

1 Introduction
The preparation of polymers from renewable resources is attracting increasing attention due to problems associated with the cost, security of supply, sustainability and environmental impact of petrochemicals [1]. The use of biomass (plant derived material such as cellulose, hemicelluloses or lignin) as a feedstock is particularly promising and biomass derived polyesters are already commercially available, e.g. Natureworks™ polylactide, Dupont Sorona™ and Metabolix Mirel™ polymers [2]. Polylactide (PLA) has attracted much attention as it derives entirely from renewable resources, has properties suitable for range of applications and can be degraded by hydrolysis reactions [3,4]. PLA is prepared from lactic acid, which is harvested from corn or sugar beet; the process involves formation of an oligo(lactic acid), its degradation to lactide (the cyclic dimer of lactic acid) and lactide ring opening polymerisation [5–9]. PLA has been applied for some time in specialised medical applications, such as degradable sutures, pins, matrices for tissue engineering and drug delivery excipients [10]. Furthermore, recent manufacturing improvements have enabled it to be produced on sufficient scale and at a competitive price for some commodity applications, such as packaging [11]. However, there are still a number of limitations associated with polylactide, for example it is relatively slow to degrade and this relates to its hydrophobicity [3–5,10,11]. One strategy to increase degradation rates involves the preparation of polylactide with hydrophilic end groups. In keeping with the sustainability of polylactide, the end group should also derive from renewable resources. Carbohydrate end groups are attractive as they display a multitude of functional groups, are relatively inexpensive and are naturally occurring in large quantities [12]. A number of groups have investigated the preparation of polylactide end-capped with various hexoses and pentoses [13–23]. The majority of these studies have focussed on d-glucose and its derivatives and have lead to the production of branched and multi-arm polylactides, some of which are of interest for medical applications. We recently reported a method to prepare end-functionalised polylactide from a series of hexoses, pentoses and aldaric esters and established that acetyl-protecting groups were useful for increasing the hydrophilicity of the PLA [23]. Herein, we present the preparation of a series of six new aldaric esters and their use as co-initiators in the ring opening polymerisation of lactide.
2 Results and discussion
2.1 Carbohydrate co-initiator synthesis
2,3,4,6-Tetra-O-acetyl-d-gluconolactone was prepared in quantitative yield from d-gluconolactone, according to the literature method [24]. However, the isomeric lactones, d-mannonolactone and d-galactarolactone, were not commercially available. Therefore a four-step synthesis was devised which enabled good conversion to 2,3,4,6-tetra-O-acetyl-d-galactono/mannonolactone, in overall yields of 55 and 72 %, respectively. The syntheses involved preparation of penta-O-acetyl-d-galacto/mannopyranose, in quantitative yield, by treatment of d-galacto/mannopyranose with acetic anhydride and trifluoroacetic acid for 20 hours [25]. The glycosyl bromides were prepared by reaction with hydrobromic acid, initially at low temperature but warming to room temperature and stirring for 20 h, and were isolated in 76 and 95 % yield, respectively. The glycosyl bromides were hydrolysed, using aqueous silver carbonate, to yield the tetra-O-acetyl-d-galacto/mannopyranose, in quantitative yield. The pyranoses were oxidised under Swern oxidation conditions, which enabled clean conversion to the target lactones in 83 and 76 % yield, respectively. The lactones and all intermediate compounds were fully characterised, details of which are provided in the experimental section (Scheme 1).
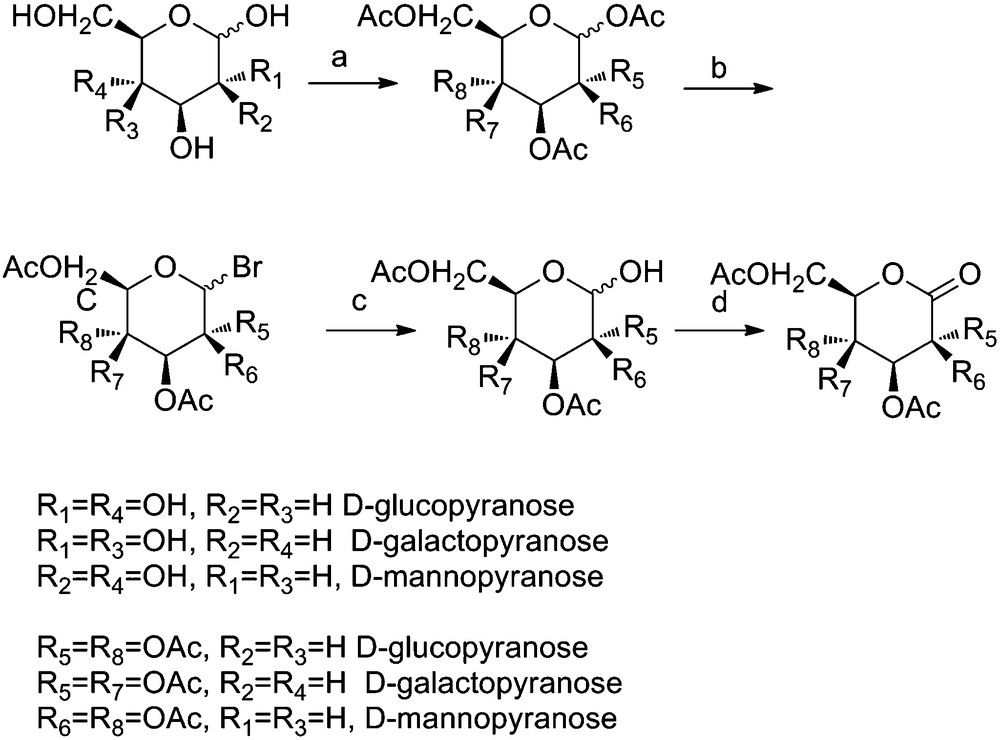
Synthesis of the acetyl protected 1,5-lactones derived from d-galactose and d-mannose. Reagents and Conditions: (a) TFA, Ac2O, 25 °C, 20 h; quantitative b) HBr (AcOH), DCM, 0 °C-25 °C, 20 h; 76 % (2,3,4,6-tetra-O-acetyl-D-galactopyranosyl bromide), 95 % (2,3,4,6-tetra-O-acetyl-D-mannopyranosyl bromide) c) Ag2CO3, acetone, water, 0 °C-25 °C, 20 h; quantitative d) i) 7 eq. (COCl)2, 13 eq. DMSO, −78 °C, 2 h ii) 5 eq. NEt3, −78 °C, 30 min, 83 % (2,3,4,6-tetra-O-acetyl-D-galactonolactone), 76 % (2,3,4,6-tetra-O-acetyl-D-mannonolactone).
The acid catalysed ring opening of the acetyl-protected 1,5-lactones, in the presence of butan-1-ol or 1,4-butanediol, yielded aldaric esters with one or two free hydroxyl groups. These alcohol groups (a single one for butoxyesters and two hydroxyl groups for hydroxybutoxyesters) were suitable as co-initiators for lactide ring opening polymerisation and enabled the preparation of polylactides with carbohydrate end groups/chain extender groups (Scheme 2).
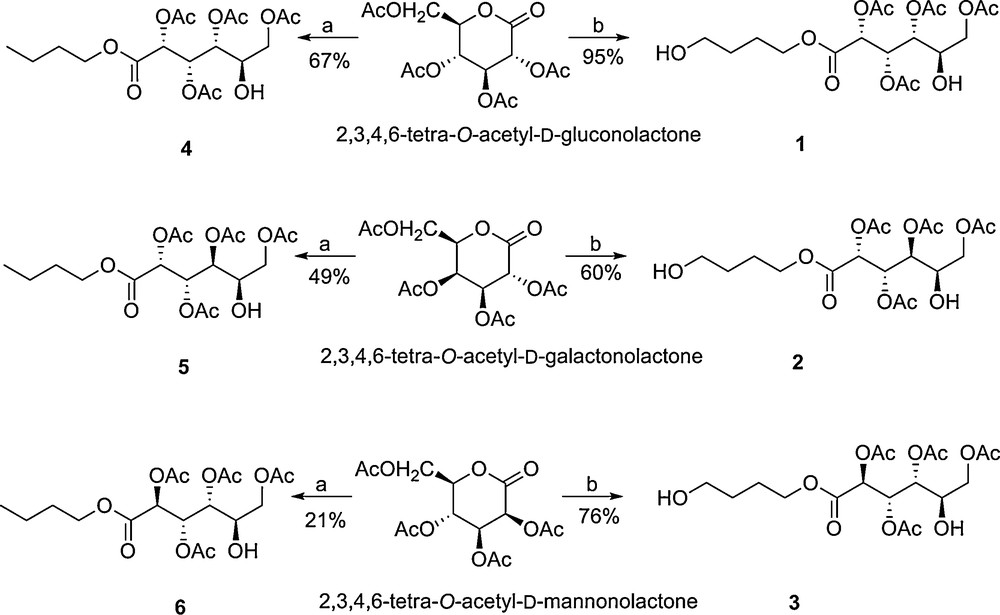
Synthesis of aldaric esters 1-6 by reaction of the per-acetylated 1,5-lactones with butan-1-ol and 1,4-butanediol. Reagents and Conditions: a) 0.1 eq. p-TSA, 1.0 eq. butan-1-ol, CHCl3, 0 °C or 25 °C b) 0.1 eq. p-TSA, 1.0 eq. 1,4-butandiol, CH2Cl2, 0 °C or 25 °C.
The rate of lactone ring opening depended on the stereochemistry, decreasing in the order: per acetylated d-galactonolactone > per acetylated D-mannonlactone > per acetylated D-gluconolactone. This could relate to the relative stability of the aldaric esters and in particular for the D-glucose derivatives there is a 1,3-steric interaction between the acetate substituents. The reaction was also reversible, although the equilibrium lay far towards the aldaric esters, however, prolonged exposure to acid led to formation of the thermodynamic product, the per acetylated 1,4-lactone. Nonetheless, by controlling the reaction time, six aldaric esters were isolated in yields from 20–95 %. These aldaric esters were grouped according to the number of free hydroxyl groups and the stereochemistry of the starting lactone, thus compounds 1–3 were all prepared using 1,4-butanediol and had two hydroxyl groups, one a primary hydroxyl and the other a secondary group, whilst compounds 4–6 had a single, secondary hydroxyl group.
2.2 Lactide ring opening polymerisations
The new aldaric esters were used as co-initiators for (S,S) and rac-lactide ring opening polymerisations (Scheme 3).
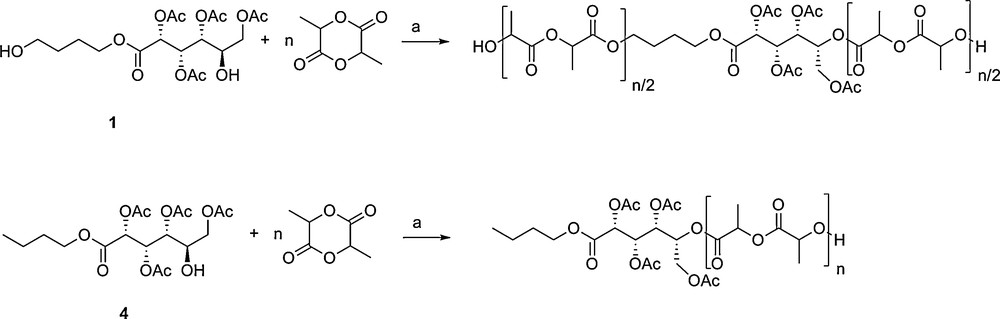
Polylactide synthesised using mono- and bi-functional carbohydrate co-initiators. Reagents and conditions: a [LA]0 = 0.7 M, [1–6]0 = 0.014 M, [LZnEt]0 = 0.014 M, CHCl3, 25 °C (n = 50). Note that although the values n/2 are used to represent the chain extended polymers formed from co-initiators 1-3, the differences in relative initiation rates between the hydroxyl groups could mean that slight differing values are obtained.
The polymerisations were all carried out at room temperature with an initiator to lactide ratio of 1:50 and at a 0.7 M lactide concentration. The true initiator was generated in situ by a stoichiometric reaction between the hydroxyl group(s) on 1-6 and a well-defined zinc complex, LZnEt (Fig. S1) [22]. The zinc complex was chosen because it had previously shown very good activity and control for lactide polymerisation using aliphatic alcohols and carbohydrate alcohols [22,23,26]. The initiating groups 1-3 gave polylactides where the carbohydrate group was a chain extender (Scheme 3), whilst the initiators 4–6 gave polylactide end-capped with the carbohydrate group (Scheme 3). The polymerisation was well controlled and by using a co-initiator:lactide loading of 1:50 it was possible to prepare polylactide with degree of polymerisation 30–50. The properties of the polylactide produced using the different initiators are compared in Table 1. The Mn was determined by SEC, using THF and with a correction factor as noted in the literature [27]. 1H NMR spectroscopy could also be used to deduce the molecular weight, by integration of the signals due to the methyne protons on the lactide repeat unit against the acetyl signals on the co-initiator. The molecular weights determined from NMR, SEC and the Mn calculated from the degree of polymerisation were all in excellent agreement. Thus, using co-initiator 1 at a conversion of 95 % (DP = 47) gave the calculated Mn of 7200, which was in close agreement with the Mn determined by SEC (6300) and that determined by 1H NMR (6700).
Polylactide prepared using co-initiators 1–6. Polymerisation conditions: [(S,S)-LA]0 = 0.7 M, [1–6]0 = 0.014 M, [LZnEt]0 = 0.014 M, CDCl3, room temperature.
| Co-initiator # | Co-initiator Chemical structure | % conva | Mn Calc.b | Mn NMRc | Mn SECd (PDI) |
| 1 | 4-hydroxybutyl 2,3,4,6-tetra-O-acetyl-D-gluconate | 95 | 7300 | 8300 | 7400 (1.52) |
| 2 | 4-hydroxybutyl 2,3,4,6-tetra-O-acetyl-D-galactonate | 94 | 7200 | 6700 | 6300 (1.24) |
| 3 | 4-hydroxybutyl 2,3,4,6-tetra-O-acetyl-D-mannonate | 98 | 7500 | 6600 | 8300 (1.66) |
| 4 | n-butyl 2,3,4,6-tetra-O-acetyl-D-gluconate | 98 | 7500 | 5500 | 5900 (1.32) |
| 5 | n-butyl 2,3,4,6-tetra-O-acetyl-D-galactonate | 62e | 4900 | 4900 | 5300 (1.31) |
| 6 | n-butyl 2,3,4,6-tetra-O-acetyl-D-mannonate | 82 | 6300 | 6500 | 5900 (1.33) |
a Determined by normalization of the integrals for the CH resonances in lactide (5.00 ppm) and polylactide (5.20 ppm).
b Mn calc = (144 × % conv. × 50) + RMM (co-initiator 1-6).
c Determined by integration of the CH signal in polylactide (5.20 ppm) versus the acetyl signal in the initiator (2.10 ppm).
d Determined by GPC, using polystyrene standards to calibrate the instrument and with a correction factor of 0.58 [27].
e Reaction was quenched before complete conversion could be attained.
The polymerisation control is further supported by the linear fit of the semi-logarithmic plots, ln{[LA]0/[LA]t} against time, for the various co-initiators (Fig. 1).

Polymerisation of S,S-lactide using co-initiators 1-6. The polymerisation conditions: [(S,S)-LA]0 = 0.7 M, [1–6]0 = 0.014 M, [LZnEt]0 = 0.014 M, CDCl3, 25 °C. The gradients give the pseudo first order rate constants (kobs) and the x axis intercept gives the initiation time (ti). 1, kobs = 0.95 h−1, ti = 1 h; 2, kobs = 0.60 h−1, ti = 2.3 h; 3, kobs = 0.74 h−1, ti = 1.6 h; 4, kobs = 0.27 h−1, ti = 2.7 h; 5, kobs = 0.37 h−1, ti = 2.2 h; 6, kobs = 0.28 h−1, ti = 2.2 h.
The gradients in Fig. 1 are the pseudo-first order rate constants, kobs for the co-initiators; the bifunctional co-initiators, 1-3, have values between 0.60 and 0.95 h−1 whilst the monofunctional co-initiators, 4–6, have values between 0.27 and 0.37 h−1. This difference can be rationalised by the active species present in the polymerisation reaction. The mechanism involves in situ generation of the initiator by reaction between LZnEt and the alcohol moieties to form a zinc alkoxide. This is followed by initiation and propagation and in order for the polymerisation to be controlled the rate of propagation should exceed the rate of initiation. Free alcohol groups are significant as they can act as chain transfer agents. Chisholm et al. have established that chain transfer occurs significantly faster than propagation in lactide polymerisation [28]. This dictates that kobs is a function of the initial hydroxyl group concentration, therefore the rate is doubled for co-initiators 1–3 because the initial hydroxyl concentration is twice that for co-initiators 4–6. This also rationalises the chain extended structure of the PLA derived from bifunctional co-initiators: chain transfer allows the polymer chain to grow from both ends of the molecule. Thus the carbohydrate unit is no longer an end group but is a chain extender, as shown in Scheme 3.
Another characteristic of these polymerisation reactions is the induction period which is manifested by a non-zero x-intercept in the semi-logarithmic plot, ln{[LA]0/LA]t}against time (Fig. 1). The induction period ranges between 1 and 3 h and reflects the time taken for the alcohol groups to react with the alkyl zinc co-initiator. The PLA produced from rac-lactide was atactic.
2.3 Degradation studies
There are a multitude of methods used to analyse the degradation of polylactide including in vitro and bulk degradation studies [29–36]. Acid catalysed degradation is useful as it is much accelerated compared to standard aqueous conditions. The polymers were dissolved in TFA(aq.)/CHCl3 (2 % v/v) and were analysed by SEC (polystyrene standards, CHCl3) until complete degradation had occurred. Analysis of the degradation was based on the widely accepted random chain scission mechanism for acid catalysed degradation [30–39]. Liu et al. [34] have shown the relationship between the molecular number and the degradation rate constant, kd, can be simplified:
M0/Mt − 1 = − M0kdtwhere M0 and Mt are the molecular number at time 0 and t, respectively. The plots of [M0/Mt − 1]/M0 versus time were linear, supporting a random chain scission degradation pathway, the gradients correspond to the degradation rate constant, kd. The polylactide prepared from rac-lactide underwent significantly more rapid degradation than the isotactic polylactide derived from S,S-lactide (Fig. 2) [29]. Fig. 3 compares the degradation of PLA initiated using compounds 1, 4 and iPrOH. The carbohydrate moieties clearly increase the degradation rate compared to the i-propoxyester terminated polylactide. The carbohydrates 1 and 4 both derive from D-glucose, and differ only in the polymer structure (Scheme 3). The polylactide initiated using compound 1 degrades more rapidly; this could be because the hydrophilic carbohydrate group is in the centre of the PLA chain, which facilitates breakdown of the chain into smaller and more rapidly degradable units. There is no particular influence of the carbohydrate stereochemistry on the degradation kinetics with similar results being obtained for polylactide prepared from 2, 3, 5 and 6 (data not shown). However, it is clear that the carbohydrate groups increase the degradation rate of polylactide compared to unfunctionalised alcohol end groups. This is probably due to the hydrophilicity of the acetyl-protected carbohydrate moieties, which are expected to increase the rate of hydrolysis of the polylactide chain and thereby increase the degradation rate.
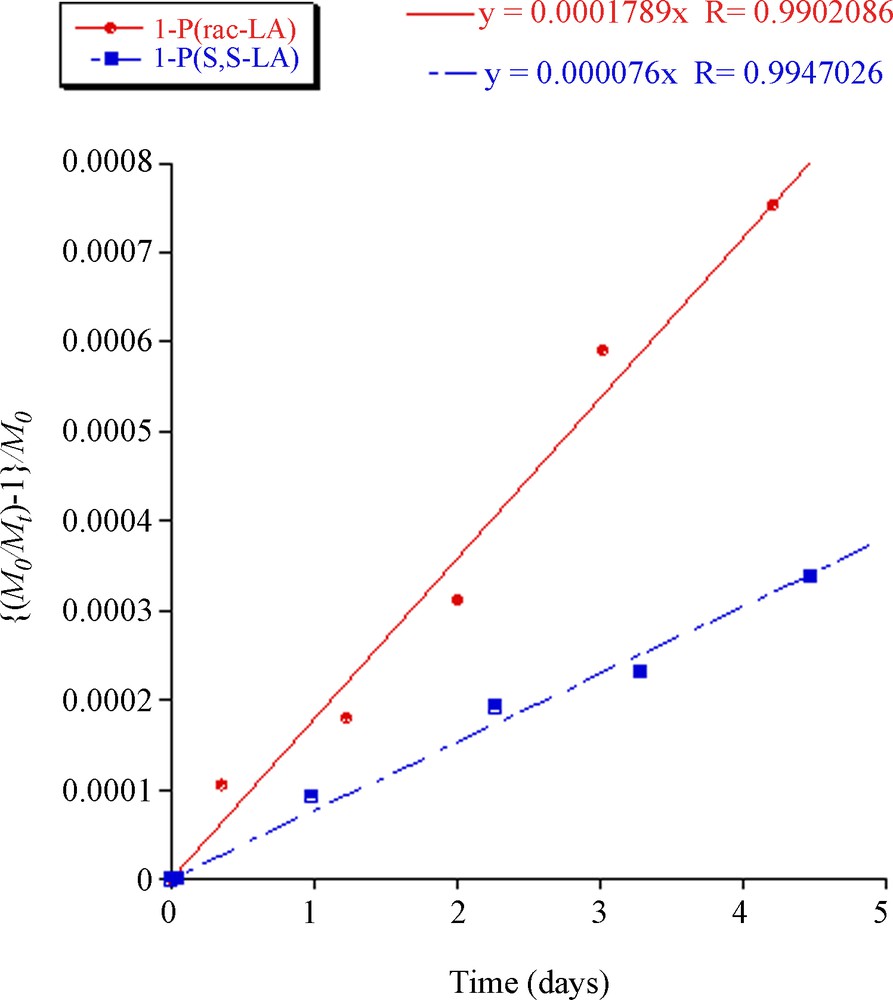
Polylactide degradation assessed by plotting (M0/Mt-1)/M0 versus time for polymer prepared from initiator 1 and rac-lactide (M0 = 7300; kd = 1.79 × 10−4 day−1) or S,S-lactide (M0 = 7400; kd = 0.76 × 10−4 day−1).
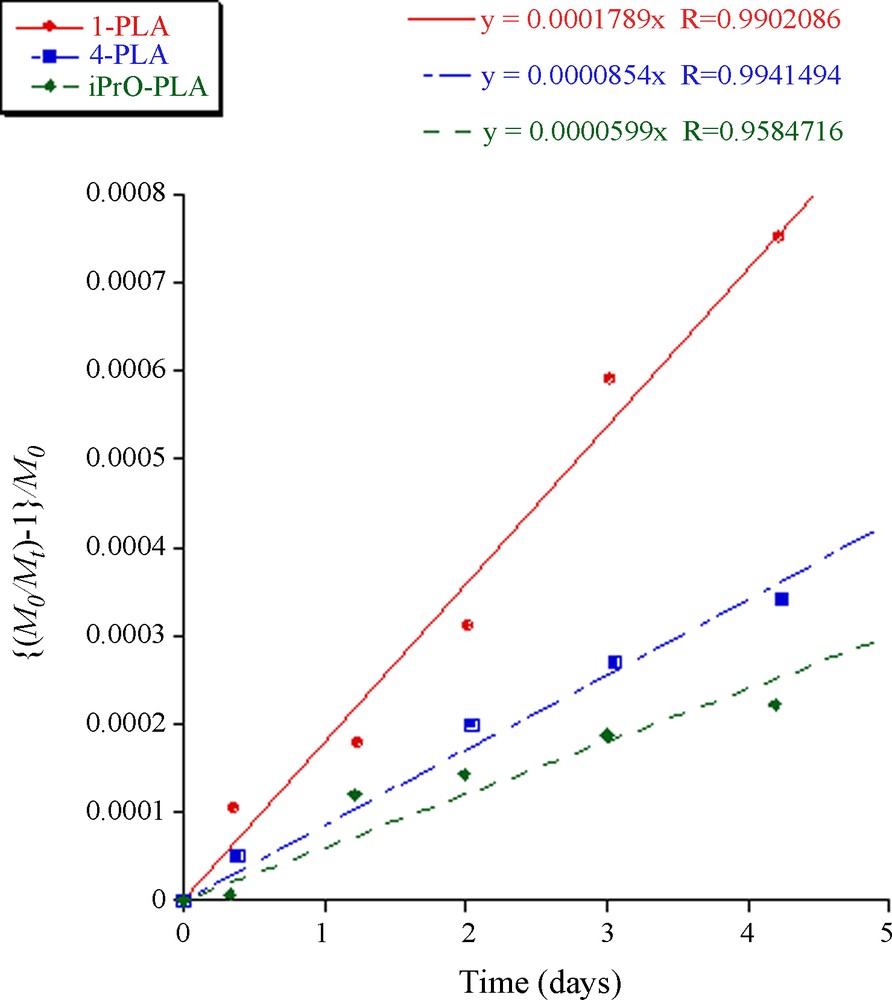
Plot of (M0/Mt-1)/M0 versus time for the degradation of polylactide prepared from rac-lactide and initiators 1 (M0 = 7300; kd = 1.79 × 10−4 day−1), 4 (M0 = 7500; kd = 0.85 × 10−4 day−1), and iPrOH (M0 = 2500; kd = 0.60 × 10−4 day−1).
3 Conclusions
In conclusion, a series of six aldaric esters were prepared in good yields, from the hexoses d-gluconolactone, d-galactose and d-mannose. The synthesis involved preparation of per-acetyl protected lactones and ring opening of the lactones, with either butanol or 1,4-butanediol, to yield aldaric esters with one or two free hydroxyl groups, respectively. The aldaric esters were used as co-initiators, with a zinc complex, for the ring opening polymerisation of lactide. The polymerisations were all well controlled, as shown by the good agreements between theoretical and experimental molecular weights and the linear reaction kinetics. The aldaric esters with a single hydroxyl group formed polylactide with a carbohydrate end group, whilst the aldaric esters with two hydroxyl groups formed polylactides chain extended by the carbohydrate moiety. The polymerisation kinetics were analysed, under pseudo first order conditions, and showed that the rates of polymerisation depended on the concentration of free hydroxyl groups, consistent with chain transfer occuring more rapidly than propagation. The acid catalysed degradation of the polyesters was examined by GPC and showed that the carbohydrate groups increased the degradation compared to polymers with unfunctionalised end groups. This is because the acetyl protected aldaric ester groups are hydrophilic and thus improve water uptake by the polyester and thereby increase the degradation rate. The strategy of introducing carbohydrate functionality into polylactide is therefore valid as it maintains the renewability of the material whilst increasing the degradation rate.
4 Experimental section
1H NMR spectra were collected on a Bruker AV400 instrument. 13C{1H} NMR spectra were collected on a Bruker AV500 instrument. CDCl3 was used as the NMR solvent and reference compound. The GPC measurements were performed on a Polymer labs GPC 60 instrument with two Polymer labs mixed D columns and THF at a flow rate of 1 mLmin-1 as the eluent. Narrow molecular weight polystyrene standards were used to calibrate the instrument and a correction factor of 0.58 was applied to the Mn obtained as outlined in the literature [27]. [S,S]-Lactide and rac-lactide were donated by Purac Plc, they were purified by recrystallization from hot toluene followed by repeated sublimations in vacuo (3 times). 2,3,4,6-Tetra-O-acetyl-d-glucono-1,5-lactone was prepared according to the literature in a single step from d-gluconolactone [24]. The other two lactones were prepared according to a four stage procedure involving per actylation, bromination, hydrolysis and oxidation. The starting materails: 1,2,3,4,6-penta-O-acetyl-α-d-galactopyranose and 1,2,3,4,6-penta-O-acetyl-α-d-mannopyranose were prepared according to the literature [25].
4.1 General procedure for the synthesis of glycosyl bromides
The procedure for the synthesis of glycoside bromides from their corresponding peracetylated pyranosides was adapted from the one reported by Mitchell et al. [40]. The reactions were allowed to proceed for 20 h instead of 1 h at r.t. as reported in the literature. Reactions were monitored by TLC. The resulting products were in the form of syrups.
4.1.1 2,3,4,6-Tetra-O-acetyl-α-d-galactopyranosyl bromide
76 % yield. TLC Rf 0.50 (EtOAc:Hexane; 1:1). 1H NMR (400 MHz, CDCl3) δ ppm 6.68 (1 H, d, J = 3.9 Hz, H-1), 5.50 (1 H, m, H-4), 5.39 (1 H, m, H-3), 5.03 (1 H, dd, J = 10.5, 3.9 Hz, H-2), 4.47 (1 H, m, H-5), 4.17 (1 H, dd, J = 11.4, 6.3 Hz, H-6), 4.10 (1 H, dd, J = 11.4, 6.8 Hz, H-6’), 2.14, 2.10, 2.05, 2.00 (4 x 3 H, 4 x s, 4 x (COCH3)). 13C{1H} NMR (126 MHz, CDCl3) δ 170-169 (COCH3), 88.07 (C-1), 71.01 (C-5), 67.95 (C-3), 67.72 (C-2), 66.93 (C-4), 60.81 (C-6), 20.72, 20.62, 20.56, 20.54 (COCH3) ppm. MS (CI, ammonia): m/z (%) 428 (100 %, M + NH4, Br-79), 430 (100 %, M + NH4, Br-81).
4.1.2 2,3,4,6-Tetra-O-acetyl-α-d-mannopyranosyl bromide [41]
95 % yield. TLC Rf 0.38 (EtOAc:Hexane 1:1). 1H NMR (400 MHz, CDCl3) δ 6.32 (1 H, br s, H-1), 5.74 (1 H, dd, J = 10.1, 3.4 Hz, H-3), 5.47 (1 H, dd, J = 3.4, 1.6 Hz, H-2), 5.39 (1 H, apt, J = 10.2 Hz, H-4), 4.36 (1 H, dd, J = 12.4, 4.9 Hz, H-6), 4.25 (1 H, ddd, J = 10.2, 4.9, 2.0 Hz, H-5), 4.16 (1 H, dd, J = 12.5, 2.1 Hz, H-6’), 2.20, 2.13, 2.10 (3 H, s, (COCH3)), 2.03 (3 H, s, (COCH3)) ppm. NMR data in agreement with literature data [41].
4.2 General procedure for the hydrolysis of glycoside bromides
The procedure for the hydrolysis of glycoside bromides to their corresponding pyranoses was adapted from one reported by Grummitt et al. [42] using Ag2CO3. The reactions were allowed to proceed for 20 h, instead of 4 h 0 as reported in the literature. Reactions were monitored by TLC.
4.2.1 2,3,4,6-Tetra-O-acetyl-α-D-galactopyranose
88 % yield. TLC Rf 0.20 (EtOAc:Hexane 1:1). 1H NMR (400 MHz, CDCl3) δ 5.50 (1 H, d, J = 3.4 Hz, H-1), 5.46 (1 H, m, H-2), 5.39 (1 H, m, H-3), 5.13 (1 H, dd, J = 10.8, 3.6 Hz, H-4), 4.46 (1 H, m, H-5), 4.15 (2 H, m, H-6, H-6’), 2.14, 2.13, 2.09, 2.08 (4 x 3 H, s, (COCH3)) ppm. 13C{1H} NMR (126 MHz, CDCl3) δ 170-169 (COCH3), 90.67 (C-1), 68.24 (C-4), 68.15 (C-2), 67.18 (C-3), 62.54 (C-5), 61.81 (C-6), 20.82-20.55 (4 x (COCH3)) ppm. MS (CI, ammonia): m/z (%) 366 (M + NH4, 100 %).
4.2.2 2,3,4,6-Tetra-O-acetyl-α-D-mannopyranose [42]
Quantitative yield. TLC Rf 0.22 (EtOAc:Hexane 1:1). 1H NMR (400 MHz, CDCl3) δ 5.39 (1 H, dd, J = 10.1, 3.3 Hz, H-3), 5.28 (1 H, d, J = 9.88, H-4), 5.26 (1H, dd, J = 3.3, 2.0 Hz, H-2), 5.25 (1H, br, H-1), 4.28 - 4.16 (2 H, m, H-5, H-6), 4.15–4.08 (1 H, m, H-6’), 2.14, 2.08, 2.03, 1.98 (4 x 3 H, 4 x s, 4 x (COCH3)) ppm. 13C{1H} NMR (400 MHz, CDCl3) δ 170.8, 170.1, 170.0, 169.8 (4 x COCH3), 91.9 (C-1), 70.1, 68.8, 68.1, 66.1, (C-2 to C-5), 62.5 (C-6), 20.9, 20.8, 20.6, (COCH3) ppm. MS (CI, ammonia): m/z (%) 366 (M + NH4, 100 %). 1H NMR data in agreement with the literature [42].
4.3 General procedure for the Swern oxidation of pyranoses
Oxalyl chloride (3.4 mL, 39 mmol, 7 eq.) was dissolved in dry DCM (100 mL) and cooled to -78 °C. A solution of DMSO (5.4 mL, 76 mmol, 13 eq.) in DCM (15 mL) at −78 °C was added dropwise to the oxalyl chloride solution under N2. After 15 min a solution of the funcitonalised pyranose (5.7 mmol, 1 eq.) in DCM (50 mL) at −78 °C was added dropwise. After 30 min NEt3 (4.0 mL 29 mmol, 5 eq) was slowly introduced and the reaction mixture was stirred at 78 °C for a further 30 min (until this point, the reaction temperature was not allowed to rise beyond −60 °C). The reaction mixture was then allowed to warm to −30 °C and washed with ice-water (200 mL), 1 M HCl (200 mL), saturated NaHCO3(aq) (200 mL), water (200 mL) and brine (200 mL). The organic layer was dried (MgSO4) and concentrated in vacuo to yield the product as a syrup.
4.3.1 2,3,4,6-Tetra-O-acetyl-d-galactono-1,5-lactone
(1.66 g, 4.79 mmol, 83 %). TLC Rf 0.33 (EtOAc:Hexane 1:1). [α]D25 = +43.2° (c, 1.25, CHCl3) νmax(neat) 1766 cm-1 (C = O).1H NMR (400 MHz, CDCl3) δ ppm 5.70 (1 H, dd, J = 2.76, 1.5 Hz, H-4), 5.47 (1 H, dd, J = 10.28, 2.9 Hz, H-3), 5.28 (1 H, d, J = 10.3 Hz, H-2), 4.82 (1 H, m, H-5), 4.25 (2 H, m, H-6, H-6’), 2.20 (3 H, s, COCH3), 2.19 (3 H, s, COCH3), 2.10 (3 H, s, COCH3), 2.08 (3 H, s, COCH3) ppm. 13C{1H} NMR (126 MHz, CDCl3) δ 170.18, 170.12, 169.75, 169.46 (COCH3); 164.95 (C-1); 74.71 (C-5); 69.16 (C-3); 68.44 (C-2); 66.22 (C-4); 60.98 (C-6); 20.56, 20.52, 20.44, 20.41 (COCH3) ppm. MS (CI, ammonia): m/z (%) 364 (100 %, M + NH4). Anal Calcd for C14H18O10: C, 48.56 %; H, 5.24 %. Anal Found: C, 48.65 %; H 5.37 %.
4.3.2 2,3,4,6-Tetra-O-acetyl-d-mannono-1,5-lactone [43]
(1.50 g, 4.34 mmol, 76 %). TLC Rf 0.25 (EtOAc:Hexane 1:1). [α]D25 = 30.4°(c, 1.25, CHCl3). νmax(neat) 1748 cm−1 (C = O). 1H NMR (400 MHz, CDCl3) δ 5.75 (1 H, d, J = 3.9 Hz, H-2), 5.52-5.46 (1 H, m, H-3), 5.07–5.03 (1 H, m, H-4), 4.62 (1 H, ddd, J = 8.6, 5.8, 3.2 Hz, H-5), 4.37 (1 H, dd, J = 12.4, 3.1 Hz, H-6), 4.27 (1 H, dd, J = 12.5, 5.8 Hz, H-6’), 2.15 (3 H, s, COCH3), 2.16 (3 H, s, COCH3), 2.18 (3 H, s, COCH3), 2.12 (3 H, s, COCH3) ppm. 13C{1H} NMR (100 MHz, CDCl3) δ 170.39, 169.41, 169.04, 168.97 (COCH3), 164.81 (C-1), 75.37 (C-5), 70.38 (C-3), 69.10 (C-4), 66.53 (C-2), 62.26 (C-6), 20.63 - 20.38 (COCH3) ppm. MS:(CI - NH3) m/z: 364 (M + NH4, 60 %). Anal Calcd. for C14H18O10: C, 48.56 %; H, 5.24 %. Found: C, 48.70 %; H, 5.12 %.
4.4 General procedure for preparation of initiators 1-6
The 1,5-lactone (300 mg) was dissolved in CHCl3 (15 mL) and to it was added a 1.0 M solution of the alcohol (1 eq.) containing p-TSA (0.1 eq.) After the appropriate time (15 mins -2 h), the reaction was quenched with NaHCO3(aq) (50 mL), washed with water (2 × 50 mL) and dried (MgSO4). The organic layer was concentrated in vacuo to yield the aldaric ester as a syrup. n-Butyl aldaric esters were prepared using butan-1-ol and 4-hydroxybutyl aldaric ester were prepared using 1,4-butanediol.
4.4.1 4-Hydroxybutyl 2,3,4,6-tetra-O-acetyl-d-gluconate 1
Reaction time: 2 h. (0.37 g, 0.89 mmol, 95 %). TLC Rf 0.43 – some decomposes to lactone on TLC plate (EtOAc:Hexane 1:1). [α]D25 = +45.2° (c, 1.15, CHCl3). νmax(neat) 3485 cm−1 (OH), 1738 cm−1 (C = O). 1H NMR (400 MHz, CDCl3) δ 5.74 (1 H, m, H-2), 5.29 (1 H, m, H-3), 5.20 (1 H, dd, J = 8.3, 3.6 Hz, H-4), 4.22–4.12 (6 H, m, J = 14.3, 7.2 Hz, C(O)OCH2CH2CH2CH2OH, H-6), 3.84(1 H, m, H-5), 3.01 (1 H, d, J = 5 Hz, OH), 2.16 (3 H, s, COCH3), 2.14 (3 H, s, COCH3), 2.10 (6 H, s, 2 x COCH3), 1.27 (4 H, m, C(O)OCH2CH2CH2CH2OH) ppm. 13C{1H} NMR (126 MHz, CDCl3) δ 171.15, 170.58, 169.76, 166.76 (COCH3, C-1), 71.81 (C-3), 70.56 (C-4), 69.50 (C-2), 68.41 (C-5), 64.65, 62.16 (C(O)OCH2CH2CH2CH2OH, C-6), 20.78, 20.58, 20.47 (COCH3), 14.00 (C(O)OCH2CH2CH2CH2OH) ppm. MS: (CI - NH3) m/z: 454 (M + NH4, 25 %). Anal Calcd. for C18H28O12: C, 49.54 %; H, 6.47 %. Anal Found: C, 49.62 %; H, 6.38 %.
4.4.2 4-Hydroxybutyl 2,3,4,6-tetra-O-acetyl-d-galactonate 2
(0.76 g, 1.11 mmol, 60 % yield). TLC: decomposes on silica. [α]D25 = +8.00°(c, 0.5, CHCl3). νmax(neat) 3519 cm−1 (OH), 1749 cm−1 (C = O). 1H NMR (400 MHz, CDCl3) δ 5.78 (1 H, dd, J = 9.8, 2.0 Hz, H-2), 5.21-5.16 (1 H, m, H-3), 4.22–4.06 (4 H, m, H-4, H-5, H-6, H-6’), 4.03-3.82 (2 H, m, C(O)OCH2CH2CH2OH), 3.72–3.63 (2 H, m, C(O)OCH2CH2CH2OH), 2.18 (3 H, s, COCH3), 2.14 (3 H, s, COCH3), 2.13 (3 H, s, COCH3), 2.09 (3 H, s, COCH3), 1.81–1.68 (2 H, m, C(O)OCH2CH2CH2CH2OH), 1.32–1.23 (2 H, m, C(O)OCH2CH2CH2CH2OH) ppm. 13C{1H} NMR (126 MHz, CDCl3) δ 170.98–169.35 (COCH3), 168.03 (C-1), 69.709 (C-2), 68.531 (C-3), 68.21, 68.29, 68.21, 63.51 (C-4, C-5, C-6, C-6’), 68.27 (C(O)OCH2CH2CH2CH2OH), 25.38 (C(O)OCH2CH2CH2CH2OH), 21.08–19.66 (COCH3), 12.97 (C(O)OCH2CH2CH2CH2OH) ppm. MS: (CI - NH3) m/z: 454 (M + NH4, 100 %). Anal. Calcd. for C18H28O12 C, 49.54 %; H, 6.47 %. Anal. Found: C, 49.50 %; H, 6.57 %.
4.4.3 4-Hydroxybutyl 2,3,4,6-tetra-O-acetyl-d-mannonate 3
(0.96 g, 2.20 mmol, 76 % yield). TLC: decomposes to lactone on silica. [α]D25 = +13.09°(c, 2.75, CHCl3). νmax(neat) 1761 cm−1 (C = O). 1H NMR (400 MHz, CDCl3) δ 5.66 (1 H, dd, J = 8.6, 2.2 Hz, H-3), 5.27 (1 H, dd, J = 9.1, 2.1 Hz, H-4), 5.17 (1 H, d, J = 7.8 Hz, H-2), 4.29–4.07 (3 H, m, H-5, H-6, H-6’), 4.01 (2 H, m, C(O)OCH2CH2CH2CH2OH), 3.68 (2 H, m, C(O)OCH2CH2CH2CH2OH), 2.15 (3 H, s, COCH3), 2.13 (3 H, s, COCH3), 2.09 (6 H, 2 x s, COCH3), 1.75 (2 H, m, C(O)OCH2CH2CH2CH2OH), 1.27 (2 H, m, C(O)OCH2CH2CH2CH2OH) ppm. 13C{1H} NMR (126 MHz, CDCl3) δ 171-169 (COCH3, C-1), 66.44 (C-3), 69.55 (C-4), 69.55 (C-2), 64.21, 62.87, 61.96 (C-5, C-6, C-6’), 64.65 (C(O)OCH2CH2CH2CH2OH), 61.96 (C(O)OCH2CH2CH2CH2OH), 25.42 (C(O)OCH2CH2CH2CH2OH), 20.02 (COCH3), 20.02 (COCH3), 20.48 (COCH3), 20.48 (COCH3), 13.78 (C(O)OCH2CH2CH2CH2OH), ppm. MS: (CI - NH3) m/z: 454 (M + NH4, 29 %). Anal Calcd for C18H28O12 C, 49.54 %; H, 6.47 %. Anal Found C, 49.68 %; H, 6.39 %.
4.4.4 n-Butyl 2,3,4,6-tetra-O-acetyl-d-gluconate 4
(0.28 g, 0.66 mmol, 67 % yield). TLC: decomposes on silica. [α]D25 = +24.9° (c, 1.85, CHCl3). νmax(neat) 3523 cm−1 (OH), 1741 cm−1 (C = O). 1H NMR (400 MHz, CDCl3) δ 5.73 (1 H, apt t, J = 3.7 Hz, H-3), 5.30 (1 H, d, J = 3.9 Hz, H-2), 5.20 (1 H, dd, J = 8.3, 3.6 Hz, H-4), 4.23–4.02 (4 H, m, H-6. H-6’, C(O)OCH2CH2CH2CH3), 3.89–3.80 (1 H, m, H-5), 2.16 (3 H, s, COCH3), 2.13 (3 H, s, COCH3), 2.09 (6 H, s, 2 x COCH3), 1.66–1.57 (2 H, m, C(O)OCH2CH2CH2CH3), 1.45–1.31 (2 H, m, C(O)OCH2CH2CH2CH3), 0.93 (3 H, apt t, J = 7.4 Hz, C(O)OCH2CH2CH2CH3) ppm. 13C{1H} NMR (126 MHz, CDCl3) δ 171.13, 170.51, 169.73, 166.87 (COCH3, C-1), 71.75 (C-2), 70.78 (C-4), 69.51 (C-3), 68.41 (C-5), 65.94, 64.66 (C-6, C(O)OCH2CH2CH2CH3) 30.39 (C(O)OCH2CH2CH2CH3), 20.74, 20.54, 20.44 (COCH3), 18.93 (C(O)OCH2CH2CH2CH3), 13.61 (C(O)OCH2CH2CH2CH3) ppm. MS: (CI - NH3) m/z: 438 (M + NH4, 100 %). Anal. Calcd. for C17H26O11: C, 51.42 %; H, 6.71 %. Anal. Found: C, 51.33 %; H, 6.78 %.
4.4.5 n-Butyl 2,3,4,6-tetra-O-acetyl-d-galactonate 5
(0.15 g, 0.35 mmol, 49 % yield). TLC: decomposes on silica. [α]D25 = +6.7° (c, 1.8, CHCl3). νmax(neat) 3430 cm-1 (OH), 1751 cm-1 (C = O). 1H NMR (400 MHz, CDCl3) δ 5.76 (1 H, m, H-2), 5.30 (1 H, dd, J = 3.9, 2.0 Hz, H-4), 5.16 (1 H, m, H-3), 4.25–4.15 - 4.06 (4 H, m, C(O)OCH2CH2CH2CH3, H-6), 3.97 (1 H, dd, J = 11.5, 7.4 Hz, H-5), 2.15 (3 H, s, COCH3), 2.11 (3 H, s, COCH3), 2.07 (3 H, s, COCH3), 2.06 (3 H, s, COCH3), 1.72–1.52 (2 H, m, C(O)OCH2CH2CH2CH3), 1.45–1.20 (2 H, m, C(O)OCH2CH2CH2CH3), 0.88 (3 H, m, C(O)OCH2CH2CH2CH3) ppm. 13C{1H} NMR (126 MHz, CDCl3) δ 170.99, 170.51, 170.18, 169.36 (COCH3), 167.76 (C-1), 69.62 (C-2, C-4), 68.65 (C-3), 67.71 (C-5), 66.01 (C(O)OCH2CH2CH2CH3), 65.86 (C-6), 30.3 (C(O)OCH2CH2CH2CH3), 20.74, 20.71, 20.55, 20.43 (COCH3) 18.95 (C(O)OCH2CH2CH2CH3) 13.6 (C(O)OCH2CH2CH2CH3) ppm. MS: (CI - NH3) m/z: 438 (M + NH4, 100 %). Anal. Calcd. for C18H28O11: C, 51.42 %; H, 6.71 %. Anal. Found: C, 51.33 %; H, 6.60 %.
4.4.6 n-Butyl 2,3,4,6-tetra-O-acetyl-D-mannonate 6
(0.13 g, 0.30 mmol, 21 % yield). TLC: decomposes on silica. νmax(neat) 3406 cm−1 (OH), 1749 cm−1 (C = O). 1H NMR (400 MHz, CDCl3) δ 5.67 (1 H, dd, J = 8.5, 2.2 Hz, H-3), 5.28 (1 H, dd, J = 9.1, 2.2 Hz, H-2), 5.09 (1 H, d, J = 8.5 Hz, H-4), 4.29 - 4.14 (2 H, m, C(O)OCH2CH2CH2CH3), 4.11 (2 H, td, J = 11.0, 5.3 Hz, H-6/H-6’), 3.80 (1 H, ddd, J = 11.6, 8.1, 3.4 Hz, H-5), 2.15, 2.13, 2.09 (12 H, 4 x COCH3), 1.69 - 1.52 (2 H, m, C(O)OCH2CH2CH2CH3), 1.38 (2 H, td, J = 15.0, 7.4, Hz, C(O)OCH2CH2CH2CH3), 0.94 (3 H, t, J = 7.4 Hz, C(O)OCH2CH2CH2CH3) ppm. 13C{1H} NMR (126 MHz, CDCl3) δ 171.14, 170.85, 169.64, 169.55 (COCH3), 167.57 (C-1), 69.59 (C-4), 69.55 (C-2), 68.97 (C-3), 67.62 (C-5), 64.78 (C-6), 65.91 (C(O)OCH2CH2CH2CH3), 30.36 (C(O)OCH2CH2CH2CH3), 20.77, 20.62, 20.39 (COCH3), 18.94 (30.36 (C(O)OCH2CH2CH2CH3)) 13.6 (30.36 (C(O)OCH2CH2CH2CH3)) ppm. MS: (CI - NH3) m/z: 438 (M + NH4, 100 %).
4.5 Lactide Polymerisation Protocol
To a solution of LZnEt (0.1 mL of 0.07 M solution in CDCl3, 7.00 μmol, 1 eq.) and the aldaric ester (0.1 mL of 0.07 M solution in CDCl3, 7.00 μmol, 1 eq.), in a Young's tap NMR tube, was added lactide (50.4 mg, 0.35 mmol, 50 eq.) and CDCl3 (0.3 mL). Monomer conversion was monitored by 1H NMR spectroscopy. Once complete conversion was achieved, the polymer was isolated by repeated precipitation into hexane (isolated yield = 90 % in all cases) and was analyzed by 1H NMR spectroscopy and GPC.
4.6 Degradation Protocol
The polymer sample (20 mg), which had previously been dried in vacuo was dissolved in CHCl3 to form a 3 mg/mL solution. To this was added TFA(aq)/CHCl3 (2 % v/v) (0.1 mL per mL of polymer solution). Aliquots were taken on a daily basis and analysed by GPC (CHCl3, polystyrene standards).