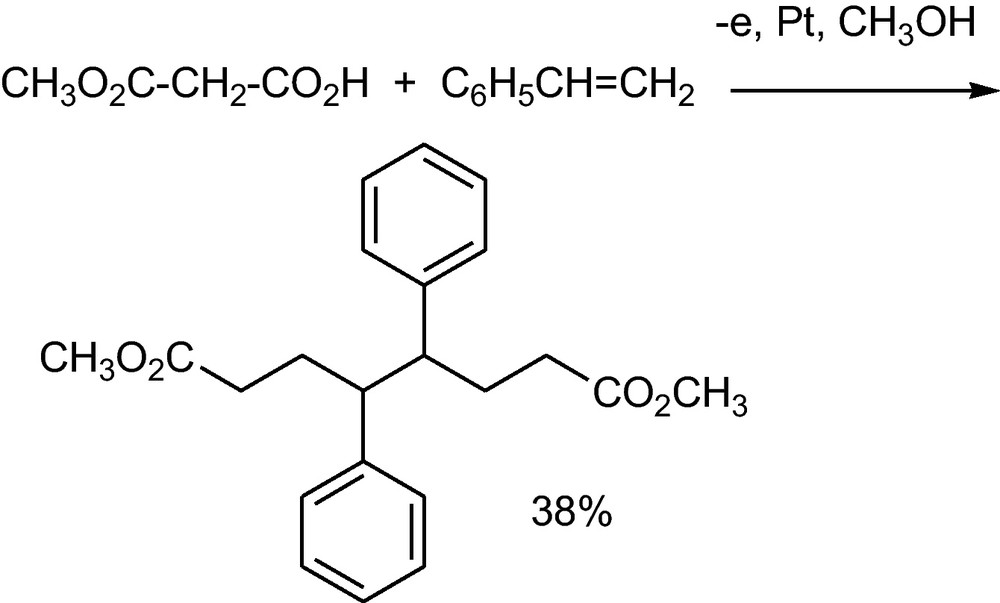
Additive dimerization of styrene with a methoxycarbonylmethyl radical generated from methyl malonate1 [37].
In intermolecular addition, the yields are much better with more reactive olefins like styrene or butadiene than with less reactive olefins. However, good yields are also obtained with less reactive olefins in intramolecular additions. Here the Kolbe-radical adds to the double bond in a 5-exo-trig-reaction to form cyclic products, for example tetrahydrofurans (Scheme 18) [38]. The reaction has an alternative scope when compared to the cyclization of 5-alkenyl bromides with tributyltin hydride. At the anode, two C–C bonds are formed, whereby the second carbon chain can be flexibly changed via the choice of the coacid. Furthermore contrary to tributyltin hydride used in the non-electrochemical reaction, the electrode is non-toxic. With the double bond in a suitable position, consecutive additions are possible. In this way, a tricyclic product is formed in a one-pot reaction from a readily available starting compound in a tandem cyclization (Scheme 19) [39].

Intramolecular addition of Kolbe radical to form a prostaglandin precursor [38].

Radical tandem cyclization via Kolbe radicals [39].
The homo- and heterocoupling and the olefin addition of radicals generated by anodic decarboxylation of carboxylates has found widespread application. But also anions formed by deprotonation of CH-acidic compounds such as nitroalkanes or anionic organometallics like Grignard reagents have been dimerized at the anode (Schemes 20 and 21) [40,41].
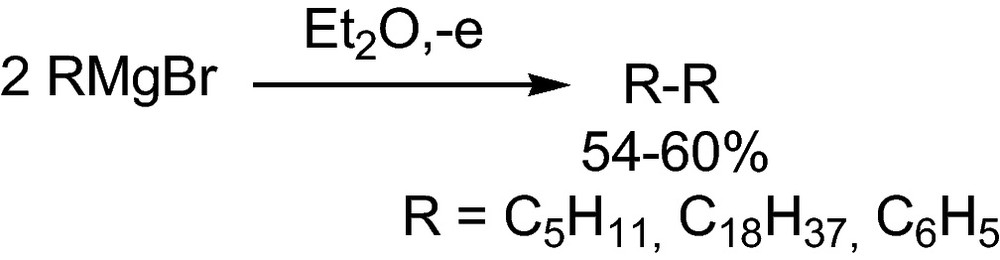
Anodic dimerization of Grignard reagents1 [40].

Anodic coupling of nitroalkanes [41].
Dimethyl malonate has been deprotonated and the anion oxidized in the presence of styrene to afford the orthoester of a γ-butyrolactone (Scheme 22) [42]. Here presumably the first formed radical adduct, a benzyl radical, is further oxidized to a benzyl cation that reacts with the oxygen atom of the ester carbonyl group; the subsequent methanolysis of the cyclic cation leads to the orthoester.
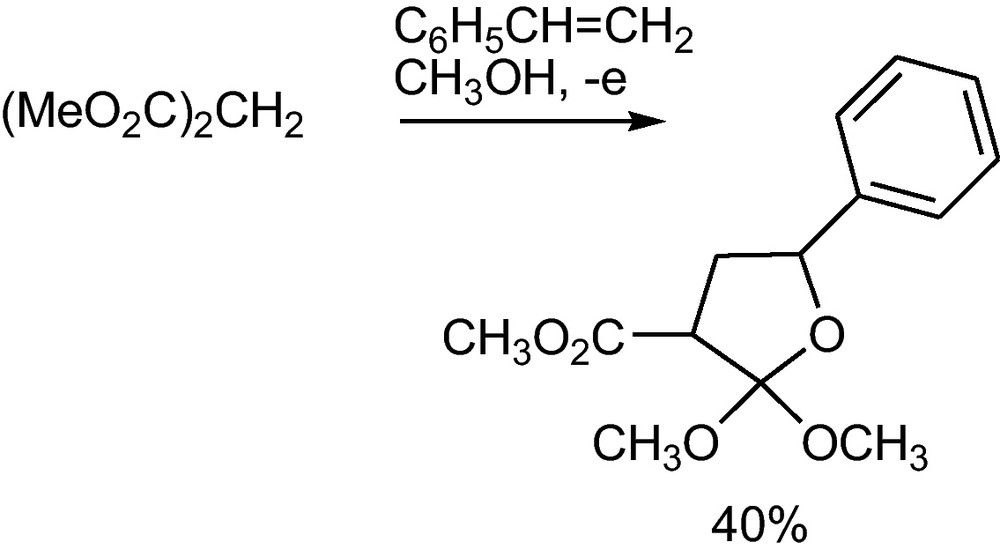
Anodic addition of dimethyl malonates to styrene1 [42].
5 C–C bond formation at the cathode
5.1 Activated C–C double bonds and organic halides
Olefins with electron withdrawing substituents can be hydrodimerized at the cathode via anion radicals (Scheme 23) [43–45]. Acrylonitrile is hydrodimerized in an industrial scale (300,000 t per year) to produce adipodinitrile, which is used as intermediate for the production of Nylon 66 [45]. The hydrodimerization of alkyl cinnamates indicates the structural complexity that can be achieved in high stereoselectivity in a one-pot reaction. Here the intermediate anion undergoes a subsequent intramolecular ester condensation to form a five membered ring in high yield and diastereoselectivity. Additionally the C–C coupling proceeds with high enantioselectivity, when the alkyl group of the ester is chiral (Scheme 24) [43,46].

Cathodic intermolecular coupling of activated olefins1 [43–45].

Cathodic hydrodimerization of alkyl cinnamates and subsequent intramolecular ester condensation1 [43(a),46].
N-Oxoalkyl-pyridiniumsalts can be coupled intramolecularly in aqueous sulfuric acid to quinolizidines with partially high diastereoselectivity (Scheme 25) [47]. The pyridinium salt is first reduced at the carbonyl group to afford a nucleophilic carbon-centered radical that adds to the pyridinium group to form a delocalized cation radical. Further reduction and protonation of the cation radical leads to an iminium cation; the iminium cation is finally reduced to the quinolizidine.

Cathodic cyclization of N-oxoalkyl-pyridinium salts to quinolizidines1 [47].
Cathodic umpolung of compounds with two electron acceptors allows one-step cyclizations [2]. At this instance one of the acceptors is reduced to a donor anion radical and this way the reaction between both groups of originally equal polarity becomes possible (Scheme 26) [48].

Cathodic intramolecular coupling of activated olefins1 [48].
At the cathode ethyl 2-bromo-3-propargyloxy-propionate cyclizes in the presence of Ni(II) complexes as catalysts in DMF and in the environmental friendly protic solvents: ethanol, butanol or ethanol-water mixtures to form as main product 2-aryl-3-ethoxycarbonyl-4-exo-methylene-tetrahydrofuran in 85% yield (Scheme 27) [49].

Cathodic cyclization of ethyl 2-bromo-3-propargyloxy-propionate with Ni(II) as catalyst [49].
The toxic pollutants carbon tetrachloride and chloroform have been electrochemically carboxylated to trichloroacetate (TCA) and dichloroacetate (DCA). Galvanostatic electrolysis in an undivided filter-press flow reactor consisting of a planar Zn cathode and a planar Al sacrificial anode afforded with 90% current efficiency in 0.1 M Bu4NClO4 in acetonitrile 85% of TCA and 69% of DCA, respectively [50].
Cathodic reductions, including transition metal catalyzed reactions of organic halides with subsequent intermolecular and intramolecular addition to C–C double and C–C triple bonds, to aromatic systems, carbonyl groups and epoxides have been summarized in a review [51].
The electroreduction of styrenes or alkyl methacrylates in the presence of aliphatic acid anhydrides or N-acylimidazoles in an undivided cell with zinc electrodes afforded in a twofold carbon-acylation 1,4-diketones in 33–85% yield [52].
5.2 Carbonyl compounds
Aldehydes and ketones are hydrodimerized at the cathode to form pinacols. Often, in a slightly acidic medium, the carbonyl group is reduced at a graphite cathode in an aqueous alcoholic solvent to a hydroxyalkyl radical that dimerizes (Scheme 28) [53,54]. In a chemical reduction, stoichiometric amounts of a reducing metal are needed and the corresponding amounts of metal salts are formed as waste.

Cathodic coupling of carbonyl compounds to pinacols1 [53,54].
The electrochemical pinacol coupling of acetophenone has been performed in a small scale of 3 mmol and a low current density of 1 mA/cm2 in the ionic liquid [BMIM][NTf2] (1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide). A d,l: meso ratio of 53: 47 is found in [Me3BuN][NTf2] (trimethylbutylammonium bis(trifluoromethylsulfonyl)imide), this increases considerably to 77: 23 in [Et3BuN][NTf2] (triethylbutylammonium bis(trifluoromethylsulfonyl)imide). The stereoselectivity and the reaction rate are significantly influenced by the interactions of the ionic liquid with the intermediates [55].
N-acyliminium ions, generated at the anode by the “cation pool” method, could be reduced at platinum electrodes in THF to free carbon radicals. These underwent homocoupling in up to 75% yield. With SmI2 or Zn as reducing agent, the dimer was obtained in only low yield. With activated olefins, e.g. methyl acrylate, adducts of the radicals were obtained in up to 84% yield [56].
Crossed cathodic coupling between aldehydes, ketones and α,β-unsaturated carbonyl compounds is possible in aqueous or aprotic solvents to form γ-lactones (Scheme 29) [54,57].

Cathodic cross coupling between aldehyde and α,β-unsaturated ester1 [54,57].
The intramolecular cathodic cross coupling between a carbonyl group and a double bond produces cyclopentanes in high diastereoselectivity; in this cyclization possibly radicals or anion radicals are involved (Scheme 30) [58].
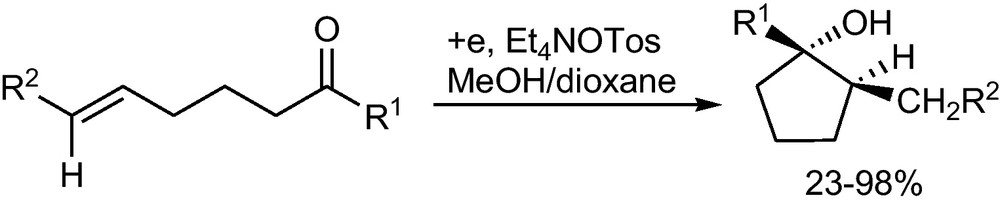
Intramolecular anodic cross coupling between double bond and carbonyl group1 [58].
5.3 Electrogenerated bases
C–C bond formation at the cathode can also occur via an EGB, which can be a radical anion or anion that is formed at the cathode [59]. EGBs can efficiently promote base catalyzed reactions as Michael additions or aldol reactions or can be used for deprotonations as in alkylations of CH-acids or of phosphonium salts in the Wittig-olefination (see also 7.5.). The renewable feedstock trimethyl citrate can be dehydrated to trimethyl aconitate, this dimerizes at the cathode in 75% yield to a cyclic hexamethyl ester (Scheme 31) [60]. The polyester is probably formed by deprotonation with an EGB produced from trimethyl aconitate. The generated anion undergoes an intermolecular Michael addition with trimethyl aconitate followed by an intramolecular Michael addition to form the product.

Cathodic dimerization of trimethyl aconitate to a cyclic hexamethyl ester [60].
6 C–H substitution and functional group interconversion at the anode
6.1 Anodic substitution
Nonactivated C–H bonds can be substituted for a nucleophile in electrolytes with a high anodic limit (acetonitrile, dichloromethane [DCM], trifluoroacetic acid). In this manner, the C–H bond is oxidized to a cation radical that is transformed by deprotonation and further oxidation to a cation, which reacts with a nucleophile. In this way, trans-decaline forms decalyl acetates with a remarkably high selectivity for the tertiary C–H bond (Scheme 32) [61].

Anodic substitution of nonactivated C-H bonds1 [61].
Benzylic C–H bonds can be substituted by acetate, acetamide or other nucleophiles, when by careful choice of the reaction conditions nuclear substitution is avoided. The selective oxidation of toluenes to aldehydes is even possible in industrial scale (Scheme 33) [45,62].

Anodic substitution of benzylic C-H bonds1 [45,62].
The enantioselective organocatalyzed alkylation at the α-C–H bond of aldehydes with xanthene was achieved by anodic oxidation in the presence of chiral cyclic amines. The components were electrolyzed current controlled in 0.1 M TBAClO4 in DCM at platinum electrodes. The best yield (80%) and highest enantioselectivity was obtained with the shown organocatalyst at 4 °C. The authors propose on the basis of cyclovoltammetry and DFT calculations the coupling of a xanthene radical with an enamine cation radical, both are generated at the anode (Scheme 34) [63].

Organocatalyzed anodic α-alkylation of aldehydes with xanthene [63].
The combination of electrochemistry and asymmetric organocatalysis is demonstrated in a further example by a direct intermolecular α-arylation of aldehydes with aniline leading to a meta-alkylation; this selectivity cannot be achieved by a Friedel-Crafts reaction. It is proposed that in a catalytic cycle, the aldehyde is converted first with a chiral auxiliary to an enamine. This adds to a p-iminoquinone that is generated by anodic oxidation from a 4-hydroxyaniline to form the meta-substituted aniline with regeneration of the chiral auxiliary. The electrolysis is performed current controlled in an undivided cell in 0.1 M NaClO4 in CH3CN/H2O. From five different aldehydes the products are obtained in 69–87% yield and 81–96% enantioselectivity. Similar yields can also be obtained with iodosobenzene diacetate as oxidant (Scheme 35)[64].

Part of the catalytic cycle in the α-arylation of aldehydes with aniline by combining electrochemistry with asymmetric organocatalysis [64].
Aromatic C–H bonds can be substituted by anodic oxidation of the aromatic compound to the cation radical. This reacts with a nucleophile to form a radical, whose oxidation and deprotonation leads to the substituted aromatic compound. In some cases, the side chain substitution is competing with the nuclear substitution. A favourable nucleophile is acetic acid, which is more stable against oxidation than alcohols; furthermore the acetate can be easily hydrolyzed to form phenols. The anodic acetoxylation of naphthalene is of technical interest, as this way an isomerically purer α-naphthol is obtained to produce azo pigments with a higher colour purity (Scheme 36) [62].
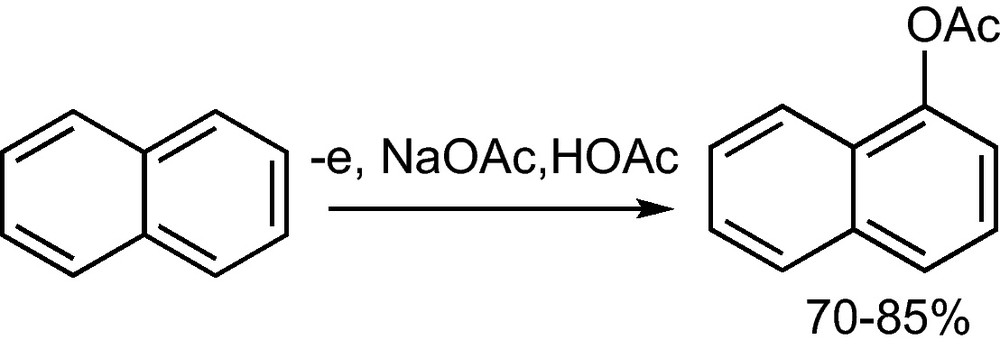
Anodic nuclear aromatic substitution of naphthalene1 [62].
The oxidative α-methoxylation or α-acetoxylation of N-acyl amines is a versatile and robust electrochemical reaction, which leads to valuable intermediates for synthesis (Scheme 37a) [65]. In carboxylic acids that are α-branched or contain an electron donating substituent in α-position the CO2H-group can be replaced by a nucleophile or a double bond by an anodic decarboxylation (non-Kolbe electrolysis) (Scheme 37b) [66].

Anodic substitution a) of a hydrogen adjacent to a C-N bond or b) by replacement of a carboxyl group via anodic decarboxylation1 [65,66].
2-Phenylthioacetate, cyclic ethers, lactones, cyclic carbonates and phenylthioglycoside have been regioselectively fluorinated by anodic substitution in the ionic liquids: Et4NF·4HF, Et3N·5HF and [EMIM][BF4], Et3N·5HF [67].
With an o-iminoquinone as a biomimetic mediator a primary aliphatic amine is selectively oxidized in the presence of a second amine used as the alkylating agent. This allows the rapid synthesis of various secondary amines in moderate to good yields [68]. The process appears to be a greener alternative to the preparation of secondary amines with alkyl halides being potentially toxic and producing salt waste.
N-Cyano substituted cyclic amines are regioselectively methoxylated at the higher substituted α-carbon in good overall yield. The regioselectivity is correlated with the higher stability of the intermediate iminium cation found by DFT-calculations. The methoxy group can be then substituted by the cyano or allyl group [69].
C1-cyano substituted tetrahydroisoquinolines were prepared by anodic cyanation using controlled potential electrolysis in methanol. By subsequent alkylation, decyanation and hydrogenolysis the syntheses of rac-carnegine, rac-norlaudanosine and rac-O-dimethylcoclaurine have been achieved in few steps, good overall yield and high diastereoselectivity [70].
The cross-dehydrogenative coupling between nitromethane and N-phenyltetrahydroisoquinoline with molecular oxygen was achieved in a 0.2 mmol scale in the ionic liquid 1-butyl-3-methylimidazolium tetrafluoroborate ([EMIM][BF4] as solvent and copper(II) bromide (6 mol %) as catalyst to yield 80–99% of the ß-nitroamine. Both the ionic liquid and the copper catalyst were recycled nine times with almost no loss of activity. The iminium ion could be also generated electrochemically as a stable intermediate in a divided cell. Thereby the ß-nitroamine and α-aminophosphonate could be obtained in high chemical and current yield in a 0.15 mmol scale with deprotonated nitromethane and diethylphosphite as nucleophiles (Scheme 38). A mechanism was proposed and supported by cyclovoltammetry [71].

Cross-dehydrogenative coupling between nitromethane or diethylphosphite and N-phenyltetrahydroisoquinoline with molecular oxygen and at the anode [71].
Electron donating oxygen atoms in the α-position of a carboxylic acid favour in anodic decarboxylation the further oxidation of the intermediate radical to a carbocation. This can undergo a fragmentation to form the energetically stable carbonyl group. The produced exocyclic carbocation reacts with methanol to the ketal (Scheme 39). The reaction makes the otherwise difficult to prepare l-xylonolactone readily accessible from 2-oxo-α-l-gulonic acid, which is an intermediate of a vitamin C synthesis that is performed in a pilot plant-scale [1(d)].
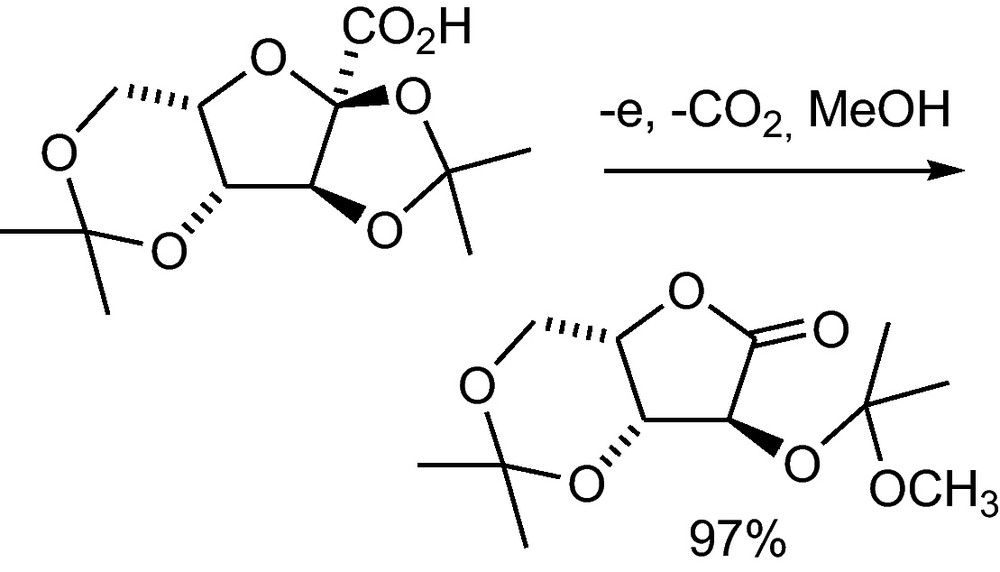
Anodic decarboxylation of 2,3:4,6-diisopropylidene-2-oxo-α-l-gulonic acid to l-xylonolactone [1(d)].
O-Protected thioglycosides were converted into O-glycosides of primary alcohols in good yields by electrolysis in an undivided cell and sodium trifluoromethansulfonate in acetonitrile as electrolyte [72].
6.2 Anodic addition
Methyl conjuenate, a plant oil product, is converted by electrolysis in an undivided cell in acetic acid and sodium acetate as electrolyte to an unsaturated 1,4-diacetate in 85% yield. The product is formed via a cation radical and an acetoxy-allyl cation as intermediates (Scheme 40) [1(d)]. The diacetate can be further converted into a 1,4-diol that is applicable for the preparation of polyesters. In addition, elimination of acetic acid forms trienes, which are isomers of α-eleostearic acid that is used as component of a water-resistant varnish.

Anodic addition of two acetoxy groups to methyl conjuenate1 [1(d)].
Current controlled anodic fluorination of various N-acetyl-3-substituted indole derivatives could be achieved in Et4NF·4HF/MeCN to provide the corresponding trans-2,3-difluoro-2,3-dihydroindoles exclusively or selectively in 15–56% yield [73].
A substituted bicyclo[2.1.0]pentane (housane) is electrochemically oxidized with tris(p-bromophenyl) amine as mediator to form in 70% yield a hydroazulene, which was transformed in few steps to the sesquiterpene daucene. The reaction proceeds in an oxidative ring opening to a cation radical that rearranges by a 1,2-carbon migration. The rearranged species is reduced by electron transfer from housane to form the product and to generate the cation radical for the next catalytic cycle. The mechanistic proposal is supported by cyclic voltammetry and quantum chemical calculations (Scheme 41) [74].

Anodic rearrangement of a substituted bicyclo[2.1.0]pentane (housane) to a precursor of the sesquiterpene daucene [74].
Methanol can be added to a hydroquinone dimethyl ether to form a benzoquinone bisacetal. A reasonable mechanism is the oxidation of the benzene ring to a cation radical, which undergoes methanolysis and further oxidation to a bisallyl cation that reacts with methanol to the product (Scheme 42) [75]. The bromide in the bisacetal can be exchanged for lithium generating this way a nucleophilic benzoquinone synthon. Further examples of anodic additions to aromatic compounds can be found in reference [76].
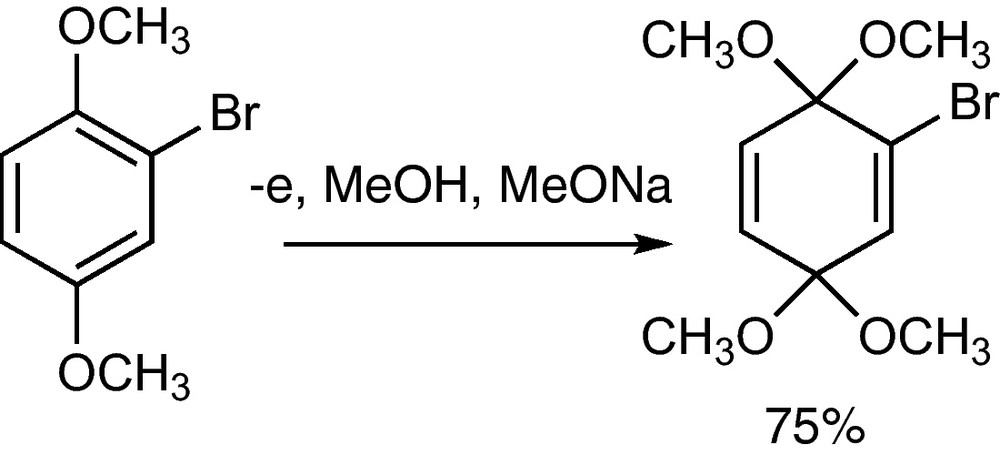
Anodic bismethoxylation of a 2-bromo-hydroquinone dimethylether1 [75].
Anodic aziridination of olefinic double bonds can be achieved by anodic oxidation of o-phthalylhydrazide in the presence of alkenes. The hydrazide supposedly is converted to a cation radical that adds to the double bond; deprotonation and oxidation of the formed radical generates a cation, which reacts with the nucleophilic nitrogen atom to form the aziridine (Scheme 43). Here the toxic Pb(OAc)4, which is the usual oxidant, has been successfully replaced by the non-toxic anode [77].
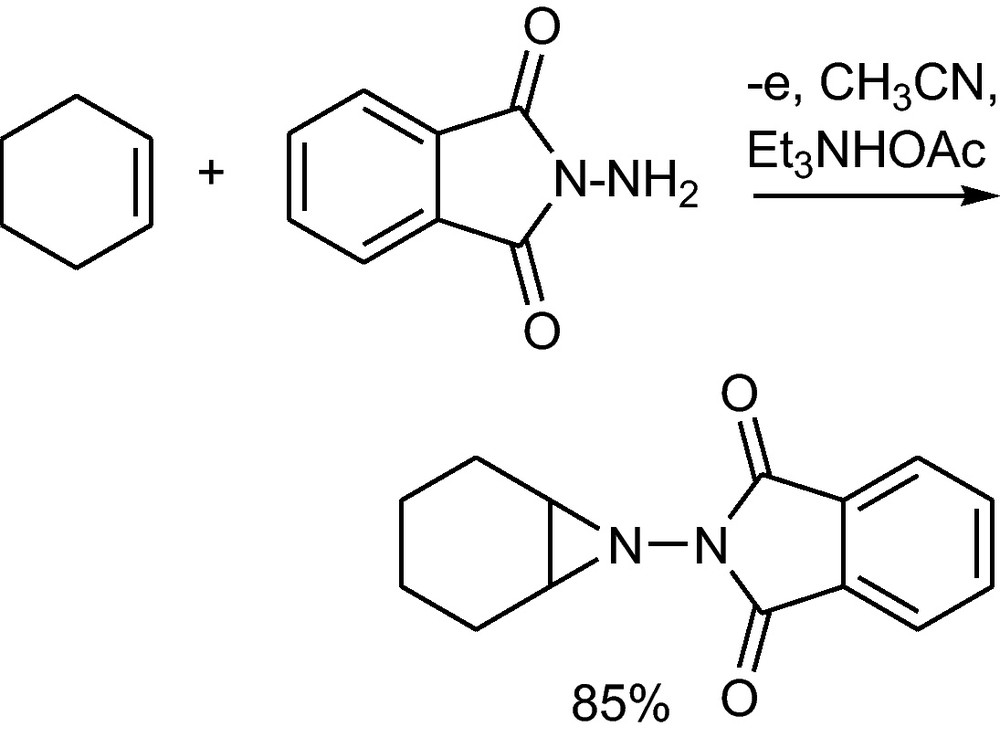
Anodic aziridination of a double bond [77].
As pointed out above, an intermediate secondary radical can be further oxidized in the Kolbe-electrolysis to a carbocation (non-Kolbe electrolysis). When the cation is substituted with a good leaving group in ß-position, e.g. the trimethylsilyl group, a regioselective ß-elimination to an olefin occurs. This non-Kolbe electrolysis and the use of ß-trimethylsilylacrylic acid as dienophile provide an acetylene-equivalent in Diels-Alder reactions (Scheme 44) [78].

Anodic elimination of carboxyl- and trimethylsilyl-group1 [78].
6.3 Anodic cleavage
In anodic cleavage, different types of C–C and C-hetero atom bonds are split by oxidation and chemical follow-up reactions of the intermediates.
Stilbenes with electron-deficient substituents could be cleaved in wet acetonitrile (0.1 M LiClO4) cleanly at +1.3 V (Ag/0.1 M AgNO3) to the corresponding aldehydes with yields of ≥ 95% in an undivided cell and with tris(2-nitro-4-methyl-aryl)amine as mediator. The oxidation potentials of the substituted stilbenes were up to 0.63 V more positive than this of the unsubstituted one. This reaction extends the scope of anodic double bond cleavage to olefins with higher oxidation potential [79].
Seven alkenes, e.g. 1-decene, methyl oleate, cyclododecene, norbornene, are cleaved by indirect anodic oxidation with IO4−/RuCl3 as double mediator to carboxylic acids. The best results were achieved with two alternative ex cell-methods. Acids and diacids are obtained in 61–91% chemical yield and good current yields. The amount of consumed periodate can be reduced to less than 5%, when compared to the same non-electrochemical cleavage. This mediated anodic cleavage of alkenes is an interesting alternative to ozonolysis [80].
Anodic bromo-formyloxylation followed by an anodic cleavage provides a two step conversion of cyclohexene to hexane-1,6-dial. Thereby anodic discharge of bromide in formic acid leads to (2-bromocyclohexyl)-formate, which is converted to cyclohexane-1,2-diol. The major part of potassium bromide is recovered for the next cycle. This electrochemical conversion appears to be an attractive option to chemical oxidations with oxygen and catalysts or with hydrogen peroxide. The diol is cleaved in high yield to hexane-1,6-dial or its acetal either directly or indirectly with periodate as mediator [81].
7 Functional group interconversion at the cathode
7.1 Cathodic cleavage
A C–X bond can be cleaved at the cathode, thereby X can be Cl, Br, I, CN, +PR3, +NR3 or SO2R. In these instances, the C–X bond is electroactive. Electron transfer leads to an anion radical, which dissociates into X− and a radical. The radical is subsequently reduced to a carbanion that is protonated. The overall result of this one-pot reaction is the replacement of a substituent X by a hydrogen atom. The advantage of the reaction is the potential selectivity of the cathodic reduction that allows removing selectively the most easily reducible leaving group (Scheme 45a) [82]. This reaction is used to decrease the number of halogen atoms in compounds to generate higher value synthetic intermediates1 (Scheme 45b) [83] and Scheme 45c [84]. In the latter reaction (Scheme 45c), the supporting electrolyte is used to control due to its reduction the potential at the working electrode. Cathodic cleavage allows preparing tertiary amines or phosphines with different substituents by alkylation and cathodic reduction of the onium-salts1 (Scheme 45d) [85,86]. Finally due to the potential selectivity the reaction is used for the mild and selective deprotection, e.g. of esters (Scheme 46a) [87], alcohols [88] or amides [89]. Furthermore cathodic elimination can be applied to achieve the selective protection of differently alkylated C–C double bonds (Scheme 46b) [90]. After the conversion of the unprotected double bond, the protected double bond can be regenerated by cathodic elimination.

Cathodic cleavages (see text).

Cathodic deprotection and protection (see text).
Deoxygenating an alcohol to the corresponding alkane represents an important conversion in organic chemistry. Alkyl 4-methylbenzoates are reduced in a divided cell at graphite electrodes in 0.15 M Bu4NBF4 in THF/NMP (9:1) at 130 °C to afford the deoxygenated alcohol in 41–85% yield. It is assumed that the ester is reduced to the anion radical, which cleaves to the 4-methylbenzoate anion and an alkyl radical that abstracts a hydrogen atom to form the product [91].
The transformation of acid hydrazides to primary amides is useful for the organic synthesis of complex molecules. Monoacylhydrazines can be electrochemically reduced to primary amides in 40–90% yield in a divided cell with a tin cathode. The method proved superior to reduction by sodium/mercury or lithium/biphenyl in terms of yield and practicability and tolerates aryl halogen and olefinic groups [92].
7.2 Cathodic generation of nucleophiles
At the cathode, carbanions or anion radicals can be generated that can react as nucleophiles with electrophilic compounds. The electroreduction of allylic halide derivatives in the presence of pinacolborane affords in a non-divided cell with an Al-anode in THF allylboronic pinacol esters in 64 to 86% yield and high regioselectivity (68–91%). Allylboronic derivatives can undergo Suzuki-type cross-coupling reactions. The low toxicity of boronic acids and their ultimate degradation to boric acid qualify these derivatives as “green” compounds. The ES may constitute an interesting synthetic alternative to conventional methods that use allyl Grignard or lithium reagents with low functional group compatibility [93].
Arylboronic acids could be converted to phenols in 77–91% yield by electrolysis in a divided cell in 0.1 M Bu4NClO4 in acetonitrile and in presence of molecular oxygen. Presumably molecular oxygen is reduced to the anion radical that substitutes one hydroxy group and after reduction to the peroxy anion induces the rearrangement of the aryl group to oxygen. Hydrogen peroxide and KO2 react with lower yield and selectivity [94].
7.3 Cathodic hydrogenation
The C = C- and C = X-bond can be hydrogenated with a variety of chemical reagents and also at the cathode. The electrochemical method offers the advantage of being able to provide a potential controlled hydrogenation thereby avoiding reactions of double bonds with more negative reduction potential (Scheme 47a1 [62,95] and Scheme 47b1 [96]). Furthermore one can achieve either a trans-hydrogenation via an anion radical or a cis-hydrogenation with electrogenerated hydrogen at the palladium or platinum cathode1 (Scheme 47c) [97].

Cathodic hydrogenation of C-C- and C-X double bonds (see text).
7.4 Cathodic elimination
Cathodic reduction of two leaving groups in 1,1- (Scheme 48a)1 [98], 1,2- (Scheme 48b)1 [99], 1,4- (Scheme 48c) [100] and 1,6-position (Scheme 48 d) [101] affords carbenes, olefins, reactive dienes and trienes, respectively. The intermediate o-quinodimethane in Scheme 48c can be trapped with an maleic anhydride (MA) that also operates as a mediator for the reaction. The p-bis(dibrommethyl)benzene forms poly-(phenylene vinylenes) [PPVs] in a subsequent cathodic elimination from the first formed polymer (Scheme 48d). The ES of poly(p-xylylenes) (PPXs) and poly(p-phenylenevinylenes) (PPVs) by the direct and mediated cathodic reduction of (halomethyl)arenes has been reviewed [101]. Compared to chemical methods, the advantages are mild (non-thermal) conditions and toleration of a wide range of substituents in the (halomethyl)arene precursors. Copolymers are obtained by co-electrolyses and aprotic and aqueous conditions may be used. For the preparation of the soluble PPVs a 100 g or more scale is possible using an undivided flow cell with constant current at a lead cathode and aqueous DMF. PPXs and PPVs can be prepared also via an indirect electrolysis using Ni(II)Cl2 dppe, Ni(salen) or dimethylterephthalate as mediators.
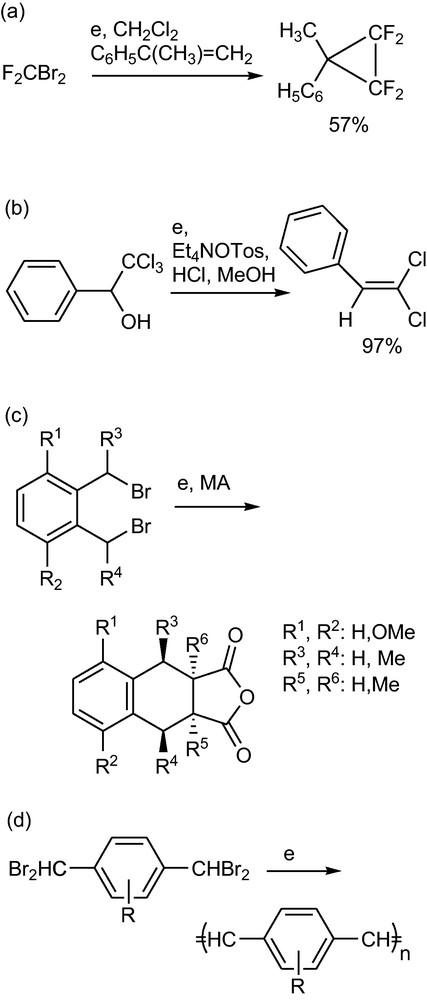
Cathodic eliminations (see text).
7.5 Electrogenerated bases
Electrogenerated anion radicals or anions are also bases, called EGBs, which can be used for deprotonations and to initiate base catalyzed reactions (see also Scheme 31).
Imidazolium-based room temperature ionic liquids (RTILs) are cathodically reduced current controlled in a divided cell to carbenes that catalyze the addition of nitromethane to aldehydes (Henry reaction). Best selectivities are obtained with 1-methyl-3-ethyl- or 1-methyl-3-butyl-imidazolium tetrafluoroborates or hexafluorophophates. With substituted benzaldehydes, cinnamaldehyde, 2-furaldehyde, cyclohexanecarbaldehyde and octanal the corresponding nitroalcohols are obtained in 79–100% yield. The RTIL can be used in five subsequent runs with slowly decreasing yields from 81 to 49%. The RTILs may be regarded as green solvents as well as precursors of N-heterocyclic carbenes [102]. Further uses of EGBs have been reviewed in [1(g),51,59,103].
8 Indirect electrolysis
In the indirect electrolysis, a selective chemical oxidizing or reducing reagent, called mediator (M) is added in understoichiometric or even catalytic amounts. The mediator oxidizes or reduces the substrate and is then regenerated at the electrode (Scheme 49a). Due to an often more specific interaction between the chemical reagent and the substrate the chemical oxidation or reduction can be more selective. This way the oxidation or reduction with stoichiometric amounts of expensive chemical reagents becomes more or less catalytic and this improves the economy and ecology of the conversion. The extent of the indirect electrolysis can be limited through deactivation or insufficient solubility of the mediator. Applications of indirect electrolysis are for example the oxidation with Ce4+ as mediator (Scheme 49b) [104]. A very useful mediator is the nickel electrode in alkaline medium. At the electrode immobilized Ni(III)-oxides are formed and regenerated that are useful for a wide variety of indirect oxidations. This immobilized mediator causes no solubility and separation problems and under alkaline conditions does not release nickel ions into solution (Scheme 49c) [105]. At a nickel anode in 3 M sodium hydroxide and 145 °C up to 150 g spruce lignosulfonate has been oxidized at up to 12 A to form vanillin as main product. Yields of vanillin were similar to those obtained industrially using chemical oxidants (about 5–7% w/w) [106].

Indirect anodic oxidations and cathodic reductions (see text).
Another mediator is TEMPO being converted at the anode into the selective oxidant TEMPO+, which oxidizes selectively primary hydroxy groups in diols or polyols. This selectivity is used to convert exclusively the primary hydroxy group and this at the anomeric center in unprotected carbohydrates to the carboxylic acid (Scheme 49d) [107].
Oxidations with mediators in organic ES have been reviewed covering the literature of the last 15 years in nearly 300 references [108]. The mediators allow transformations at lower oxidation potentials than in direct conversions. Extended tables present the mediated electrooxidation of side chains in alkyl substituted arenes and hetarenes with mediators as Ce4+/Ce3+, Mn3+/Mn2+, V5+/V4+, Co3+/Co2+, Ru4+/Ru2+, the oxidation of arenes to quinones with Ag2+/Ag+, Cu2+/Cu+/O2 and the aforementioned ones, the oxidative addition of CH-acids to alkenes with manganese and cerium ions, the oxidation of alcohols, carbohydrates and allyl ethers to aldehydes, ketones and carboxylic acids with RuO2/NaCl and metal ions. Further topics are oxidations at the nickel hydroxide anode, the transformation of alkenes and alkynes catalyzed by metal and selenium compounds, transformation of carbonyl compounds and their derivatives mediated by halide ions, synthesis of esters and polycarboxylic acids from CH-acids mediated by halide ions. Another subject are indirect oxidations mediated by IO4−, NO3−, H2O2 and metal ions and indirect oxidations with organic mediators as trisarylamines and TEMPO. Furthermore mediated reactions at the anode and the cathode have been reviewed with nearly 600 references in [109].
Metal-catalyzed ES through activation by transition-metal catalysts has been mostly found at the cathode. It involves the reaction of the substrate with an electrogenerated transition metal species, which is followed by a cathodic activation of the formed organometallic species. This double chemical and electrochemical activation allows new reactions to proceed. This topic has been reviewed with regard to application in synthesis [110] and the mechanisms involved [13(a)].
Ni(0)-complexes formed by cathodic reduction of nickel(II) salt [13(a)] are used as mediators in e.g. the carboxylation of benzyl halides to produce anti-inflammatory compounds (Scheme 49e) [111,112]. The cathodic cyclization of unsaturated organic halides in the presence of Ni(II) complexes as catalysts was performed in aprotic DMF and in protic solvents such as ethanol, butanol or ethanol-water mixtures. The presence of the alcohols enhanced the rate of recycling of the catalytic species (Scheme 27)[49]. The conjugate addition of aryl, heteroaryl, or alkenyl halides to electron deficient olefins has been performed at the cathode with nickel complexes as catalyst, and the use of the reaction for the synthesis of fine chemicals has been reviewed [113].
Palladium or nickel catalyzed cross-couplings use frequently phenylboronic acids or heteroarylstannanes as components. The electrochemical method uses cheaper, readily available and less toxic reagents like aryl halides, 3-chloro-6-methoxypyridazine and 3-chloro-6-methylpyridazine. The electrolysis is conducted in DMF, at a constant current with an iron rod as the anode, 10% of NiBr2bpy as the catalyst, and a stoichiometric amount of the two reagents to yield the cross-coupling products in moderate to good yields [114].
Recently, transition-metal-catalyzed regioselective halogenations of aromatic C-H bonds have been developed using several halogenating agents. Electrochemical oxidation using aqueous HCl and HBr and a palladium salt enables direct C–H halogenation without generating organic by-products [115].
Indirect electrolyses are applied in large variety in laboratory and in industry [45,116]. In enzymatic syntheses with oxidoreductases cofactors are regenerated by way of indirect electrolysis [13(b,c)].
9 Industrial electrosynthesis
In industry electrolysis is applied on a large scale for the cathodic hydrodimerization of acrylonitrile to adipodinitrile (300,000 t per year) (Scheme 50a)1 [45], and in smaller processes (> 10,000 t per year), e.g. for the production of dihydrophthalic acid (Scheme 47a), piperidines, aromatic aldehydes by indirect and direct anodic oxidations (Scheme 50b) [45], naphthoquinone (Scheme 50c) as part of a anthraquinone production [45] and of calcium gluconate from glucose. Several comprehensive and authoritative reviews on industrial electrochemistry have been written [45,62,117]. The latest review [117(b)] reports on classical industrial routes like chloralkali, aluminium, p-aminophenol, adipodinitrile, ethylene glycol, anthraquinone, perfluorinated products, glyoxylic acid and l-cysteine. Furthermore the paper deals with emerging inorganic and organic processes, e.g. ES in ionic liquids (see also 3.5.) and mediated electrochemical processes.

Industrial electrosyntheses (see text).
Recently the first paired ES carried out on an industrial scale has been reported, [117(a),118]. A paired electrolysis is of special value with regard to economy and ecology, because simultaneously at the cathode and the anode products of interest are created. In this way two processes need only one cell and use about the same electrical energy that is needed to synthesize one compound. Scheme 50d shows the industrial paired electrolysis by BASF with a simultaneous cathodic hydrogenation of dimethyl phthalate and the anodic substitution of 4-t-butyltoluene to form the dimethyl acetal of the corresponding benzaldehyde. Both products are higher value industrial intermediates that are used for the production of fragrances and agents for crop protection. They are obtained in industrial scale in together 180% yield. Many other examples with different types of paired electrosyntheses have been summarized in [1b].
In the large-scale processes of industry the reaction conditions are inherently in better accord with the rules of green chemistry than in laboratory reactions, because there neglect of these rules could stop the process.
10 Summary and outlook
The selection of older and recent electrochemical reactions described above shows that organic ES enables one to conduct a broad range of C–C bond formations and FGIs. These conversions are achieved in large scope, selectively and in good yield. Access to further examples of organic ES can be found in references [10(c),12(a),13(a),43(c),119].
The use of the electron as reagent leads in all these cases to advantages; these are atom economy, redox-umpolung, FGIs alternative to these in non-electrochemical reactions and no need for auxiliaries. Furthermore, the reaction conditions that are inherent to the reagent electron are in accord with many of the rules of green chemistry (rules 1–9, see Chapter 3.).
The reaction conditions in ES are generally simple, solvents and reagents of usual purity are sufficient, molecular oxygen and small amounts of water can mostly be tolerated. The electrolysis cell and the further equipment can be flexibly used for a variety of different reactions; most conversions can be performed current controlled for which an adjustable direct-current source is sufficient. The scale of the reaction can be easily increased by expanding the electrode area.
It is to be expected that in future the scope and diversity of reactions for ES will be further increased, the possibilities of redox-umpolung to save reaction steps and to make better use of synthetic building blocks will be expanded, yields and selectivities will be improved. Increased selectivities are also to be expected from indirect electrolyses with designed ligands for chemical catalysts, or from robust redox enzymes that can be electrochemically regenerated, or from transition metal catalysts in higher oxidation states.
The toxic electrode material mercury that has been often used in classical cathodic reductions because of its efficiency to suppress an unwanted proton reduction in protic solvents will be more and more replaced. Non-toxic electrode materials that have a similar or even higher overpotential for proton reduction like graphite or the BDD electrode are suitable substitutes. The same holds for the replacement of less green solvents by greener ones, while still retaining their support of a high selectivity. The concentration of the supporting electrolyte in organic ES is continuously decreased even to zero by using very small electrode gaps [4,120]. The amount of solvent may be decreased by using the substrate with small additives simultaneously as solvent [20], possibly also ionic liquids are useful for this purpose. Another possibility for decreasing the amount of solvent is dispersing the substrate in a three dimensional working electrode [3]. Very valuable information on techniques, electrodes, supporting electrolytes, solvents, microreactors and reactions can be found in an excellent recent review on modern strategies in organic ES [10(c)].
A final point is, to teach and use theory and practice of ES increasingly in undergraduate, graduate and Ph.D. courses. For the experiments in teaching or to start an investigation an undivided electrolysis cell (Fig. 1), a set of few electrodes made from different materials, a purchasable reference electrode, an adjustable direct-current source and a high resistance voltmeter are sufficient. This inexpensive equipment should be easily accessible in each laboratory doing synthesis to integrate the reagent: electron more efficiently into the search for new green syntheses.
1 Many electrochemical conversions shown in the Schemes are compared with the conditions of the non-electrochemical reaction leading to the same or a similar molecule. The comparison is done in the supporting information under the corresponding Scheme number.