

1 Introduction
Asymmetric catalysis plays a pivotal role in modern synthetic organic chemistry [1,2]. The practicing organic chemist has the choice between chiral synthetic transition metal complexes [1–3], organocatalysts [4–6] and enzymes [7–9]. No catalyst can be truly universal for a given type of transformation. It is therefore wise to develop a large toolbox in which all three types of catalysts are represented, often functioning in a complementary fashion. Enzymes have been exploited in organic chemistry for more than one hundred years [7–9], including a number of notable industrial examples [8–10]. However, biocatalysis has traditionally suffered in many cases from stringent limitations relating to narrow substrate scope, poor enantioselectivity and insufficient robustness observed. With the advent of directed evolution of enantioselective enzymes in 1997 [11], it became possible to eliminate some of these disadvantages and limitations of biocatalysis. Indeed, 12 years after proof-of-principle was published [11], this unconventional approach to asymmetric catalysis is being increasingly used in numerous academic and industrial laboratories, covering almost all classes of enzymes [12–27]. The underlying concept is based on repeated cycles of gene mutagenesis, expression and screening (or selecting) for enantioselectivity (Scheme 1).
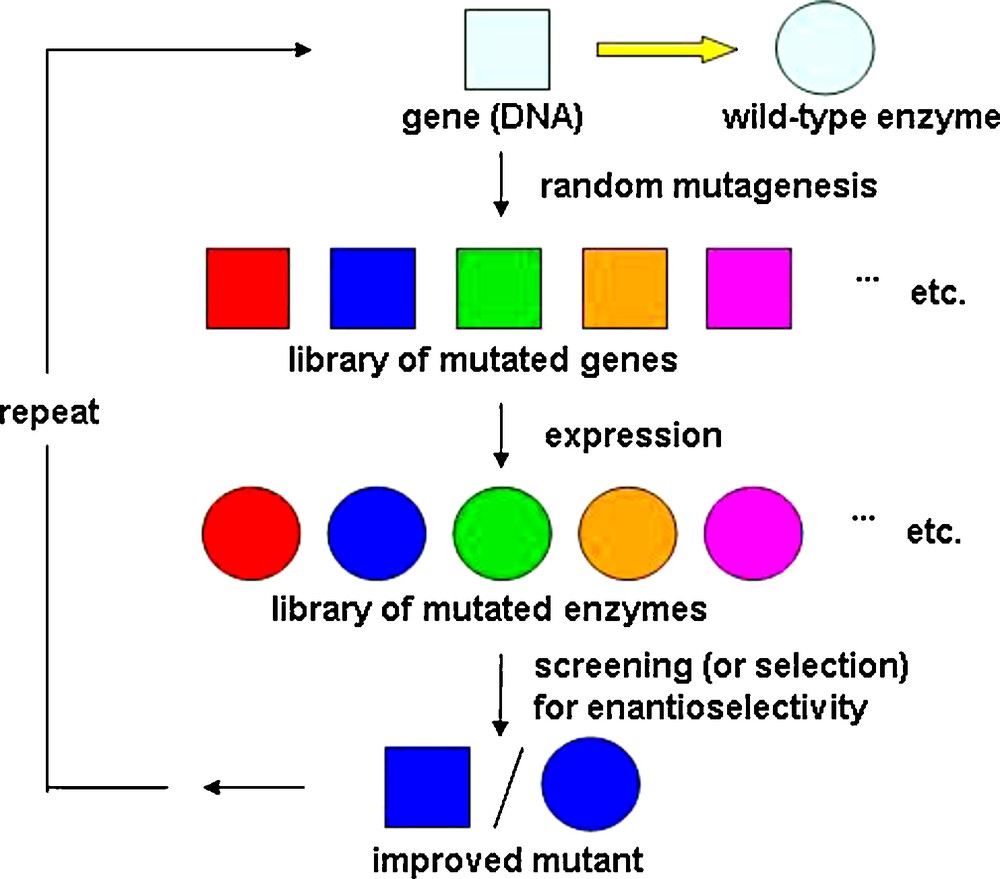
Concept of directed evolution enantioselective enzymes [11,17,19,20].
Prior to the announcement of this concept [11], directed evolution of the stability of proteins had been introduced [18]. In all studies of this type of protein engineering, the most often used gene mutagenesis methods are error-prone polymerase chain reaction (epPCR), saturation mutagenesis and DNA shuffling [12–17]. These methods are the ones that were employed in the original proof-of-principle study involving the hydrolytic kinetic resolution of rac-1, catalyzed by the lipase from Pseudomonas aeruginosa (PAL) (Scheme 2) [11]. The wild-type (WT) PAL shows poor enantioselectivity in slight favor of (S)-2, the selectivity factor amounting to only E = 1.1.
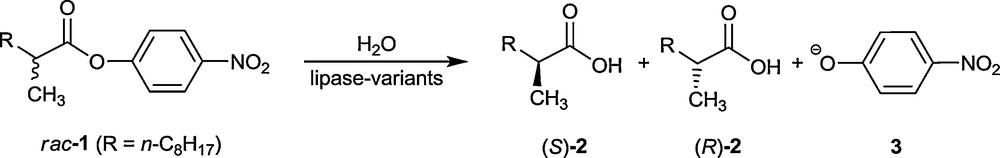
Model reaction used in the directed evolution of an enantioselective lipase (PAL) [11].
After four rounds of epPCR at low mutation rate with the introduction of four cumulative single point mutations, enantioselectivity increased stepwise to E = 11 as shown in Scheme 3 [11].
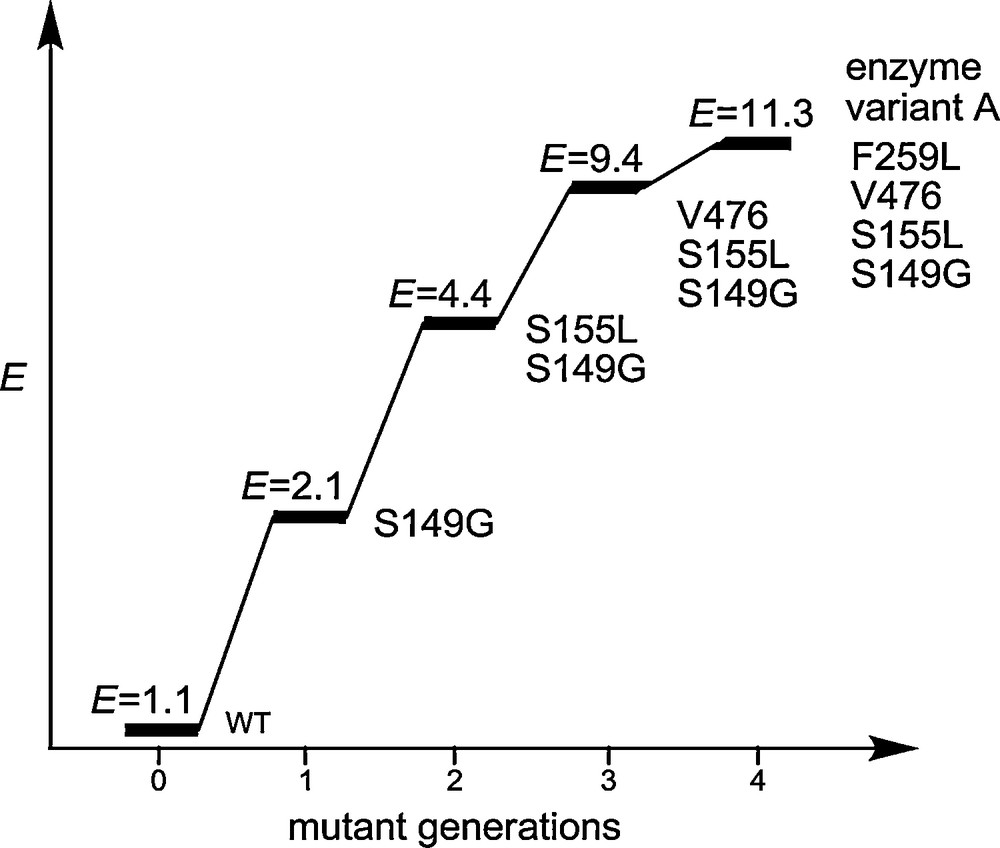
Results after four cycles of epPCR/screening in the model reaction involving the PAL-catalyzed kinetic resolution of rac-1 [11].
This early study clearly demonstrates the Darwinian principle as a means to tune a (bio)catalyst, but an E-value of 11 is hardly practical. A fifth round of epPCR resulted in E = 13, but this in turn was a signal that a better strategy had to be devised. Therefore, for several years various ideas were tested, such as saturation mutagenesis at hot spots previously identified by epPCR [19,20]. The best results were finally achieved by epPCR at high error rate, saturation mutagenesis at a four residue site next to the binding pocket with formation of what is now called a focused library, as well as DNA shuffling [21].
Following the screening about 50,000 transformants generated by these mutagenesis methods, the best mutant with six point mutations was evolved which showed a good selectivity factor of E = 51 [21]. Five of the six mutations were found to be located on the surface, and only one was next to the binding pocket, which came as a surprise. A detailed QM/MM study revealed that only two of the point mutations are necessary, and that a relay mechanism is operating [22,23]. The double mutant was made and found to be even more enantioselective (E = 63). This was a triumph of theory, but it also demonstrated that the applied strategies, although successful (including inversion of enantioselectivity [19,24]), were not efficient as one would like them to be. Picking up superfluous mutations cause unnecessary screening work and prevents “fast” directed evolution (Scheme 4).
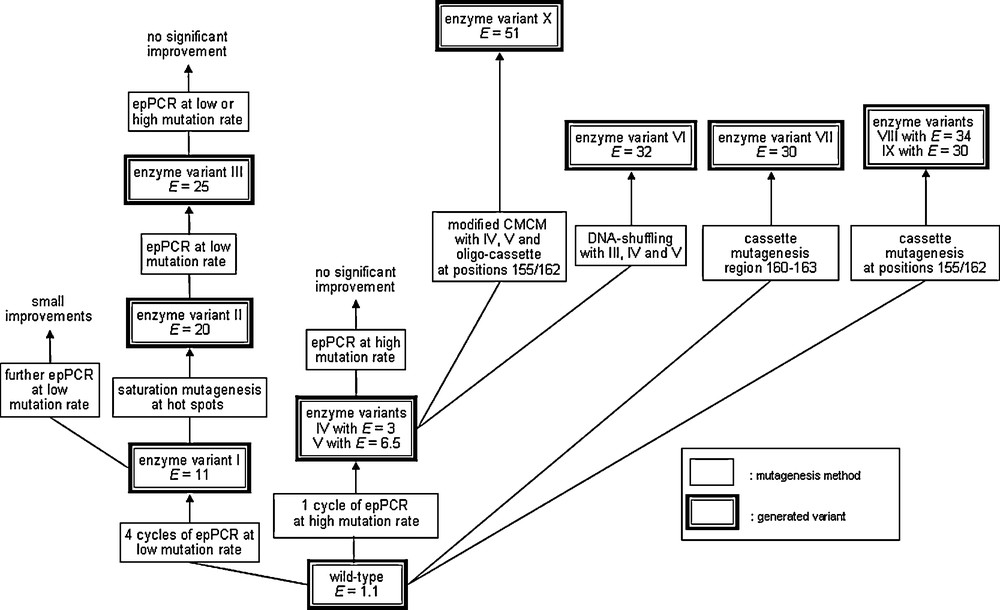
Summary of early work regarding the directed evolution of enantioselective PAL mutants as catalysts in the kinetic resolution of rac-1 [26].
The methods and strategies employed in these early studies [11,19–21] were subsequently applied to other enzymes by the Mülheim group and by other academic and industrial labs [17,25–27]. An example is the asymmetric Baeyer-Villiger (BV) reaction catalyzed by Baeyer-Villiger Monooxygenases (BVMOs). The first such attempt concentrated on the oxidative desymmetrization of 4-hydroxycyclohexanone using the cyclohexanone monooxygenase (CHMO) from Acinetobacter. WT CHMO catalyzes this synthetically interesting transformation with poor enantioselectivity (ee = 9%) [28]. Using the conventional epPCR strategy as in the original proof-of-principle lipase-study [11], (R)- and (S)-selective mutants were evolved showing enantioselectivities of ≈ 90% ee [28]. One of the mutants, Phe432Ser, was then tested as a catalyst in the desymmetrization of a set of other structurally diverse substrates without performing any additional mutagenesis/screening experiments (Scheme 5) [29]. The results show that “it is possible to get more than what you screen for”, even when employing the conventional epPCR-based strategy. The extensive contributions of other groups regarding directed evolution of enantioselective enzymes have been summarized in several reviews [25–27].
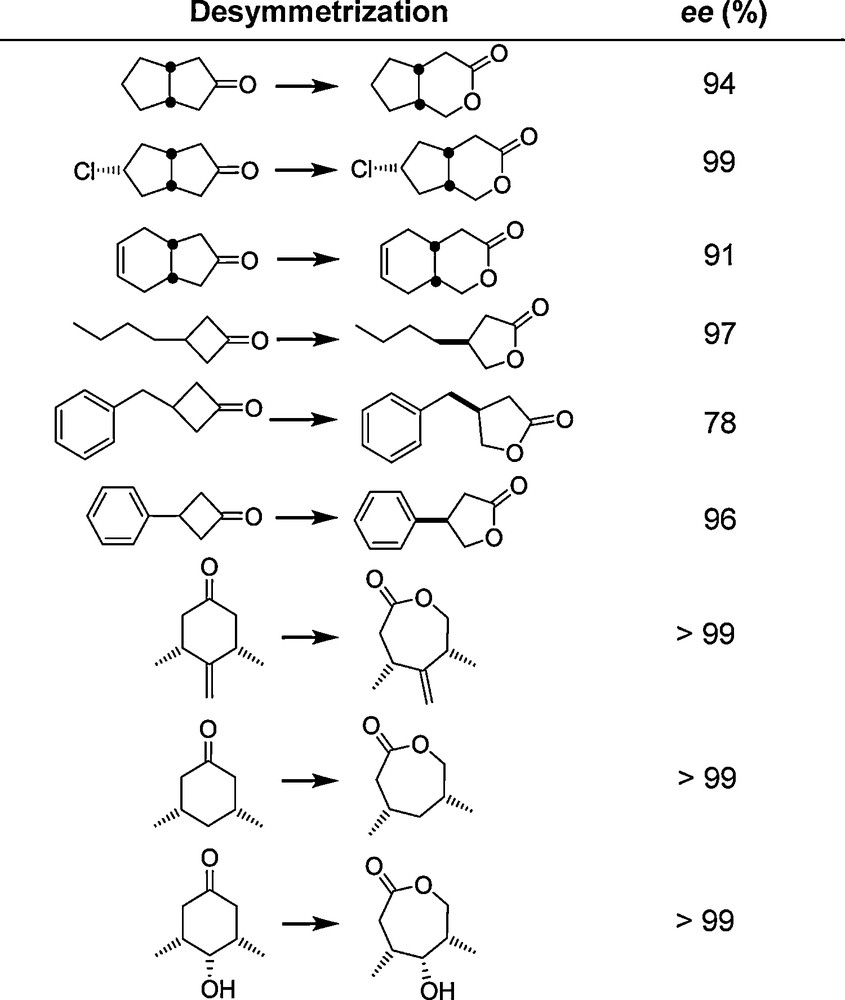
The substrate scope of CHMO mutant Phe432Ser [29] originally evolved for the desymmetrization of 4-hydroxycyclohexanone (conversion in all cases > 85%) [28]; last entry: unpublished data of M. T. Reetz, C. Clouthier and M. M. Kayser.
2 Methodology development in directed evolution
2.1 Iterative saturation mutagenesis for enhanced stereoselectivity and substrate scope
It has become abundantly clear that the bottleneck of directed evolution is the screening effort [12–17,30,31]. Libraries having 103–106 members (transformants) are common, which generally means laborious analytical work in the screening process requiring high-throughput techniques. Even with advanced analytical tools, a great deal of time and money needs to be invested. Therefore, in addition to devising high-throughput ee-assays [30,31], strategies and/or methods for probing protein sequence space more efficiently than in the past are needed [32–37], allowing for small but high-quality libraries which can be screened by conventional automated GC or HPLC, preferably following a pre-screen plate test [12–17,30,31]. Quality in this context means a high frequency of hits in a given library and a maximum in improvement of a given catalytic property such as enantioselectivity, rate or thermostability [37]. This challenging goal was recently reached by the introduction of iterative saturation mutagenesis (ISM) [38]. Accordingly, proper sites A, B, C, etc., each comprising one or more amino acid positions, are chosen for saturation mutagenesis. The best hit identified in each library is then used as a template for further rounds of saturation mutagenesis at the respective other sites, and so on (Scheme 6). The second or third best hit in a library can also be used.
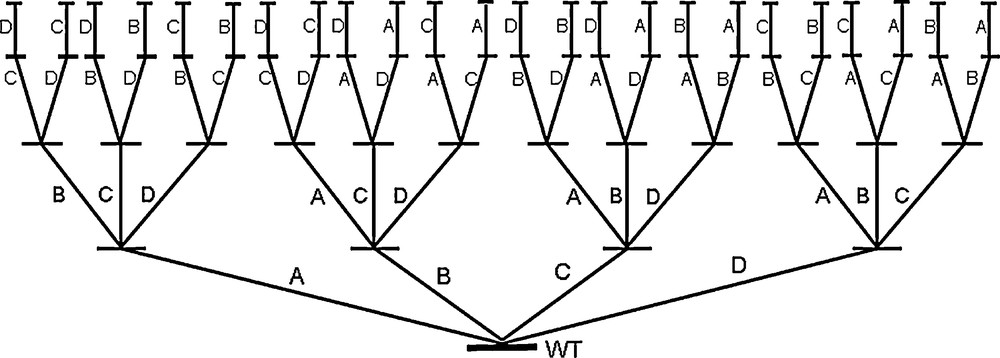
Illustration of iterative saturation mutagenesis (ISM) shown here for the case of four randomization sites A, B, C and D [38].
The decision regarding the choice of the sites at which saturation mutagenesis is to be applied is crucial, which in turn depends upon the property to be engineered. In the case of enantioselectivity and/or substrate scope (rate), the Combinatorial Active-Site Saturation Test (CAST) has proven to be unusually successful [38–41]. Accordingly, all residues with side-chains aligning the binding pocket are considered for saturation mutagenesis, which is a systematization of earlier examples of focused libraries [21]. When applying CASTing, the iterative steps often prove to be crucial [37]. The first example to be reported refers to the directed evolution of the epoxide hydrolase from Aspergillus niger (ANEH) as a catalyst in the hydrolytic kinetic resolution of substrate 4 [38]. WT ANEH leads to a selectivity factor of E = 4.6 in slight favor of (S)-5. Six sites were chosen around the binding pocket, namely A, B, C, D, E and F, each comprising either two or three residues. The chosen upward pathway B → C → D → F → E resulted in the best mutant LW202 characterized by five cumulative sets of mutations adding up to a total of nine point mutations, and showing a selectivity factor of E = 115 (Scheme 7) [38]. A total of only 20,000 transformants had to be screened, which happens to be the same number that was required in an earlier study based on epPCR which led to only E = 11 [42]. Thus, this study strongly suggested that ISM in the form of iterative CASTing provides distinctly higher-quality mutant libraries. Subsequently, the successful application of CASTing was reported by other groups as well [43,44].
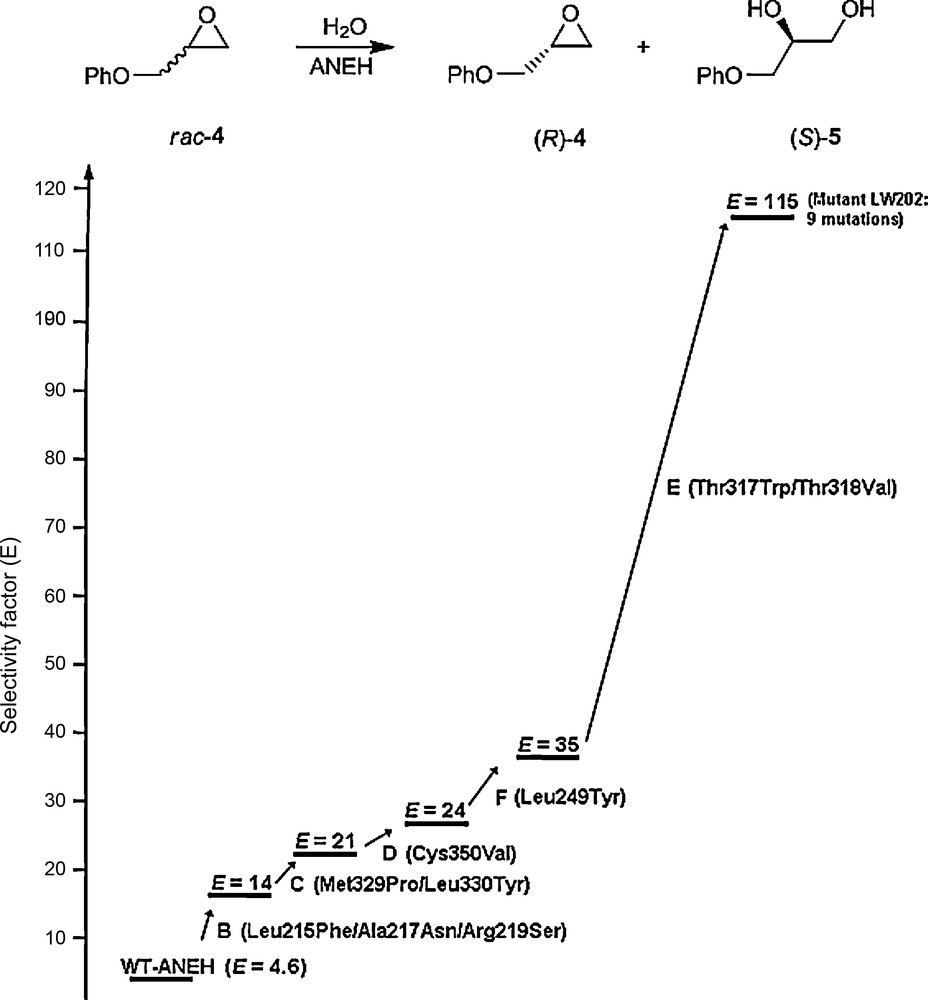
Iterative CASTing in the directed evolution of enantioselective ANEH mutants as catalysts in the hydrolytic kinetic resolution of rac-4 [38].
Several fascinating questions arose from these results which were addressed in follow-up studies. One point of interest was the source of enhanced enantioselectivity, which was addressed by a mechanistic investigation comprising kinetics, inhibitor studies, MD simulations, docking experiments and the X-ray structure of the best mutant LW202 versus that of WT ANEH [45]. Along a different line, it was of interest to construct the fitness landscape defined by 5! = 120 pathways leading from WT ANEH in five evolutionary steps to the best mutant LW202. In this systematic deconvolution study, it was found that about 50% of the trajectories are favored, meaning the absence of local minima, which is a very high score (Scheme 8) [46].
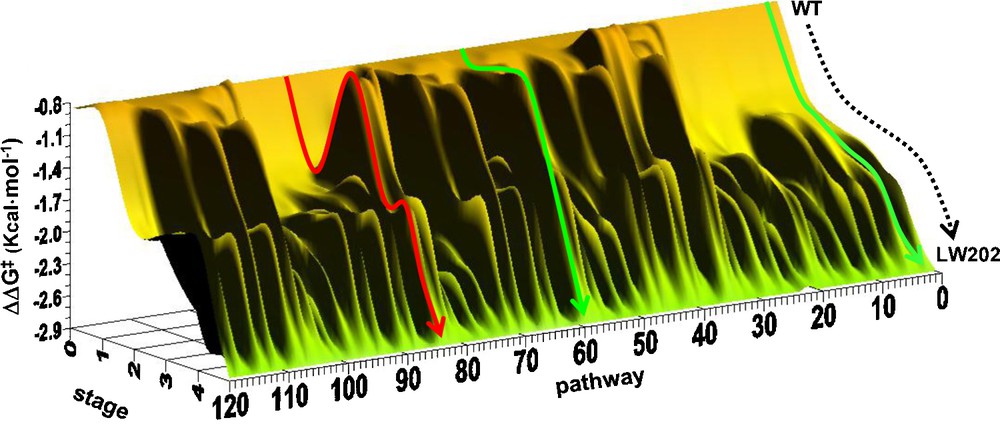
Energy profile of the two types of pathways leading from the WT to the mutant LW202: energetically favored (green) as in the original B → C → D → F → E (pathway 2) or D → C → F → E → B (pathway 60) and disfavored (red) as in E → C→ F → D → B (pathway 84).
This study also included the analysis of epistatic interactions between the five sets of mutations, strong cooperative effects (more than additivity) being identified. None of the five sets proved to be superfluous, which likewise speaks for the efficacy of ISM (Scheme 9).
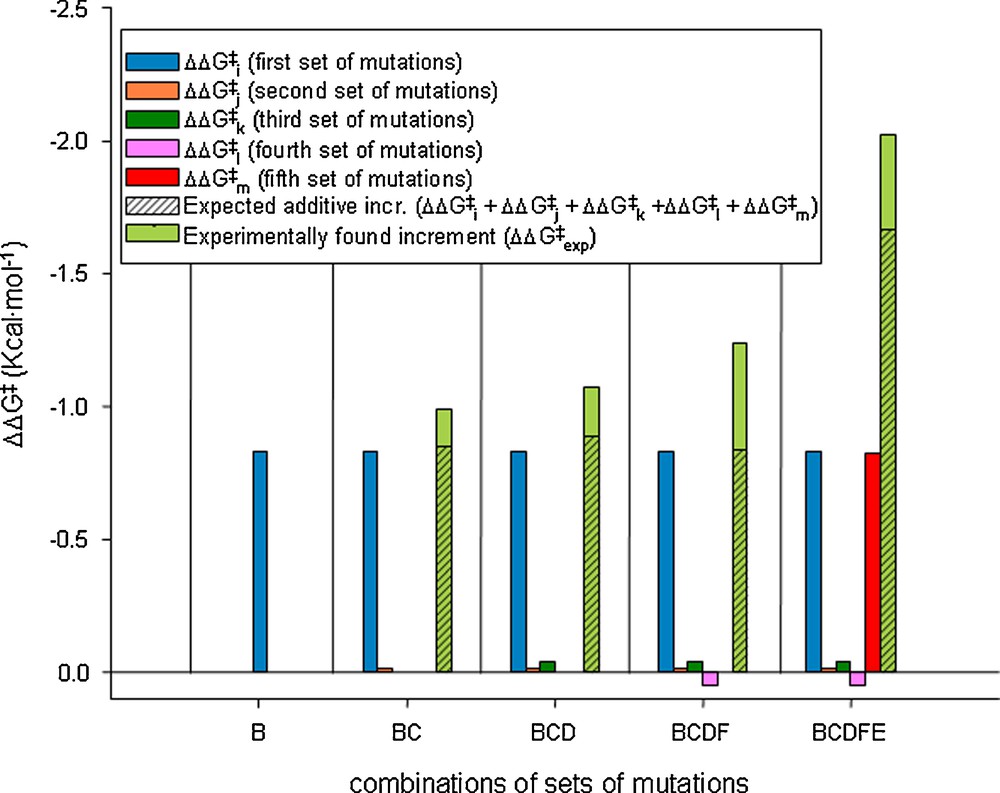
Thermodynamic cycle regarding the interaction of the sets of mutations involved at every stage along the energetically favored pathway B → C → D → F → E.
In a further study, a reduced amino acid alphabets was applied by utilizing the corresponding codon degeneracy, a very useful trick which reduces the degree of oversampling in the screening step drastically [41,47]. This is yet another tool for increasing the quality of libraries, especially when applied iteratively. A strategy in directed evolution of stereoselective enzymes yet to be tested is the use of unnatural amino acids in an expanded genetic code.
CASTing was also applied successfully to the first thermostable Baeyer-Villiger Monooxygenase (BVMO), namely Phenylacetone Monooxygenase (PAMO) [48]. The robustness of this BVMO [49–51] makes it an excellent candidate for real (industrial) applications, but unfortunately it accepts only phenylacetone and similar acyclic phenyl-substituted ketones. Using a bioinformatics approach based on the sequence alignment of eight BVMOs, a loop next to the binding pocket was targeted by CASTing, this time however using a reduced amino acid library as indicated by the conserved residues (Scheme 10) [48]. This novel strategy led to active mutants which accept 2-arylcyclohexanone derivatives with high enantioselectivity (Scheme 11). Randomization at second-sphere CAST residues was likewise successful [52].

Sequence alignment of eight BVMOs in the 441–444 loop (black box) next to the binding pocket used in the bioinformatic approach to the directed evolution of stereoselective BVMOs [48].
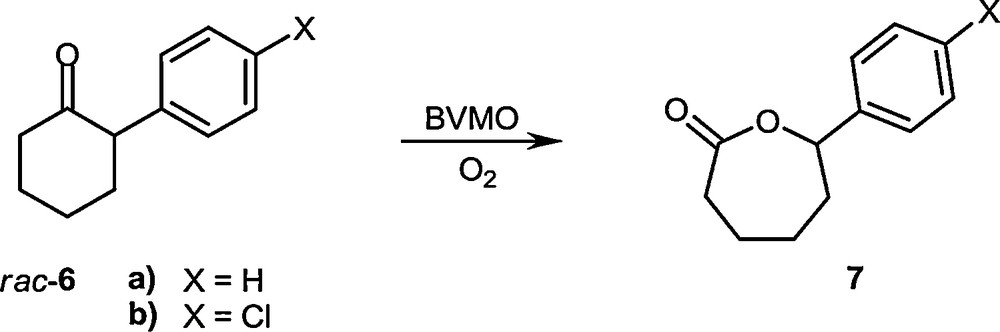
Oxidative kinetic resolution using PAMO mutants as BVMOs [48].
Iterative CASTing was also applied to the original system employed in the lipase project [11,19–21], this time with dramatically better results [53]. In the hydrolytic kinetic resolution of rac-1, six initial randomization sites were targeted, and after going into a second round and screening a total of only 18,500 transformants, a selectivity factor of E = 594 was observed [53]. The same strategy was also exploited in the directed evolution of limonene epoxide hydrolase [54]. Further extensions of the original CASTing concept were also developed, e.g., the idea of focusing on second-sphere residues. This approach was applied successfully to PAMO, resulting in a fairly broad substrate scope, high activity and pronounced enantioselectivity, while screening only 400 transformants [52]. All of these recent developments should be compared to previous studies of directed evolution of enantioselective BVMOs which involved considerably less robust representatives such as cyclohexanone monooxygenases CHMOs [28,55].
In another approach likewise based on saturation mutagenesis, randomization at the remote two amino acid site 93/94 was found to induce an allosteric effect in that two domains “move” together, leading to a reshaping of the binding pocket which in turn allows for high activity and stereoselectivity of PAMO [56] (Scheme 12).
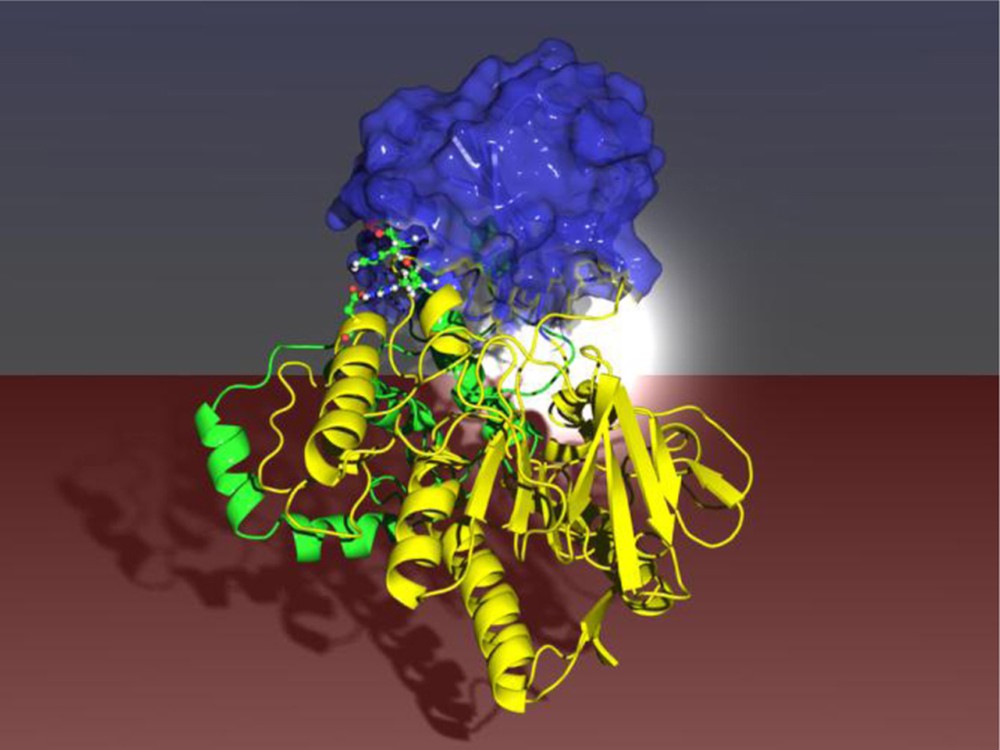
Saturation mutagenesis at a remote site (red dots) of PAMO leading to an allosteric effect with exposure and reshaping of the binding pocket (white color code) [56].
2.2 B-FIT method as a means to incease protein thermostability
Inceasing the thermostability of enzymes is an important endeavor in biotechnology, which has been accomplished by directed evolution many times [57–59]. Traditionally, such techniques as epPCR and DNA shuffling were employed. More recently, ISM has been considered, which raised the crucial question as to the appropriate choice of the saturation mutagenesis sites. The problem was nicely solved by developing the so-called B-FIT method (B-Factor Iterative Test) [60,61]. Accordingly, amino acid positions in the protein which have the highest B-factors indicating high flexibility, available from X-ray data, are chosen as the randomization sites A, B, C, etc., and then mutagenesis/screening is performed according to Scheme 6. This concept was first applied to the lipase from Bacillus subtilis (LipA), five successive steps leading to an increase in the T5060 value from 48 °C to 93 °C [60,61]. Such a dramatic effect has no precedence in the literature [57–59], yet required the screening of only 8000 transformants. In a follow-up study it was shown that strong cooperative effects are operating between the five accumulated point mutations (more than additivity), and that the evolutionary process induces the formation of a communicating amino acid network on the surface of the enzyme [62]. In unpublished biophysical work we discovered a novel effect, namely that the mutations actually prevent undesired aggregation and precipitation which would obviously impair enzyme function. B-FIT can also be employed in the unusual quest to lower protein thermostability, which in rare cases has practical applications [63]. It can also be used to enhance the robustness of enzymes in the presence of hostile organic solvents [64].
3 Conclusions
Laboratory evolution of enantioselective enzymes for use in synthetic organic chemistry and biotechnology constitutes a fundamentally new approach to asymmetric catalysis [11,17,25–27]. Traditional strategies such as epPCR and DNA shuffling, originally used in the proof-of-principle study [11,19–21], proved to be successful, although not necessarily optimal. Recent methodology development in directed evolution led to the concept of iterative saturation mutagenesis (ISM), which is accompanied by dramatically less screening effort due to the notably enhanced quality of the mutant libraries [37,38,43,44,46,47,60,61]. One of the additional tools in this approach is the use of reduced amino acid alphabets, which results in a further drastic reduction of the screening work [47]. ISM allows the experimenter to handle such features as enantioselectivity, substrate scope (rate), thermostability and robustness toward hostile organic solvents [64], these belonging to the most important catalytic parameters in biocatalysis. Applications beyond asymmetric catalysis in organic chemistry are on the horizon, as in nanobiotechnology, microbial pollution cleanup, and metabolic (pathway) engineering. Other groups have recently applied ISM for enhancing stereoselectivity, inducing enzyme promiscuity and even influencing metabolic pathways [43,44,65–70].
Acknowledgement
Thanks are due to the Fonds der Chemischen Industrie and the Deutsche Forschungsgemeinschaft (Schwerpunktprogramm 1170).