

1 Introduction
Lewis acids and Lewis bases usually quench each other when brought together in solution by formation of strong adducts [1]. This situation may be changed when these compounds become so sterically encumbered that their nucleophilic and electrophilic core centers can no longer come close enough to annihilate each other's characteristic properties by adduct formation. This situation had been encountered in the past by a small number of unsystematic examples [2,3], but the emerging field of frustrated Lewis pairs (FLPs) [4] has taken up pace only recently by the use of very strongly electrophilic and at the same time bulky C6F5 substituent containing borane Lewis acids in conjunction with rather bulky Lewis bases [5,6]. In this account we will describe a variety of examples of FLPs, mostly from my research group at Münster, and illustrate their remarkable properties by selected reactions with various small molecules.
2 Results and discussion
2.1 Reactions with dihydrogen
The activation of dihydrogen is commonly carried out with the aid of metals, whether it is achieved heterogeneously at a noble metal surface or homogeneously by using suitable well defined molecular transition metal complexes [7]. Even nature usually uses metal centers in the hydrogenase enzymes to achieve heterolytic dihydrogen activation [8]. There is a small number of examples scattered unsystematically across the chemical literature describing various types of metal-free catalytic hydrogenation reactions, most taking place at rather forcing reaction conditions [9]. In addition a few specific mono-aminocarbenes were recently shown to add H2 to the carbene carbon center to form C-H bonds, as shown by Bertrand, Schöller et al. [10]. Sander et al. had shown that the strongly electrophilic carbene difluorovinylidene reacts with H2 in a hydrogen-doped argon-matrix to form the respective alkene [11].
Stephan et al. had shown that the intramolecular frustrated P/B Lewis pair (1) adds dihydrogen reversibly to yield the zwitterionic phosphonium/hydridoborate product 2 [5,12,13,27] the phosphonium/hydridoborate salt 4, but this reaction is apparently not reversible (Scheme 1).
My group had early on contributed to this interesting new development of FLP mediated metal-free dihydrogen activation. Patrick Spiess, then a member of the group at Münster, prepared the intramolecular ethylene-bridged frustrated Lewis pair (7) by a straightforward route [14]. This system turned out to be one of the most active dihydrogen activating systems of this type. Under ambient conditions it rapidly reacts with dihydrogen to yield the intramolecular phosphonium/hydridoborate zwitterion (8). The product was characterized by X-ray diffraction (Fig. 1) (Scheme 2).
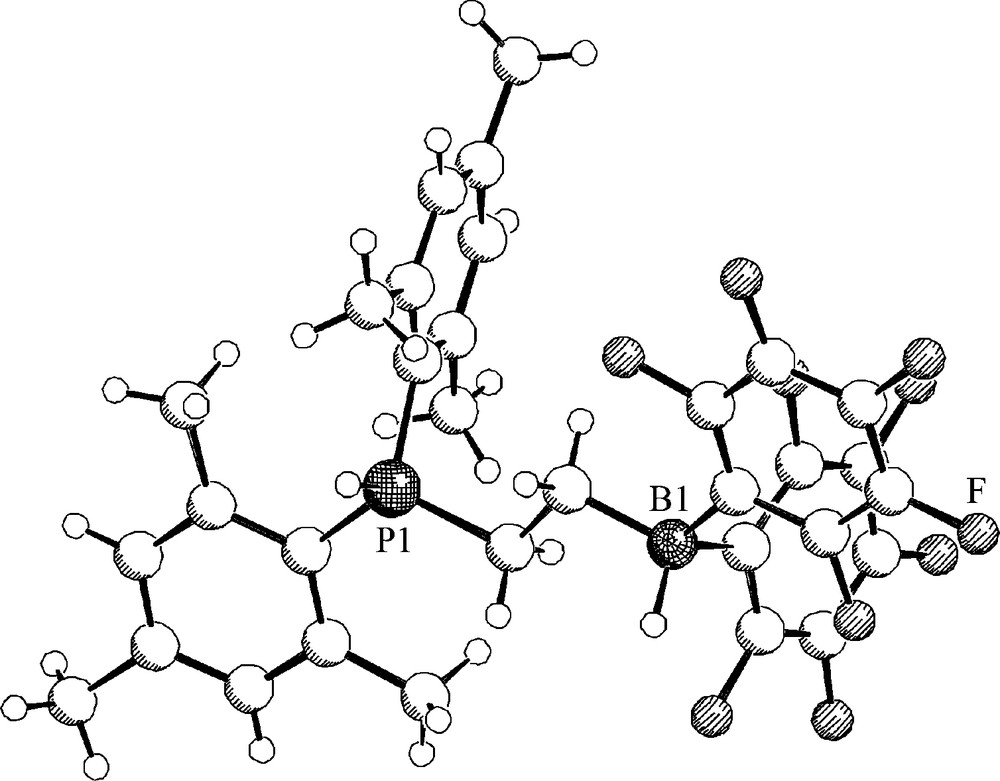
A view of the molecular structure of the zwitterion 8.
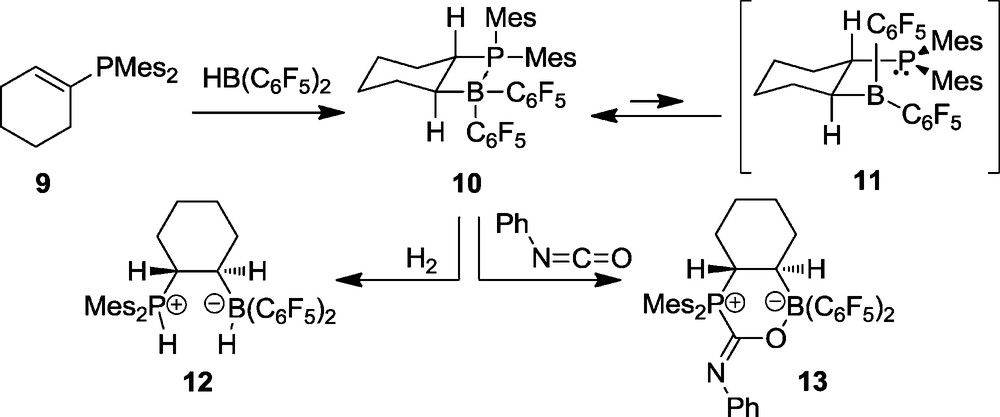
We prepared the related frustrated P/B Lewis pair 10 by “Piers’ hydroboration” of dimesityl(cyclohexenyl)phosphine (9) [15]. The X-ray crystal structure analysis of the P/B pair 10 (Fig. 2) features a four-membered heterocyclic structure annulated with the cyclohexane ring system. The P–B bond is rather long (10 features two independent molecules in the unit cell: mol. A: P–B: 2.188(5) Å, mol. B: 2.218(5) Å).
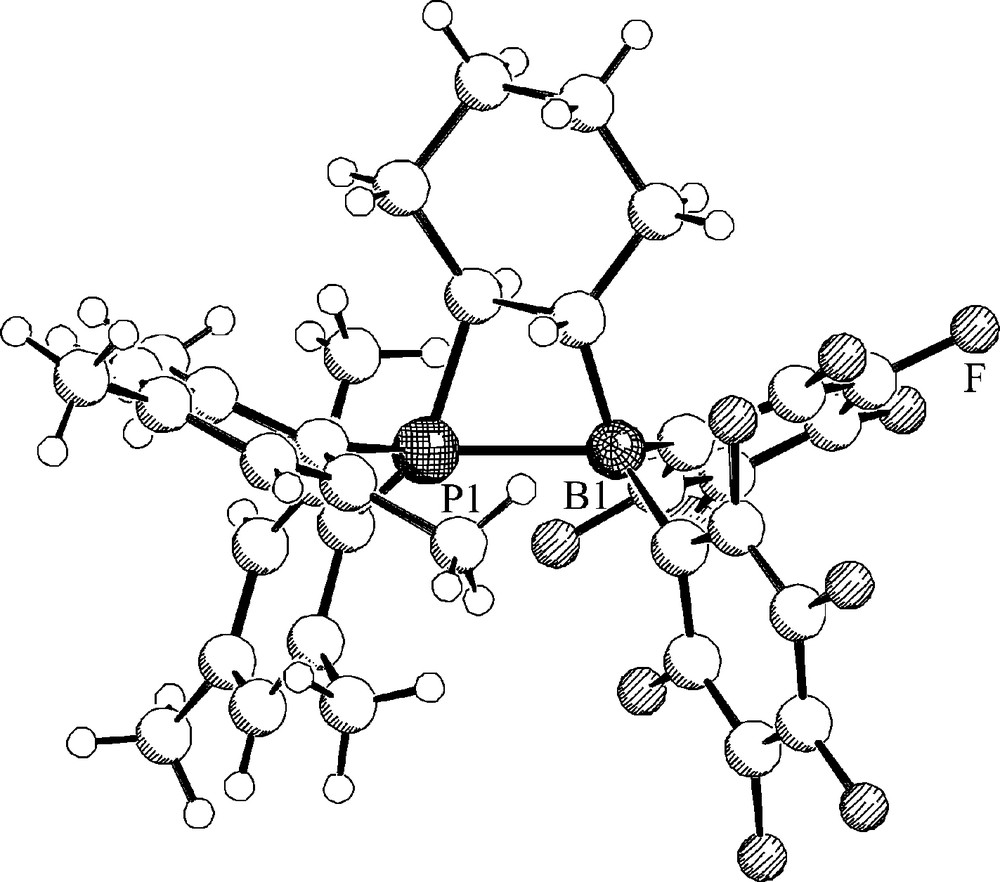
A view of the molecular structure of compound 11.
The P/B FLP 10 contains a pair of chirality centers on the bridgehead carbon atoms. Therefore, we observe the NMR features of a pair of diastereotopic mesityl substituents at phosphorus and of a pair of diastereotopic C6F5 rings at boron. Reversible rupture of the P-B linkage (to generate the non-observed high lying intermediate 11) would result in a disappearance of the B(C6F5)2 diastereotopism due to the non-prochiral nature of the trigonal planar borane moiety. This equilibrium actually reveals itself in the temperature dependent 19F NMR spectra of compound 10. From the observed coalescence of the pair of C6F5 rings in the temperature dependent 19F NMR spectra of 10 we have calculated a Gibbs activation energy of ΔGdis≠(283 K) = 12.1 ± 0.5 kcal mol−1 [15]. Consequently, the P–B bond dissociation energy in the FLP 10 must be below this value.
As expected for a rapidly opening P/B FLP system, we observe that 10 is a reactive activator of dihydrogen. Already at low temperature it splits dihydrogen heterolytically to yield the phosphonium/hydridoborate product 12. With phenylisocyanate it undergoes another typical FLP reaction, namely the 1,2-addition of the P/B pair to the reactive C=O double bond of the heterocumulene reagent to yield the product 13 (Scheme 3).
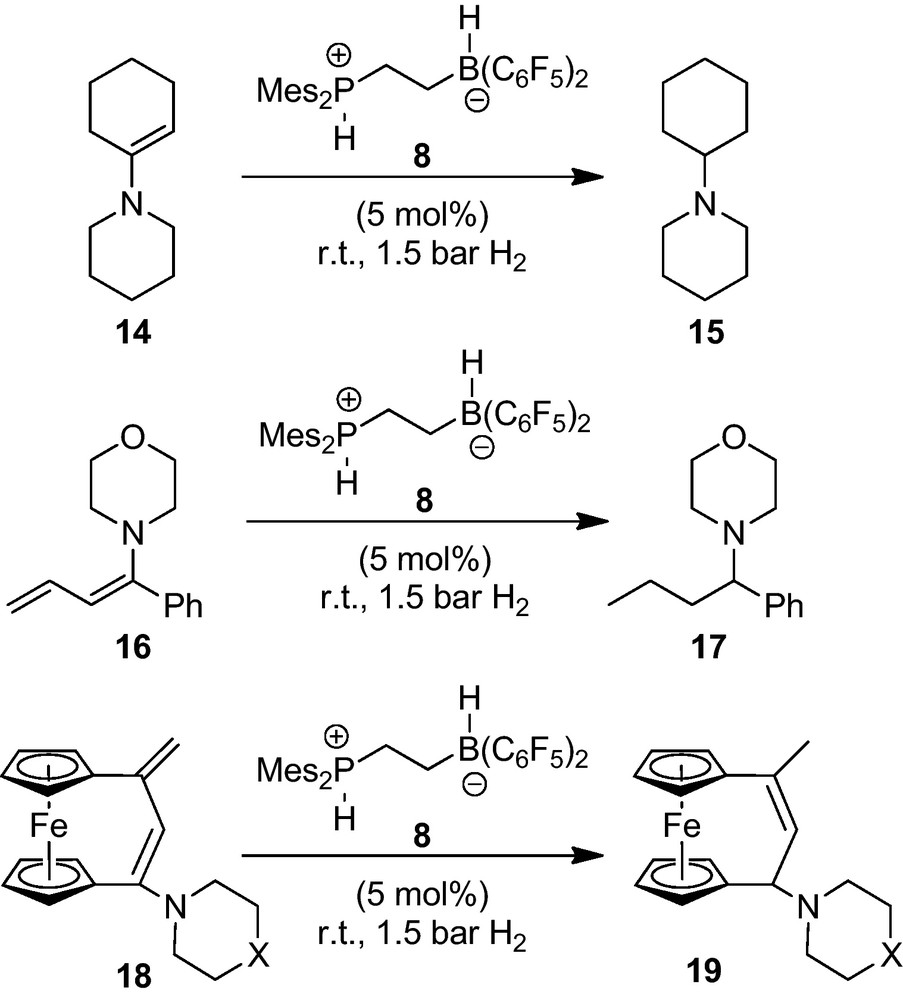
The intramolecular FLPs have served as catalysts for the hydrogenation of a variety of enamines under rather mild conditions [16]. Some representative examples are depicted in Scheme 4. Simple enamines, such as the example 14, are rapidly hydrogenated to yield the respective tert-amines. Conjugated dienamines are often fully hydrogenated. In some special cases (e.g. the [3]ferrocenophane dienamine 18) we have observed selective 1,4-hydrogenation instead, catalyzed by the system 7/8, to yield the respective allylamine product (19) [17].
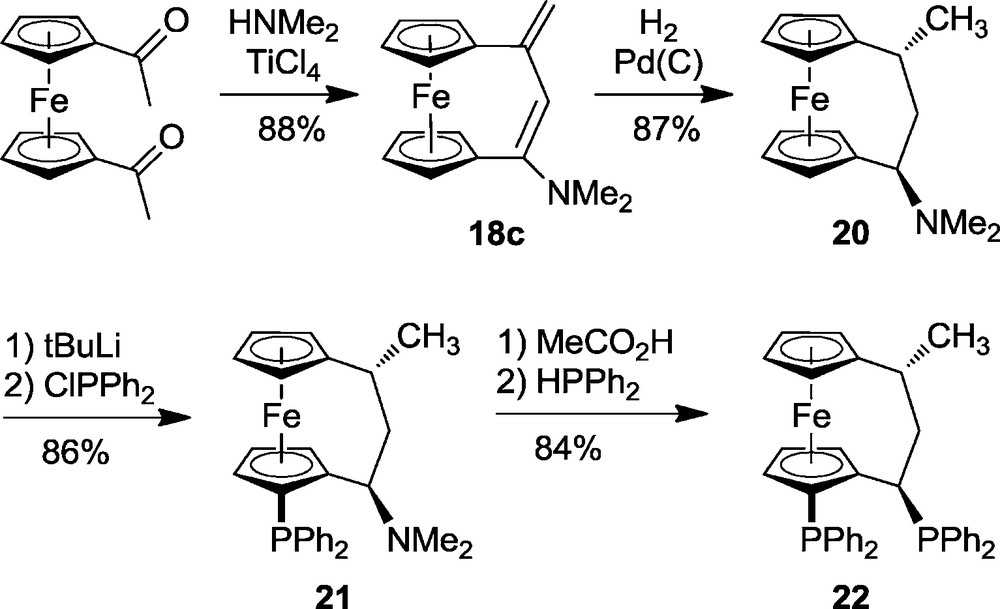
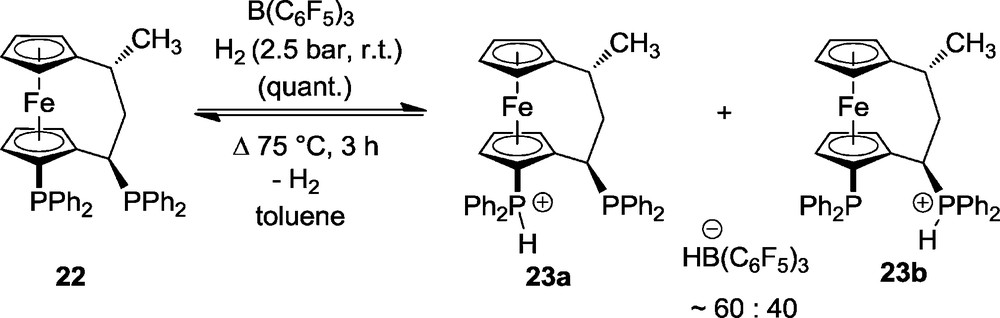
The systems 7 and 10 activated dihydrogen nicely but the heterolytic dihydrogen cleavage by these systems to yield 8 or 12, respectively, was irreversible under the applied reaction conditions. Therefore, we searched for specific systems that might allow studying FLP/H2 systems that were reversible. Some B(C6F5)3/chelate phosphine combinations were shown to do just that. Here is a typical example (Scheme 5). It comprised a chelate [3]ferrocenophane bisphosphine (22) that was synthesised in the following way. Mannich coupling of 1,1′-diacetylferrocene with dimethylamine and catalytic TiCl4 gave the unsaturated [3]ferrocenophane system 18c [18] that was subsequently catalytically hydrogenated to yield 20 (as 7:1 mixture of trans- and cis-diastereoisomers). The pair of phosphinyl substituents was then introduced by directed lithiation followed by quenching with Ph2PCl to give 21 [19]. Exchange of the -NMe2 group by -PPh2 was then carried out in the typical two step procedure at the carbon in α-position to the ferrocenophane to yield the system 22 with overall retention of configuration [20].
Compound 22 had been used as a chelate ligand either in its racemic or enantiomerically pure form [21]. With B(C6F5)3 [22] it formed a frustrated Lewis pair that rapidly activated dihydrogen at ambient conditions. It turned out that only one of the P-atoms of the phosphine was used in the H2-activation process. The other one remained untouched. The crystalline material features the proton on the “ortho”-PPh2 group (Fig. 3). However in solution we “see” both possible isomers 23a, 23b in a ratio of ca. 60:40 as judged by their typical 31P NMR spectra (Fig. 4). The heterolytic splitting of dihydrogen at this system is reversible. Heating the mixture of the mono-phosphonium/hydroborate salts 23a/23b to 75 °C in toluene solution resulted in a rapid evolution of dihydrogen to reform the frustrated Lewis pair 22/B(C6F5)3. The reaction cycle could then be closed by reacting this FLP again with dihydrogen at ambient conditions to again yield 23 [23] (Scheme 6).
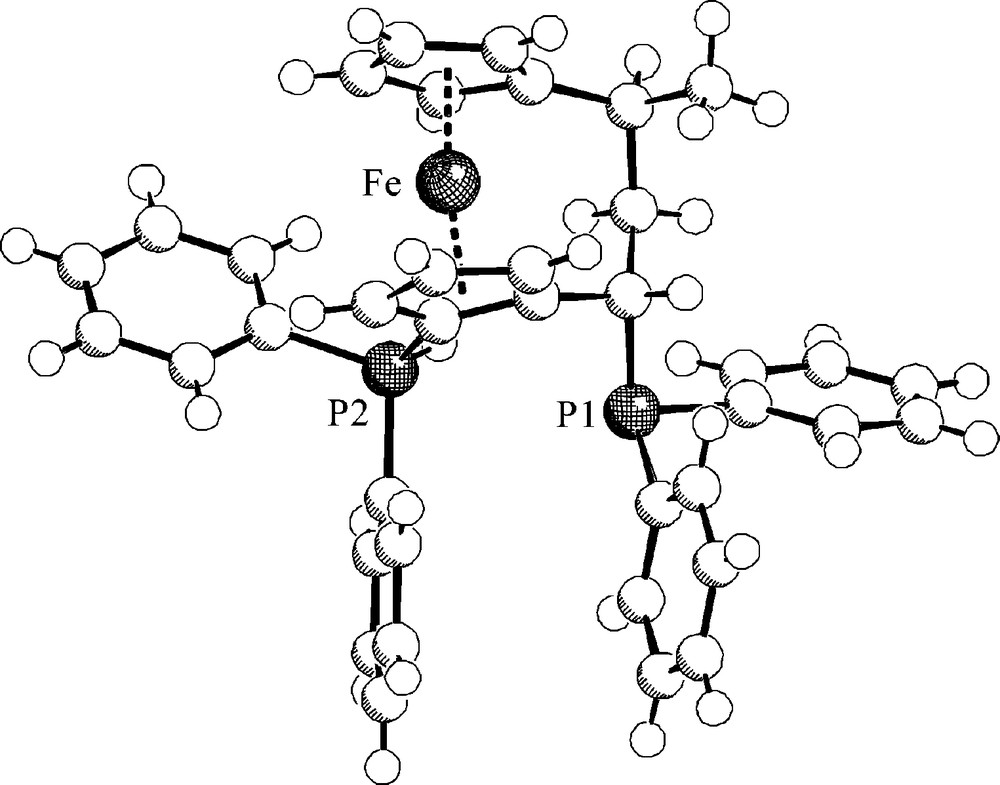
A view of the molecular structure of the [3] ferrocenophane derived phosphonium cation 23a (the [HBC6F5)3]− anion is not shown).
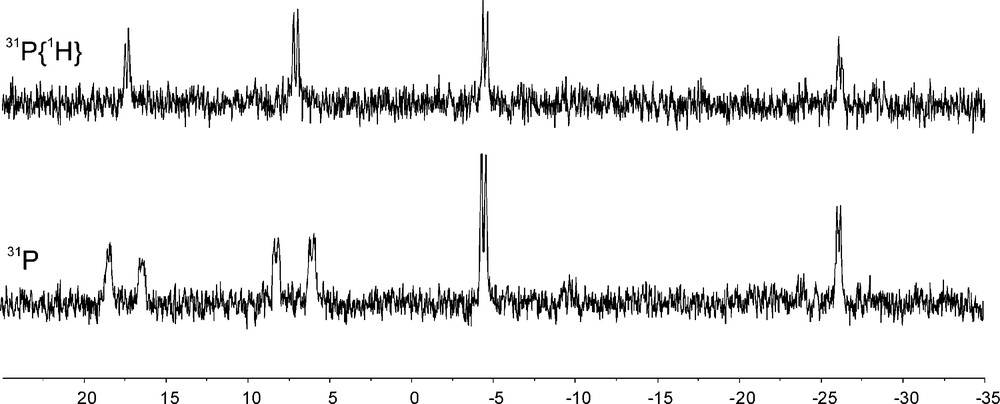
31P NMR spectra (243 MHz, 248 K, d8-toluene) of the 23a/23b mixture of isomers.
The system 23 is an active catalyst for the hydrogenation of silylenolethers, similar as it had been previously shown by us for the 1,8-bis(diphenylphosphino)naphthalene/B(C6F5)3/H2 system [24].
2.2 Reactions with carbon dioxide
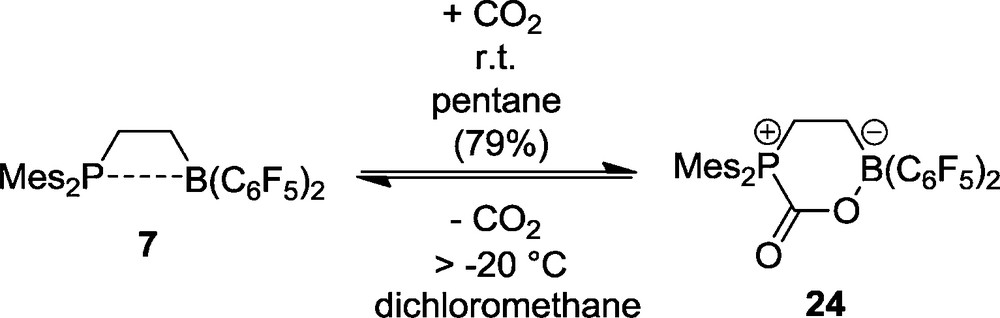
Many frustrated Lewis pairs react readily with carbon dioxide to yield the respective adducts. The very reactive intramolecular FLP 7 reacts with CO2 at ambient temperature in pentane solution to yield a white precipitate of the CO2 addition product 24. This is a rather thermolabile substance in solution, which rapidly losses CO2 again above −20 °C. In this way the CO2 uptake/CO2 elimination can be shuttled easily between the FLP 7 and the FLP-CO2 adduct 24 [25] (Scheme 7).
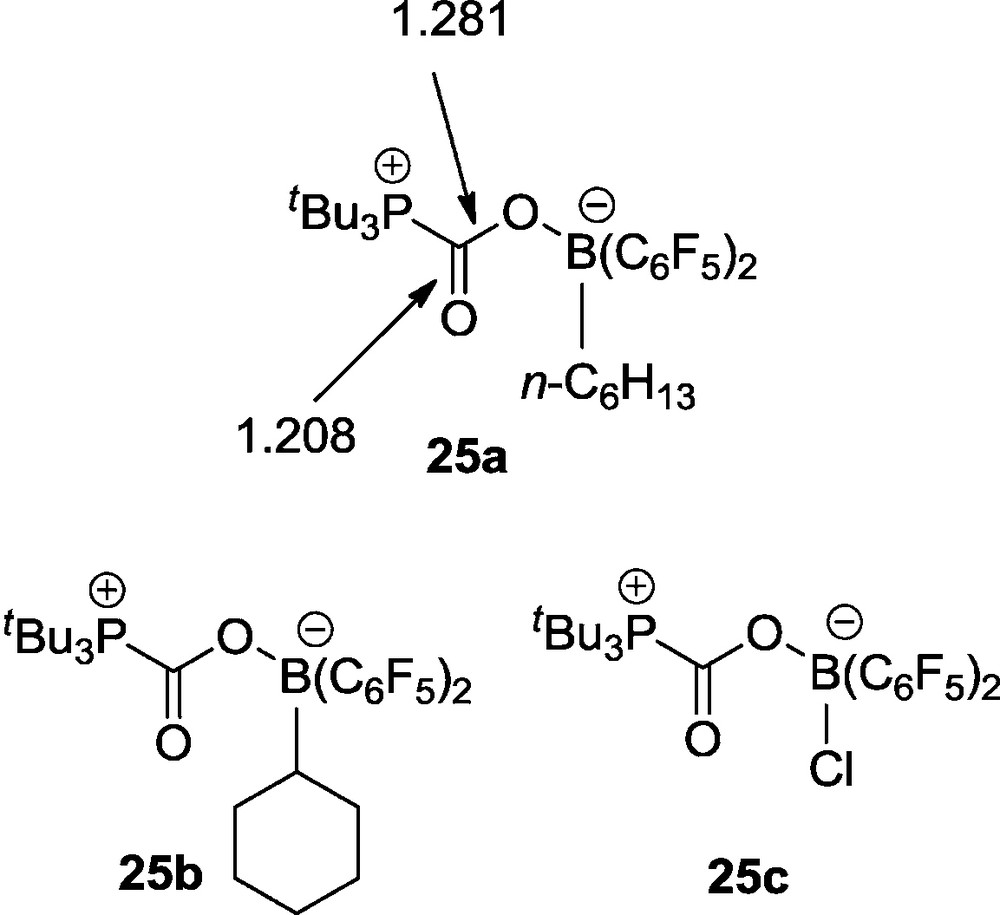
At low temperature the CO2 adduct 24 features a υ(CO) IR band at 1694 cm−1, a 13C NMR carbonyl carbon signal at δ 168.8 ppm and 31P and 11B NMR resonances at δ 0.6 ppm and δ −2.5 ppm, respectively. The X-ray crystal structure analysis of 24 shows the presence of a non-planar half-chair like six-membered heterocycle with bond length of O1-C3: 1.284(4) Å, B-O1:1.550(4) Å, a C3-O1-B angle of 130.9(3)° and a carbonyl C=O bond length of 1.209(4) Å (C3-O2) (Fig. 5).
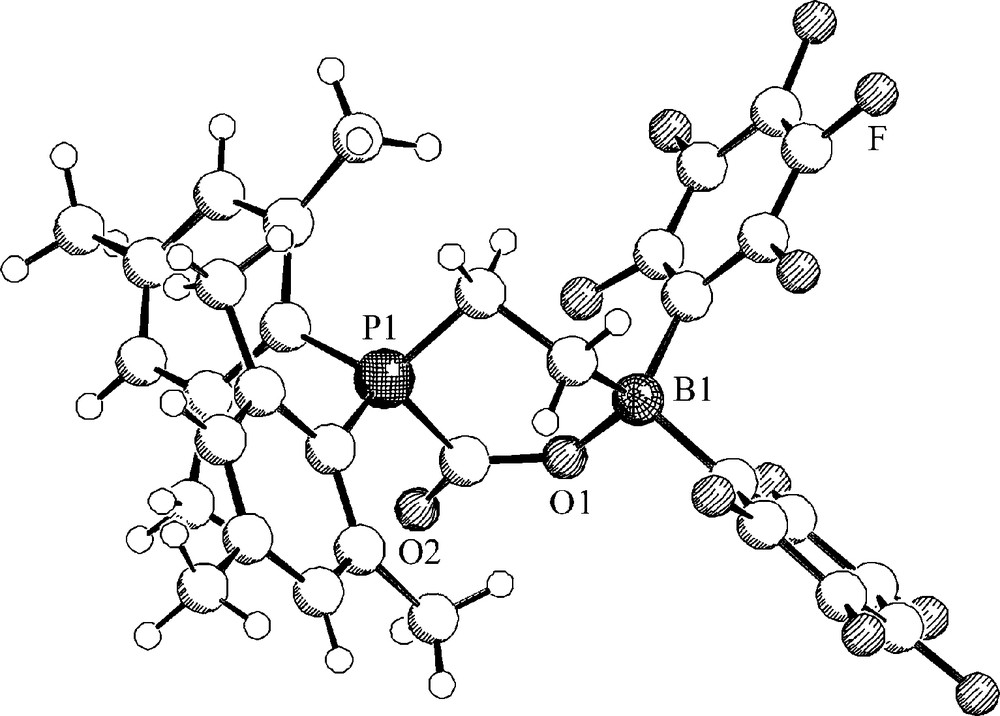
Molecular structure of the FLP-CO2 adduct 24.
Many intermolecular P/B Lewis pairs also add to CO2. Usually these adducts are thermally more robust and split off CO2 at higher temperatures [25]. Scheme 8 shows several examples of such systems that we have recently isolated and characterized [26].
2.3 Reactions with alkenes and alkynes
Frustrated Lewis pairs react with olefins. Very early examples comprise the addition of the BPh3/PPh3 pair to in situ generated benzyne or the reaction of the Ph3C−/BPh3 pair to butadiene [3]. Stephan had shown that B(C6F5)3 adds to alkenylphosphines and that the B(C6F5)3/PtBu3 pair undergoes 1,2-addition to e.g. ethylene [27].
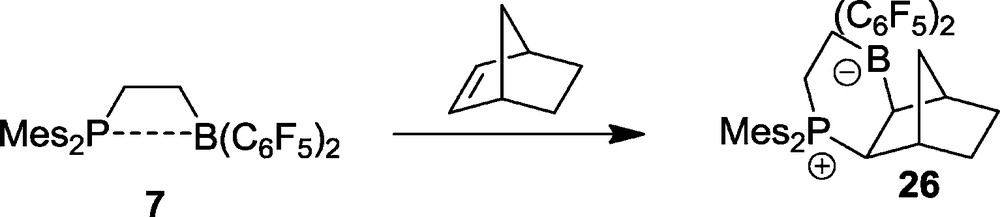
We have reacted the intramolecular P/B Lewis pair 7 with norbornene [28]. In this reaction the 2,3-exo-addition product 26 is formed exclusively. It was unequivocally characterized spectroscopically and by X-ray diffraction (Fig. 6).
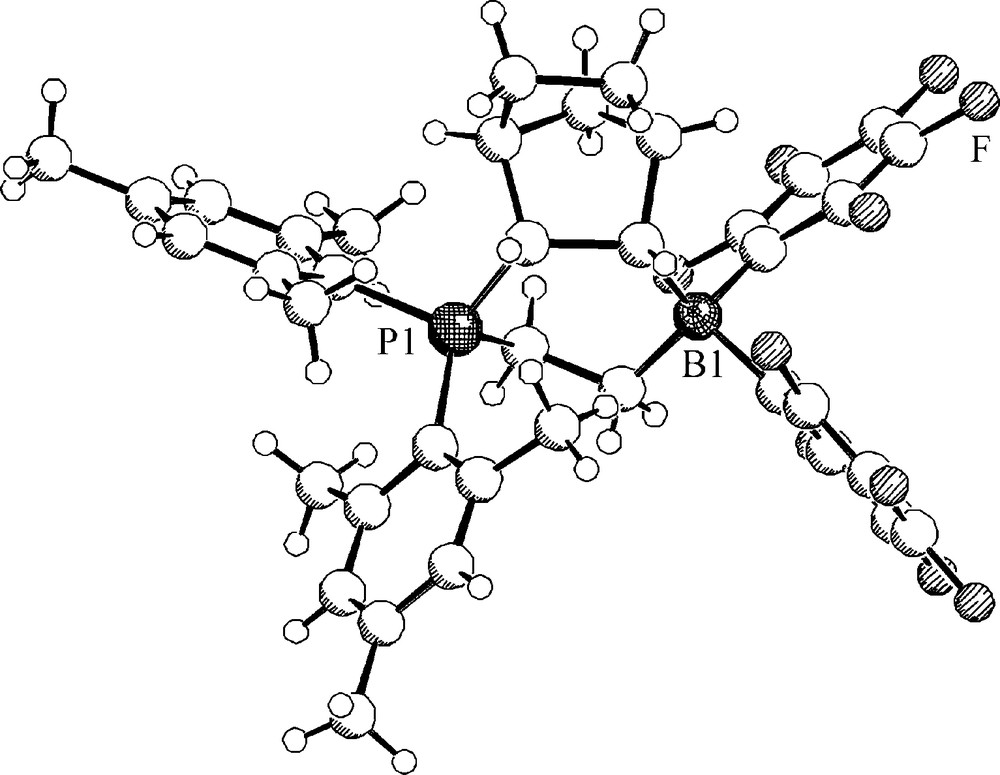
A projection of the molecular geometry of compound 26.
The theoretical analysis revealed that 26 is probably formed in a concerted but highly asynchronous reaction. In the transition state the C–B bond formation is more advanced than the formation of the C-P linkage (Scheme 9).
There are many variations of related 1,2-addition reactions to alkenes and alkynes. Typical examples are the intramolecular ring closure reactions by FLP addition to the pendant alkynyl or alkenyl units in the compounds 27 or 29 to yield the zwitterionic heterocyclic products 28 and 30, respectively (Scheme 10). Fig. 7 shows the structure of an example [29].
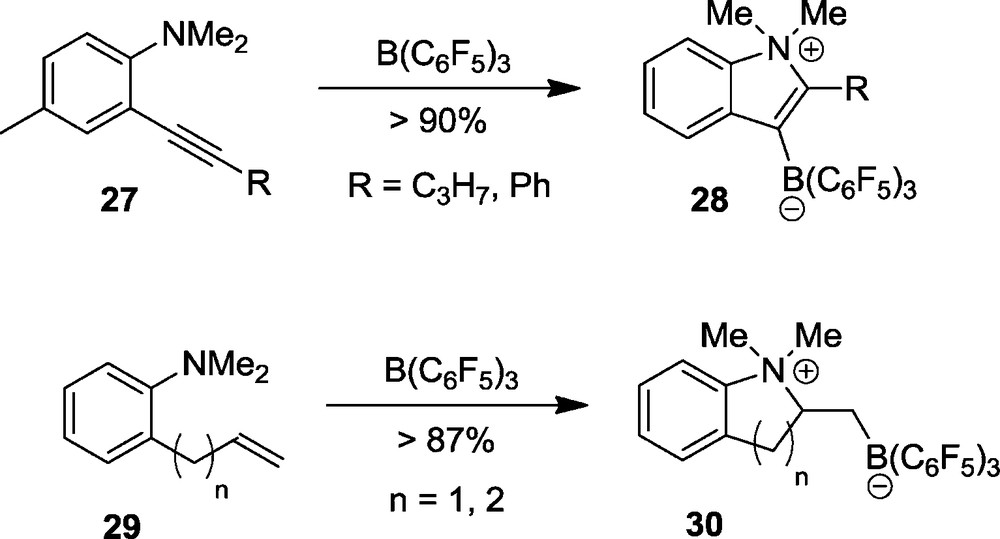
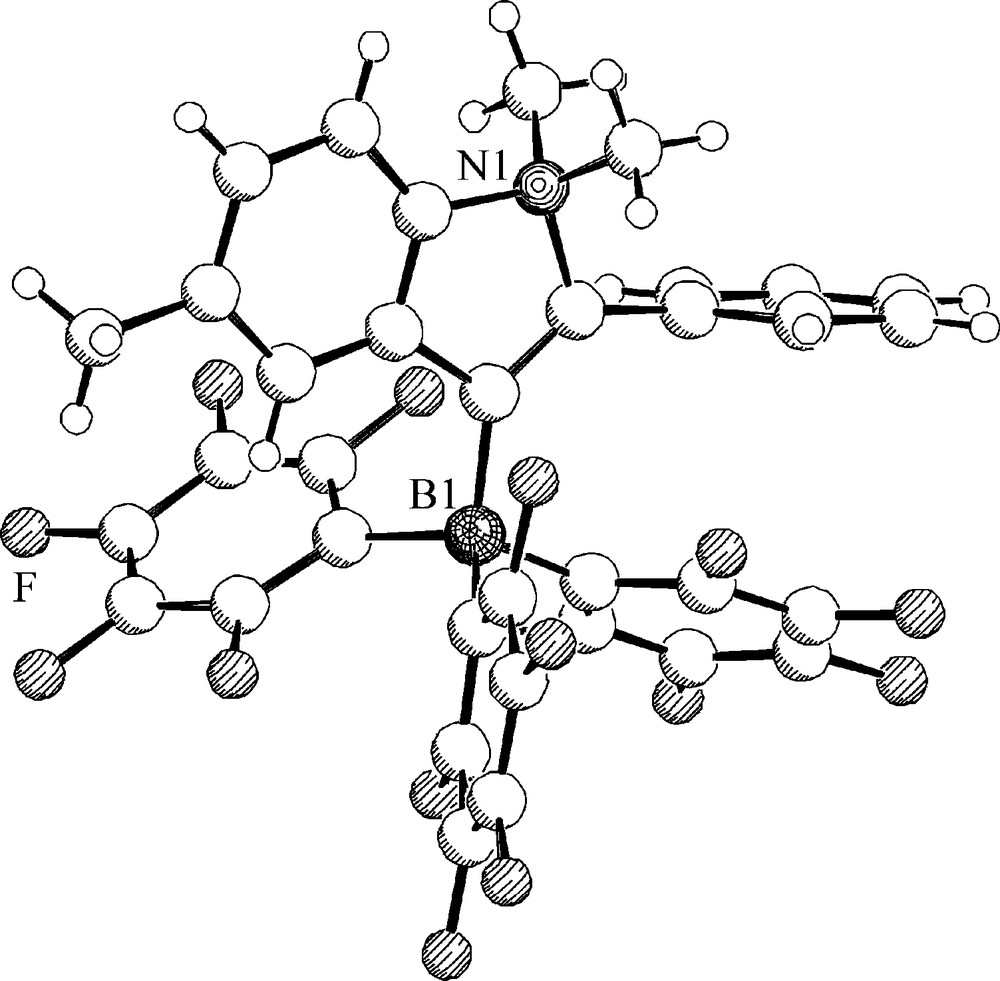
Molecular structure of the zwitterionic heterocycle 28b (R = Ph).
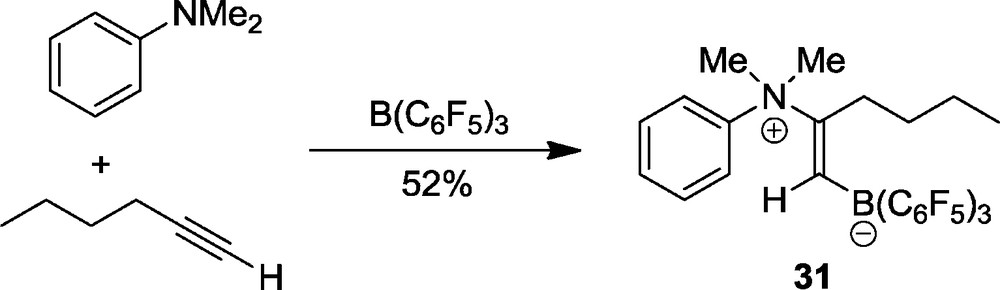
N/B FLP systems can also add to 1-alkynes. A typical example is the addition of the N,N-dimethylaniline/B(C6F5)3 pair to 1-hexyne to yield the adduct 31 (Scheme 11).
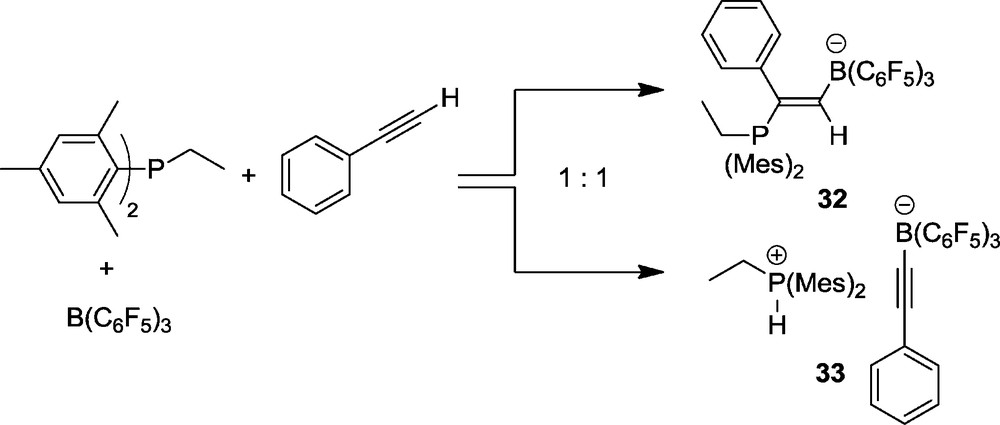
Stephan et al. had shown that intermolecular P/B pairs can either add to 1-alkynes or abstract the acidic acetylene hydrogen depending on the basicity of the phosphorus Lewis base [30]. We have found an illustrative example where these two pathways almost equally compete. The Lewis pair comprised of B(C6F5)3 and dimesitylethylphosphine reacted with phenylacetylene to yield a 1:1 mixture of the addition product 32 and the phosphonium/borate salt 33 (Scheme 12).
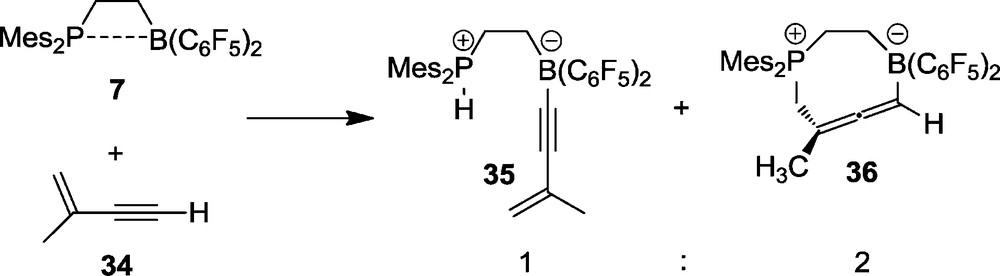
With the conjugated enyne 34 the intramolecular FLP 7 also reacts by deprotonation (to yield 35), but this is only the minor reaction pathway in this remarkable system. The major product is the eight-membered heterocyclic allene 36 [31]. This is the 1,4-addition product of the P/B FLP (7) to the conjugated enyne system 34 (Scheme 13). The product 36 is formed under kinetic control as shown by DFT. The possible six-membered heterocyclic isomer formed by alternative 1,2-P/B addition to the triple bond of 34 is thermodynamically more favourable but only the 1,4-addition to yield 36 was observed (in addition we find the acid/base reaction to occur, yielding the minor product 35). Compound 36 was characterized by X-ray diffraction. It features an almost unstrained allene moiety inside the heterocyclic eight-membered ring structure (Fig. 8).
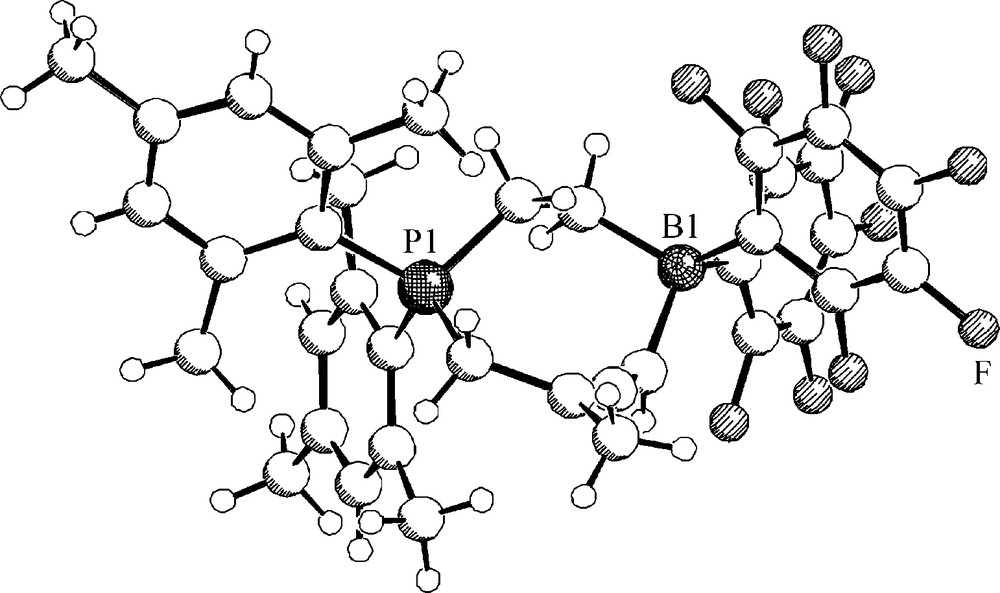
Molecular geometry of the zwitterionic eight-membered heterocyclic allene 36.
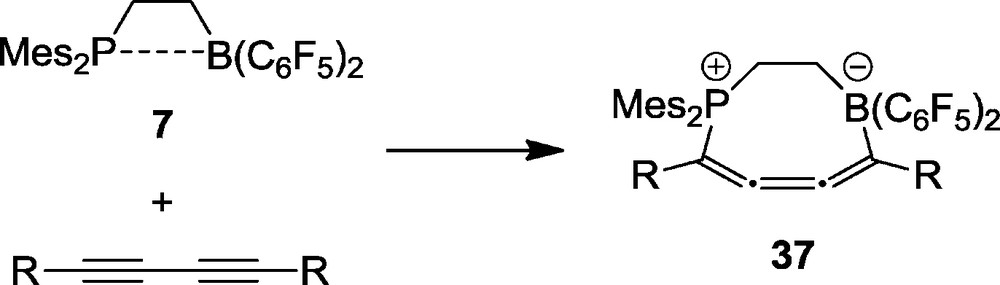
With a series of internal conjugated diynes the P/B pair 7 reacted selectively by 1,4-addition to yield the heterocyclic butatriene derivatives 37. Several examples of this remarkable class of compounds were characterized by X-ray diffraction [31]. Typically, the structure of 37b (R = n-propyl) shows a slightly bent central C=C=C=C unit (angles C4-C5-C6: 165.0(2)°, C5-C4-C3: 161.2(2)°) (Fig. 9). The framework of these systems is non-planar (Scheme 14).
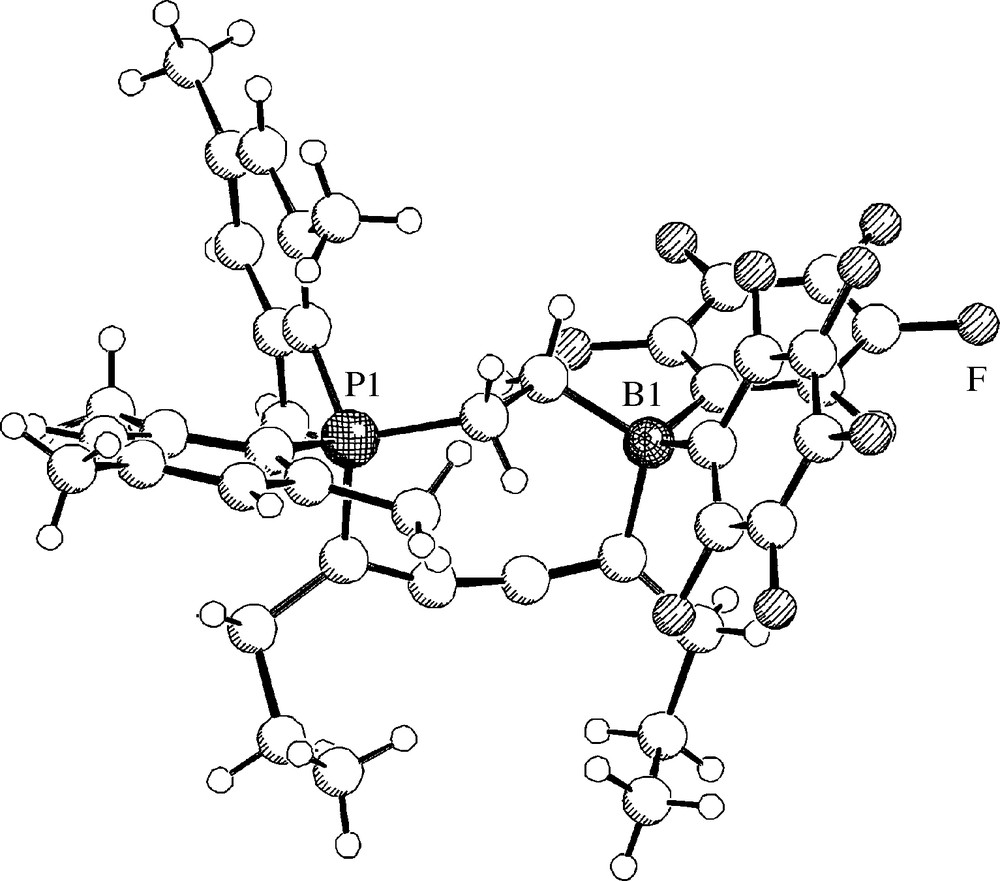
A view of the molecular structure of compound 37b (R = n-propyl).
Non-conjugated diynes may undergo different types of reactions with frustrated P/B Lewis pairs. The P(o-tolyl)3/B(C6F5)3 pair reacts readily with 1,7-octadiyne to yield the zwitterionic addition product 38 (Scheme 15 and Fig. 10) [32]. This product can be thought to be formed by addition of the strong boron Lewis acid to one of the terminal acetylene units followed by a rapid carbon-carbon coupling reaction. Eventually capture by the phosphine concluded the reaction sequence.
Molecular structure of the zwitterionic addition/C-C coupling product 38.
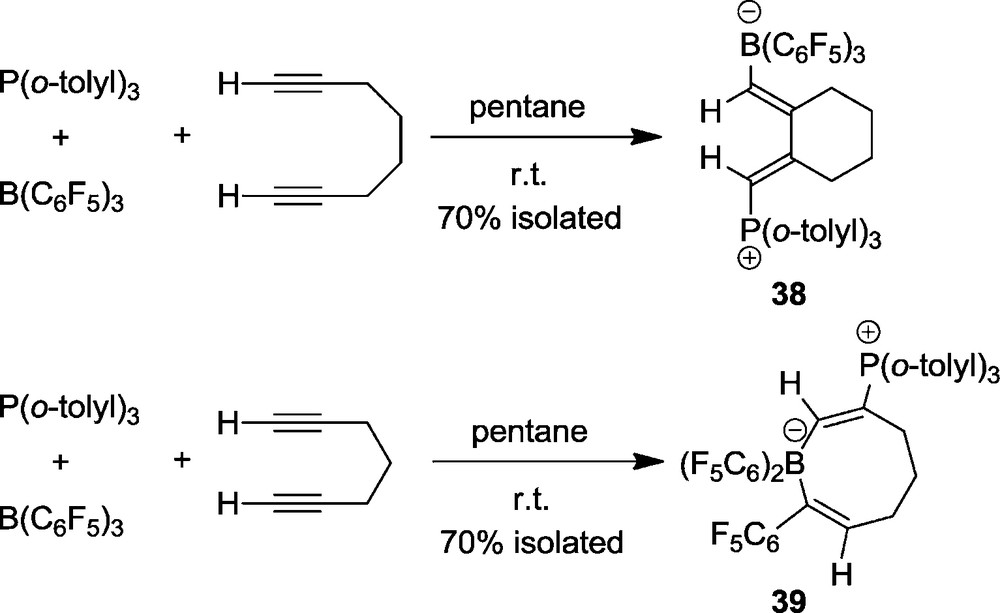
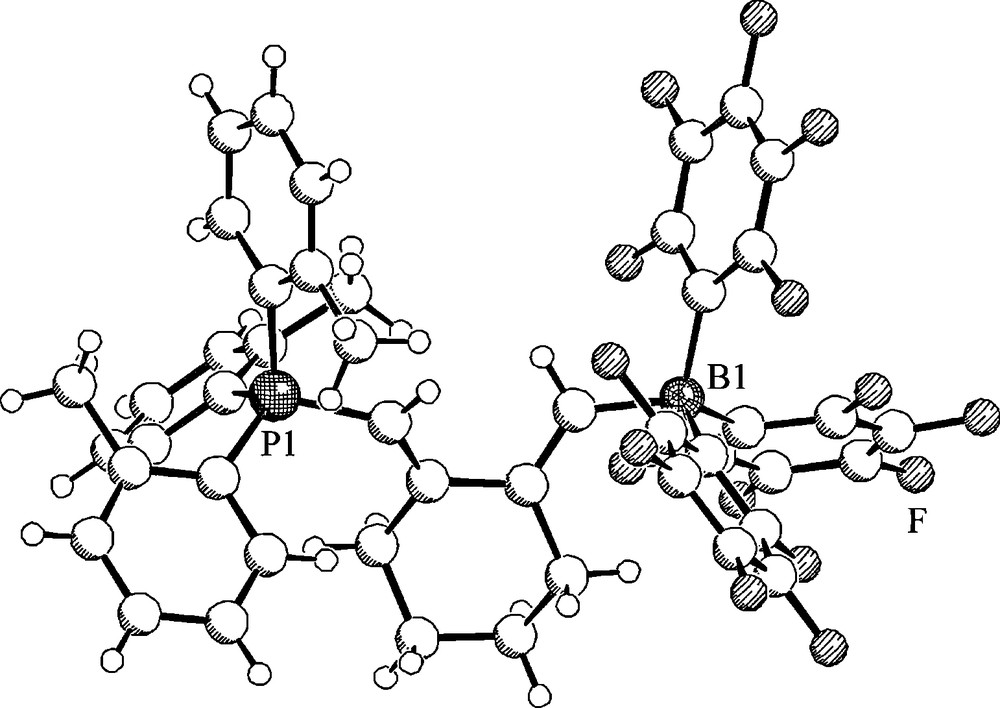
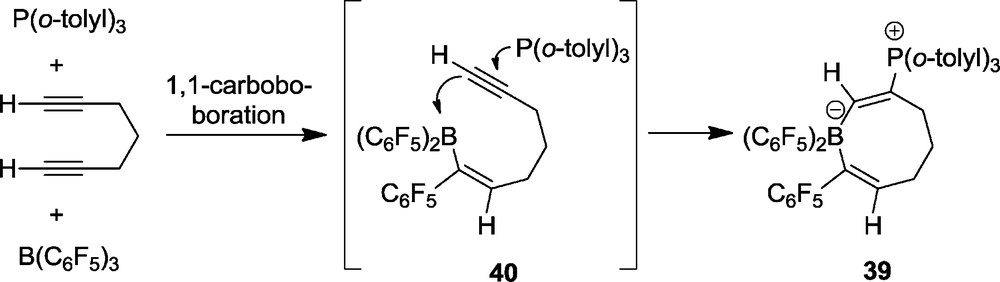
Treatment of 1,6-heptadiyne with the P(o-tolyl)3/B(C6F5)3 pair gives a different type of product. We isolated in good yield the eight-membered heterocycle 39 (Scheme 15 bottom and Fig. 11) [31]. The new product features an 11B-NMR signal at δ −13.3 and a 31P NMR resonance at δ 29.3. It shows the borate type 19F NMR signals of the endocyclic B(C6F5)2 moiety and a separate set of 19F NMR signal of the isolated single C6F5 substituent.
Molecular structure of the eight-membered heterocycle 39.
Apparently product 39 was formed by means of a two step mechanism initiated by 1,1-carboboration of a terminal alkynyl unit with B(C6F5)3. This reaction proceeds by borane addition to the terminal C(sp) carbon center coupled with 1,2-migration of a C6F5 substituent from boron to the same carbon atom and 1,2-hydrogen migration from C1 to C2 (Scheme 16). The intermediate 40 contains a strongly acidic boron Lewis acid. It then undergoes 1,2-addition with the P(o-tolyl)3 Lewis base, present in the solution, to the remaining second terminal alkynyl group of the substrate – a typical FLP reaction – to yield 39.
1,1-carboboration reactions have long been known for activated alkynes bearing silyl, stannyl, plumbyl or some transition metal containing substituents at the C(sp) center, i.e. groups that migrate well [33]. Our example (and a small group of other related reactions) [34] indicated that the 1,1-carboboration reaction of R-B(C6F5)2 systems with 1-alkynes might be a facile reaction in general. This turned out to be the case. This reaction type actually might be considered a conceptual synthetic alternative to the hydroboration reaction [35] of some alkynes.
2.4 1,1-Carboboration reactions of alkynes by the R-B(C6F5)2 reagents
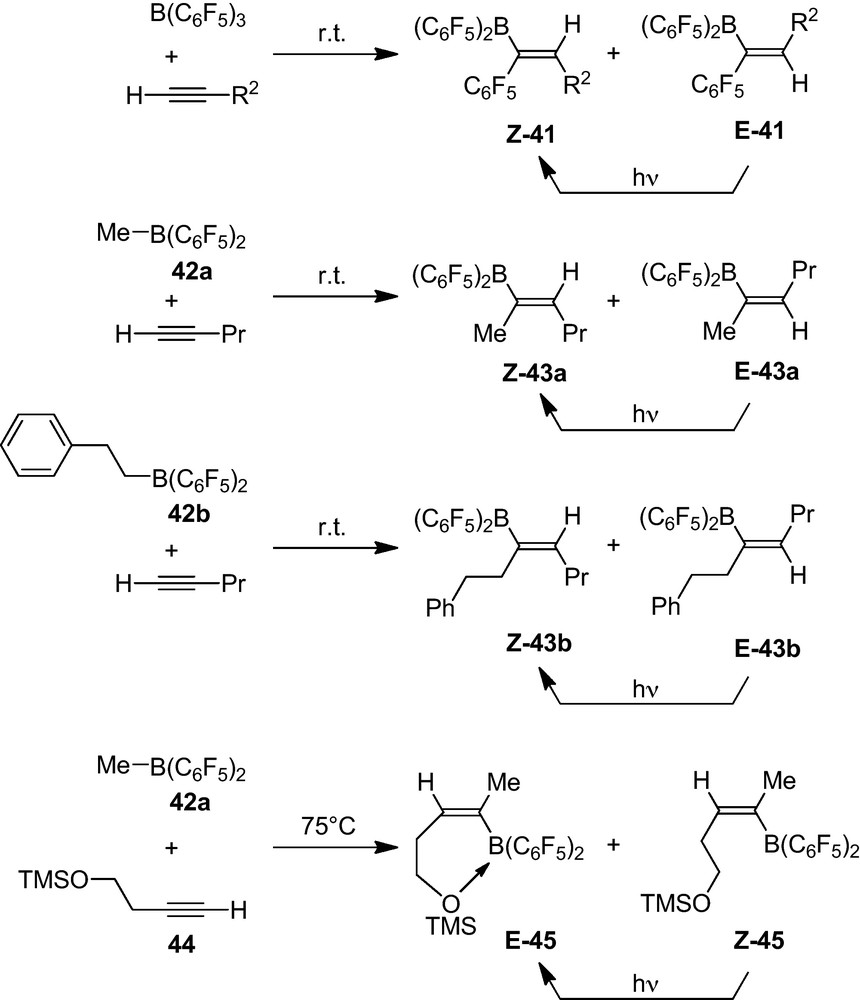
The 1,1-carboboration reaction of terminal alkynes with B(C6F5)3 proceeds rapidly at room temperature. Treatment of a series of 1-alkynes H-C≡C-R2 (with R2 = n-propyl, –(CH2)4Cl, –(CH2)3Ph, –Ph or –CMe3) rapidly gives a practically quantitative yield of the respective 1,1-carboboration products. The reaction is non-stereoselective. We obtained mixtures of the respective Z-41 and E-41 isomers (Scheme 17). However, subsequent photolysis in most cases cleanly converted the E-41 isomer to Z-41, so that highly enriched Z-41 samples were obtained in high yield. One example (R2 = Ph, Z-41d) was characterized by X-ray diffraction (Fig. 12) [35,36].
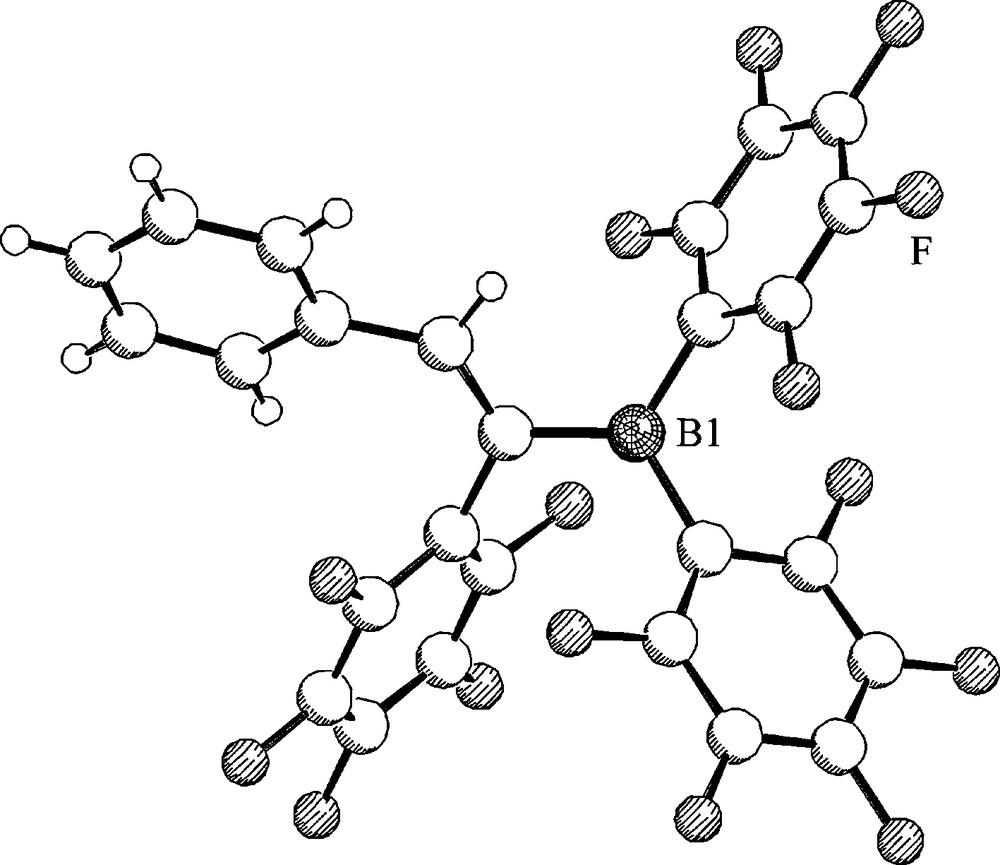
Molecular structure of the 1,1-carboboration product Z-41d.
Alkyl-B(C6F5)2 reagents (e.g. 42a, 42b) also undergo 1,1-carboboration reactions with 1-alkynes. In these cases we observed only the migration of the alkyl group from boron to carbon to selectively yield the alkyl-substituted 1,1-carboboration products 43 (Scheme 17). Again photolysis resulted in a high E-42 to Z-42 conversion.
Treatment of the–OTMS substituted 1-alkyne (43) with the Me-B(C6F5)2 reagent opened a selective pathway into the E-1,1-carboboration series. The thermally induced carboboration reaction again yielded a E-/Z-alkenylborane mixture 44. In this case the photostationary equilibrium lied on the side of the E-44 isomer [35].
The alkenylboranes are very interesting Lewis acids but they also may serve as tricoordinated borane reagents for cross-coupling reactions. Thus treatment of the product Z-41a with iodobenzene under typical Suzuki-Miyaura reaction conditions (catalytic Pd(PPh3)4/NaOH/THF) resulted in an efficient formation of the C-C coupling product, here the respective trisubstituted alkene 46a (Scheme 18). These reactions can be performed as one-pot reactions as well. Thus, the sequence starting from 42a and 5-phenyl-1-pentyne eventually gave the olefinic product 46b in an overall yield of 76%. The–OTMS functionalized systems undergo these reactions similarly [35,36].
We have even achieved the 1,1-carboboration of internal alkynes with either B(C6F5)3 or Me-B(C6F5)2, although this needs rather forcing conditions. 4-Octyne reacted with B(C6F5)3 in toluene at 110 °C over 3 days to give the 1,1-carboboration product 47 in good yield. This reaction represents a novel pathway for the activation of otherwise non-activated carbon–carbon σ-bonds. The product 47 of this remarkable C–C bond activation process can subsequently be utilized as a reagent for Pd-catalyzed cross-coupling (Scheme 19) [37].
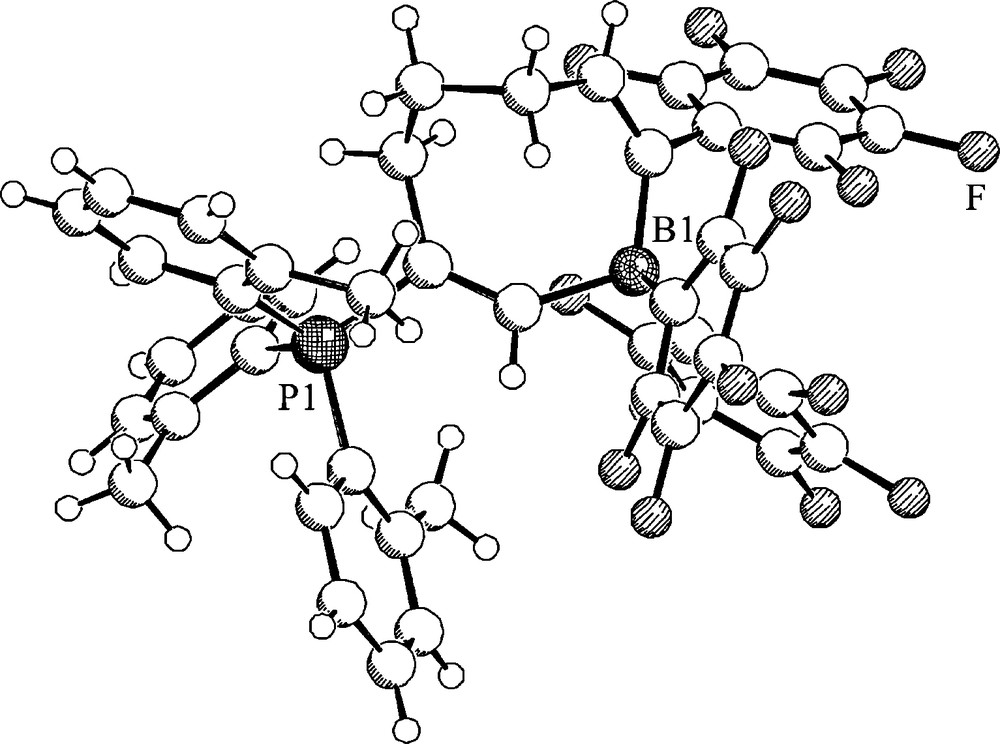
MeB(C6F5)2 (42a) reacts with 1,2-di-p-tolylacetylene during 9 days at 125 °C by means of a clean C–C bond activating 1,1-carboboration reaction to yield the product 49. This also undergoes a Suzuki-Miyaura type cross-coupling reaction with phenyliodide to yield the tetra-substituted alkene (50) that was characterized by X-ray crystal structure analysis (Fig. 13) [37].
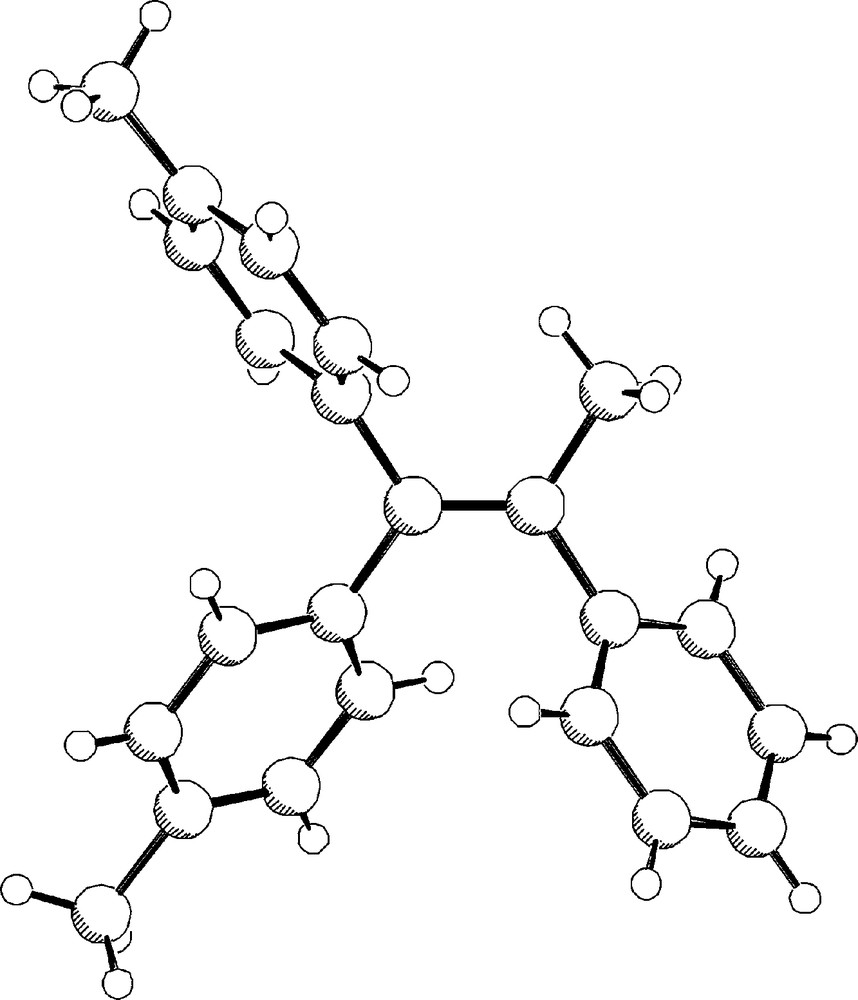
A projection of the molecular structure of the product 50.
3 Conclusion
Frustrated Lewis pair chemistry has made a rapid development during the recent years. It has led to a situation where many of these main group element systems show chemical features as they are commonly found in transition metal chemistry. Heterolytic splitting and activation of dihydrogen has certainly been one prominent feature but we are finding more and more other remarkable examples of typical FLP chemistry and characteristic FLP reactivities. These include the unusual 1,4-addition reactions to conjugated olefins and acetylenes and certainly not the least the rather common FLP addition chemistry to carbon dioxide. Although FLP chemistry still is at its early stages, it is forseeable that we will find more and more interesting novel reaction types of non-quenched Lewis acid/Lewis base pairs appear. Some of these reactions, as we have shown, are in a close competition with other very interesting new alternative reaction variants, such as the 1,1-carboboration reactions of 1-alkynes and even of internal alkynes. All this indicates that the use of the pentafluorophenyl-derived boranes have served to open a very interesting chapter in the chemistry of reactions mediated by strongly electrophilic Lewis acidic species in a variety of reaction environments.
Acknowledgement
Our part of the work described in this account was carried out by a group of very talented coworkers whose names are listed in the respective references. I cordially thank all of them. I think we all had fun and a good time working together in this exciting new field of chemistry. The financial support by the Deutsche Forschungsgemeinschaft, the Fonds der Chemischen Industrie and the Alexander von Humboldt Stiftung is gratefully acknowledged.