

1 Introduction
Avian influenza virus, especially the highly pathogenic strain H5N1, is now becoming a more and more severe threat worldwide. At the end of 2010, it has caused a death toll of more than 300 people [1] and a huge amount of property loss. Strict measurements are implemented with a great concern of virus mutation to cross species barrier and subsequently result in a dreadful global prevalence [2,3].
At present, monoclonal antibodies are needed that could be used in avian influenza, especially H5 strain diagnosis. These monoclonal antibodies should react ideally with all H5 avian influenza virus and do not have cross-reaction with other avian influenza strains like H3 and H9 [4]. So far, there are a number of monoclonal antibodies already used in avian influenza diagnosis. Although some researches focused on the epitope study of the hemagglutinin (HA) protein [5,6], detailed analysis of the antibody binding sites are still limited. New epitopes based upon subtype 5 HA remained to be further discovered and analysed especially when the epitopes have good characters like conserve, subtype-specific etc.
In this study, we report the generation of monoclonal antibodies targeting the HA1 domain of HA, which was expressed and purified from inclusion bodies of Escherichia coli. Among the monoclonal antibodies, some were selected for being able to recognize H5 subtype avian influenza virus lysate as well as the E. coli expressed HA1 protein antigen. These monoclonal antibodies are potentially diagnostic-valuable as they can be used in a sandwich enzyme-linked immunosorbent assay (ELISA) that can detect H5 subtype HA in virus lysate.
Further studies revealed that these monoclonal antibodies bound a region of amino acid residues N80 to A99 of HA and spatial structure [7] showed that this region is located upon the middle part of HA1 just below the HA's receptor-binding site [8]. Importantly, sequence alignment showed that this newly identified linear epitope is very conserved among H5 subtype HA but differs a lot from its counterparts in H3 and H9. It indicated that the monoclonal antibodies could probably bind to H5 subtype HA but not HA from other subtypes.
2 Materials and methods
2.1 Gene and reagents
HA gene and virus lysate supernatant were from the A/Goose/Guangdong/3/97(H5N1) virus strain. A reassortant inactivated avian influenza virus vaccine (H5N1 subtype, Re-1 strain) was purchased from Harbin Weike Biotech. Anti-his tag antibody and Taq DNA polymerase were purchased from Tiangen. Goat anti-mouse IgG horseradish peroxidase (HRP) conjugated antibody was purchased from Sigma-Aldrich. Ni-NTA Beads and materials for protein purification were from GE. Mouse sub-isotyping detection kit was from Bio-Rad. E. coli BL21 strain and pET28a vector were from Novagen, DH5α competent cells were preserved in our laboratory. Restriction endonucleases and T4 DNA ligase were purchased from Takara. Primer synthesis was carried out by Shanghai Sangon Co. Ltd.
2.2 Animals
New Zealand rabbits and BALB/c mice were purchased from Shanghai Laboratory Animal center, Chinese Academy of Sciences. Animals were kept in conventional conditions and were handled according to the Guidelines of Chinese Academy of Sciences for Animal Care and Use.
2.3 Expression and purification of HA1 protein
Primers used for gene amplification were as follows: sense primer: CGC GGA TCC ATT TGC ATT GGT TAC C, antisense primer: CCG CTC GAG TTC TTC TTC TCT CTC T. HA1 gene was inserted into pET28a and transformed into E. coli strain BL21. Bacterial cells were induced by isopropyl β-D-1-thiogalactopyranoside (IPTG, 1 mM of ultimate concentration) at 30 °C for 4 h in tryptone-phosphate medium. Cells were collected by centrifugation of 6000 rpm for 10 min and resuspension in phosphate saline buffer (PBS). Cells were broken up by sonication after which sediment was collected and suspended in 8 M carbamide. Extracted protein was purified by column purification with Ni-NTA beads with elution buffer of 8 M carbamide with imidazole of ultimate concentration 250 mM.
2.4 Polyclonal and monoclonal antibody generation
Rabbit immunization of the reassortant inactivated avian influenza virus vaccine was as follows: 2–2.5 kg male New Zealand rabbits were inoculated with the vaccine (1.5 ml/rabbit) subcutaneously with multi-site injection and boosted two times each with a three-week interval. One week after the last immunization, rabbit serum was collected and stored at −20 °C.
Antigen immunization for monoclonal antibody generation was as follows: six-week-old female Balb/c mice were immunized subcutaneously with 0.1 mg recombinant HA1 in emulsion 1:1 (v/v) with complete Freund's adjuvant and boosted two times each with a three-week interval of the same amount of antigen in emulsion with incomplete Freund's adjuvant. Three days before cell fusion the mouse was further boosted by 0.1 mg antigen in 0.5 ml PBS with intraperitoneal injection.
Monoclonal antibodies were raised by fusion of mouse spleen cells with murine myeloma cell line SP2/0 with 50% polyethylene glycol. Hypoxanthine, aminopterin and thymidine (HAT) was used for hybridoma selection and supernatant from wells of 96-well plates was collected for ELISA screening. A limited dilution method was used for hybridoma cloning of positive wells.
2.5 HRP labeling
HRP labeling was carried out as preciously described [9]. Briefly, Protein G purified polyclonal antibody against inactivated avian influenza virus vaccine (Weike Biotech) was labeled with HRP by 1.25% glutaraldehyde. The reaction was stopped by 0.2 M lysine for 2 h and then dialysed in PBS at 4 °C overnight. After that, the HRP conjugated polyclonal antibody was precipitated by 33% saturated ammonium sulfate and then dialyzed again in PBS. The HRP conjugated antibody was stored at −20 °C in 50% glycerol.
2.6 ELISA
ELISA was performed as preciously described [9]. Briefly, 96-well micro-titer plates were coated with HA1 protein in 0.1 M carbonate buffer (pH 9.6) with a concentration of 10 μg/ml at 4 °C overnight. Blocking buffer and dilution buffer were PBS containing 10% bovine serum and 0.1% Tween-20. After blocking of the plates at 37 °C for 2 h, the plates were washed and added with diluted sera or monoclonal antibody ascites etc. at various concentrations were added, then incubated at 37 °C for 2 h. After washing of nonspecific binding antibody, HRP-conjugated goat anti-mouse IgG antibody was diluted (with a dilution rate according to the manufacturer's instruction) and added to the plates. The plats were then incubated at 37 °C for another 1 h. Tetramethylbenzidine (TMB) was used as the substrate and the absorbance was measured at 450 nm by a microplate autoreader (Thermo).
2.7 Western blot
Protein samples were separated by sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a polyvinylidene fluoride (PVDF) membrane as previously described [9]. After blocking in 3% (w/v) bovine serum albumin (BSA) for 2 h, the membrane was incubated with anti-HA1 monoclonal antibodies at room temperature for 2 h, then incubated with HRP-conjugated goat anti-mouse IgG antibody at room temperature for another 1 h. The blots were developed using ECL detection reagents (Pierce).
3 Results
3.1 Monoclonal antibodies generated targeting E. coli expressed HA1 protein
HA1 protein contained 325 amino acid residues with a molecular weight of about 45 kDa. We obtained HA1 (with 6 × His tag) protein from inclusion bodies of E. coli and the protein was purified by Ni-NTA affinity chromatography with ultimate purity of more than 90% (Fig. 1A). Western blot also confirmed the target protein's expression by detecting the 6 × His tag (Fig. 1B). With the HA1 protein obtained, monoclonal antibodies were raised and six clones of hybridoma were obtained. To further demonstrate whether these monoclonal antibodies could react with H5 avian influenza virus lysate, a western blot was then carried out. Three clones among them were identified to be able to react with virus lysate (Fig. 2). The three clones were named 98-10, 145-9 and 157-6, respectively.

HA1 protein prepared for monoclonal antibody generation. A: Affinity chromatography purified Escherichia coli-expressed HA1 protein stained by coomassie brilliant blue. The purity of HA1 protein was > 90%. B: Western blot to confirm the HA1 protein expression. Lane 1: purified HA1 protein, lane 2: an unrelated protein with 6 × His tag.
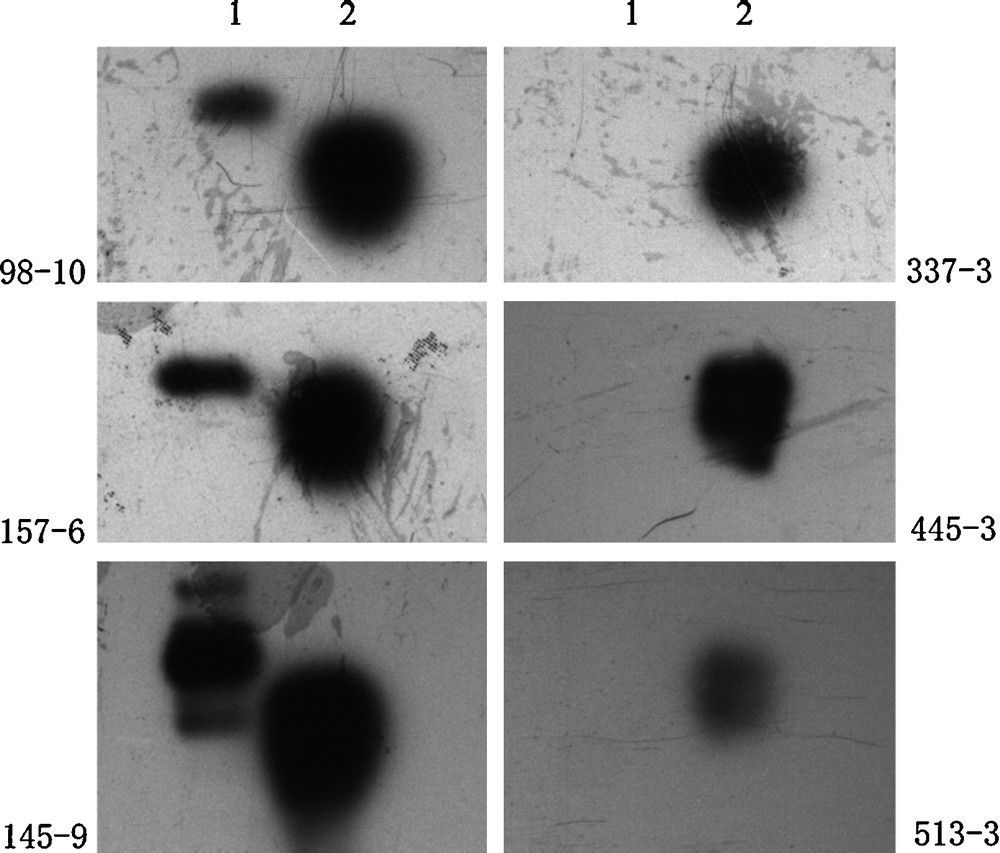
Western blot to test the anti HA1 monoclonal antibodies’ recognition with the HA1 protein antigen and the H5 virus lysate. As shown, three clones (98-10, 145-9 and 157-6) out of six (98-10, 145-9, 157-6, 337-3, 445-3 and 513-3) have binding activity to the H5 subtype virus lysate. Lane 1: sample of H5 subtype virus lysate, lane 2: sample of expressed HA1 protein.
3.2 Construction of a sandwich ELISA to detect H5 subtype avian influenza virus
Since three monoclonal antibodies that could react with H5 subtype avian influenza virus lysate were obtained, we then attempted to construct a sandwich ELISA that was able to detect H5 avian influenza virus antigen. The three monoclonal antibodies were used as coating antibody respectively and a HRP labeled polyclonal antibody generated from rabbits immunized with a reassortant inactivated avian influenza virus vaccine (H5N1 subtype, Re-1 strain) was used as detecting secondary antibody. As the results shown in Fig. 3, the three ELISA systems could detect HA antigen in H5 subtype virus lysate but not in H9. 145-9 and 157-6 were better than 98-10 for coating, for the signals were higher in these two groups. We also generated HRP labeled rabbit polyclonal antibody with immunization of the E. coli expressed HA1 protein as the secondary antibody, but the result was not as good as that used in the ELISA above (data not shown).
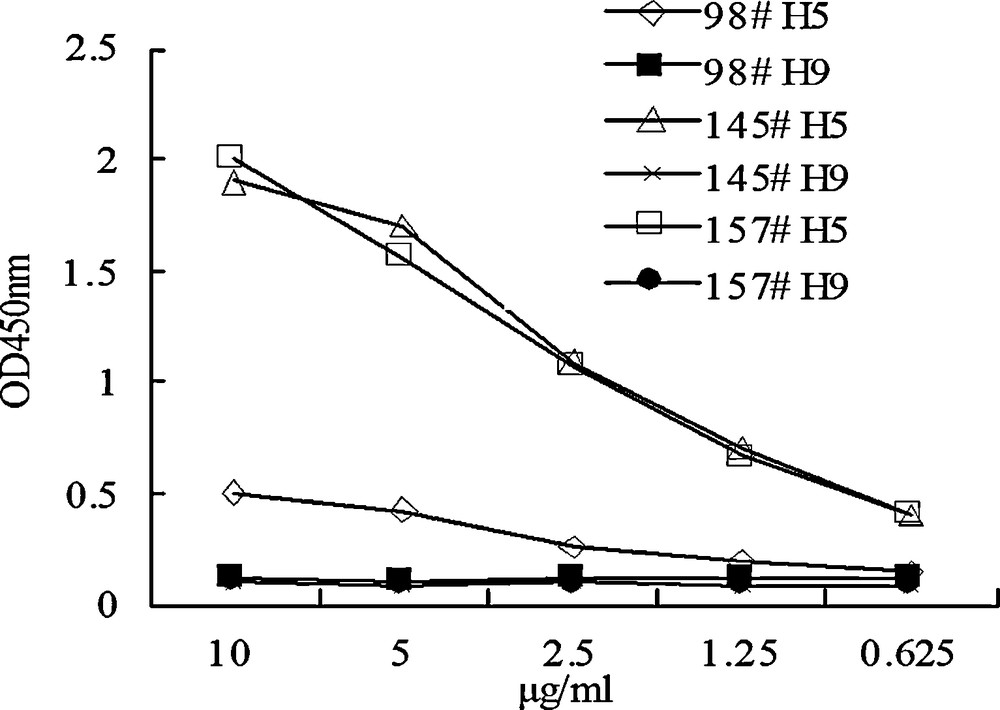
A sandwich ELISA established with the monoclonal antibodies (98-10, 145-9 and 157-6) as coating reagents to detect the virus lysate of H5 or H9 origin. A series of concentration of the virus lysate was tested. 98#, 145# and 157# represent monoclonal antibodies 98-10, 145-9 and 157-6, respectively.
3.3 Epitope analysis of the HA1 monoclonal antibodies
Based upon the data already obtained, we made further study upon the eptitopes that the monoclonal antibodies may recognize. A series of overlapping fragments that covered the entire HA1 sequence were constructed and expressed in E. coli (Fig. 4). Western blot revealed that the F1 and F2 fragments could be recognized by the monoclonal antibodies simultaneously (Fig. 5) and all the three clones of monoclonal antibody showed the same results (data not shown). This experiment indicated that the monoclonal antibodies bound the overlapping region of the F1 and F2 fragments. This binding region was located between amino acid residues 80-99 of HA and with a sequence of NPMCDEFINVPEWSYIVEKA. Sequence alignment further revealed that it was very conserved among both human and avian H5 subtype avian influenza virus and different from other virus sequences of the same location like H3 and H9 subtype.
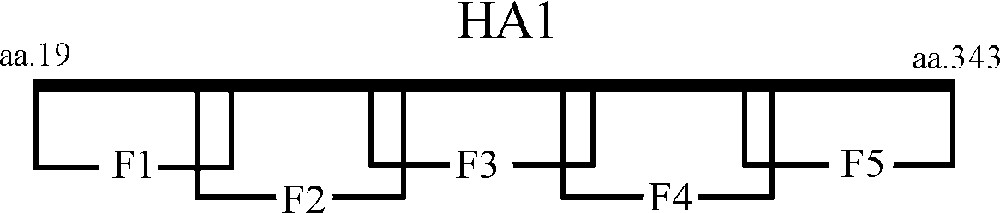
Diagram representing the overlapping fragments constructed within the HA1 sequence. F1: aa. 19-99, F2: aa. 80-160, F3: aa. 141-221, F4: aa. 202-282 and F5: aa. 263-343. Each fragment has a 20 amino acid residual overlapping region at the joint site.
Western blot to test the binding ability of the three clones of anti HA1 monoclonal antibody with the overlapping fragments. F1-5: the five fragments constructed and expressed in E. coli, U: an unrelated protein with 6 × His tag. An anti-6 × His tag antibody was used as a control and testing samples were loaded in the same amount as the control. All three clones had the same binding pattern and result of clone 157-6 was shown here.
We also marked this epitope upon the structure of the subtype 5 HA [7] (Fig. 6). As shown, this region is located in the middle part of the HA1 just below the receptor-binding site (aa. 126-278) [8]. It is upon the surface and opposite to the trimer axis face of the HA protein [8].
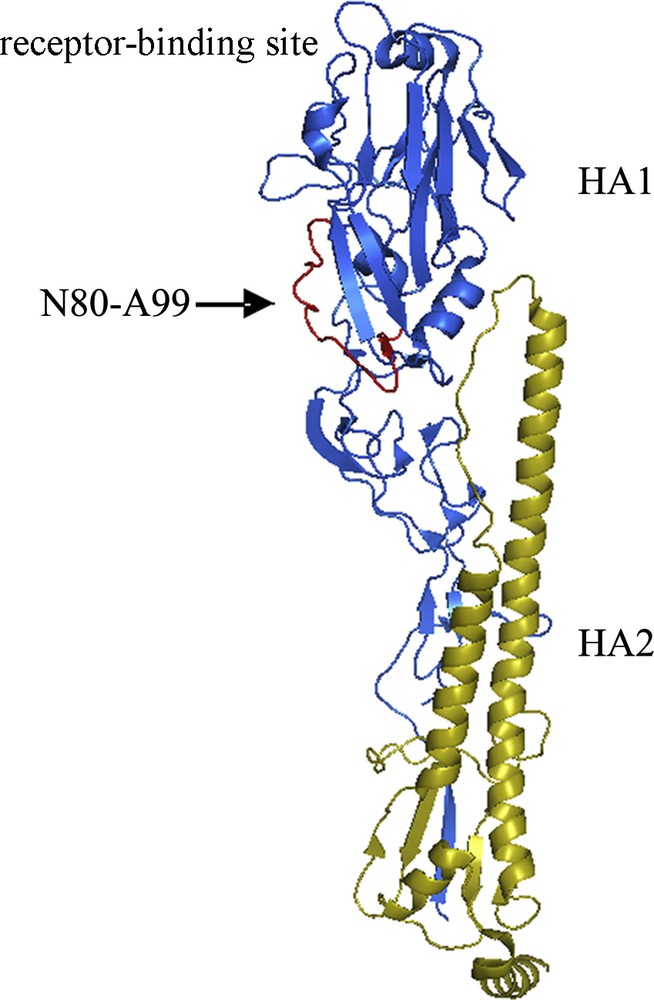
Spatial structure of the H5 subtype hemagglutinin (HA) and the epitope location. Blue: HA1 subdomain, yellow: HA2 subdomain, red and arrow marked: region of N80 to A99. N80-A99 was the epitope recognized by the three clones of monoclonal antibody 98-10, 145-9 and 157-6. The epitope was located out of the receptor-binding site region of the HA protein.
4 Discussion
In this article, we discovered that amino acid residues 80 to 99 are a linear epitope of subtype 5 HA and monoclonal antibodies targeting this region could be potentially used in H5 subtype avian influenza virus detection.
HA is an important influenza viral protein, which is crucial in viral entry process [3,10] and also a critical marker in influenza serological diagnosis owing to its strong antigenicity [11]. There are a number of antibody binding sites or epitopes, including important neutralizing epitopes, identified upon the HA especially the HA1 domain [6]. Monoclonal antibodies recognizing these epitopes have been widely used in immunoassays to detect avian influenza virus [11]. Theologically, the more conserve and subtype-specific of the antibody's recognizing epitope, the better the antibody to be used for diagnosis purpose. Therefore, epitope analysis and new epitope identification, especially for the subtype 5 HA, are very important.
There may be some differences between antigen of natural virus and virus lysate as methanal was used for virus inactivation. It is somehow difficult to obtain natural virus sample or patient sample, but further experiments are planned to test the monoclonal antibodies’ binding ability to patient samples. For the specificity of the sandwich ELISA assay, both the coating monoclonal antibody and the HRP labeled H5N1 vaccine induced polyclonal antibody may make contributions. Since the amino acid residue sequence of the epitope identified differs a lot between H5 subtype and other subtypes and additionally, the epitope is conformation-independent, the monoclonal antibodies generated could probably recognize H5 HA with high specificity.
Western blot (Fig. 2) showed that three clones out of six could react with H5 virus lysate while the other three could not, which is possibly due to E. coil expressed protein ‘creating’ some new epitope that did not exist in the virus derived antigen. The F1 and F2 fragment shown in the western blot of Fig. 5 seemed to have different reactivity with the monoclonal antibody. This might be because the F2 fragment did not contain some other amino acids just above the overlapping region that may also contribute to the antibody binding. Nevertheless the major part or the major location of the epitope was identified as described above.
Usually, it is not easy to obtain a qualified monoclonal antibody for diagnosis use by targeting an antigen obtained from regularly used E. coli protein expression system, partially due to the expressed antigen's poor ability of preserving its natural conformation. Therefore discovery of antibody binding sites or linear epitopes that are independent on the protein's natural conformation is meaningful. Such research is helpful for an efficient and more convenient target-driven monoclonal antibody generation (for example using peptide) with diagnosis purpose and contributes to a deeper understanding of the structure of the virus HA protein.
Acknowledgements
This work was supported by grant from a National Science and Technology Major Project (2008ZX10002-014, 2008ZX10004-002, 2009ZX10004-105 and 2009ZX10004-016), the National Natural Science Foundation of China (30950002, 30623003, 30721065, 30801011, 30870126, 30949019, 90713044), the Science and Technology Commission of Shanghai Municipality (08DZ2291703, 088014199, 08431903004), the Knowledge Innovation Program of Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (2010KIP304), grants from E-institutes of Shanghai Universities Immunology Division and a grant from the Li Kha Shing Foundation.