



 CC-BY 4.0
CC-BY 4.0
1. Introduction
Metal nanoparticles have been used as active sites for heterogeneous catalysts for over a century [1]. For example, the Sabatier reaction, which converts CO2 and H2 into methane and water, is catalyzed by nickel nanoparticles. A more recent field concerns the use of these same metal nanoparticles in colloidal suspension [2], that is, dispersed in an aqueous [3] or organic solvent [4] or alternatively in an ionic liquid [5]. This approach makes it possible to incorporate co-catalysts into the solvent, which can enhance the efficiency of the nanoparticles. Among them, organic ligands such as amines or phosphines, capable of adsorbing onto the particle surface, are being studied. They are likely to have a number of effects such as modification of the metal’s electronic properties, poisoning of the most reactive sites, and sometimes an improved reaction selectivity or an induction of enantioselectivity if they are themselves chiral [6]. However, care must be taken to ensure that the affinity of the ligand is not too strong, as this could lead to passivation of the surface or, worse still, leaching [7, 8], or partial dissolution of the particles through the formation of metal complexes.
In its implementation, catalysis by colloidal suspension is close to homogeneous catalysis and can therefore draw on its methods and successes. Here, we attempt to borrow a key concept of the 2000s from purely molecular organocatalysis: create frustrated Lewis pairs (FLPs) and adapt them to colloidal catalysis. This article describes our approach and gives examples of our attempts to implement this design at the crossroads of molecular chemistry and nanochemistry while providing a critical discussion concerning the competitive mechanisms to be considered.
2. Frustrated Lewis Pairs
2.1. A concept born of molecular chemistry
Chemists are familiar with the concept of Lewis acidity: a Lewis acid (e.g., BF3) has a vacant orbital capable of accepting the free pair of a Lewis base (e.g., lutidine) to form a Lewis adduct. In 1942, Brown and colleagues noted that trimethylborane (BMe3) failed to form an adduct with lutidine due to steric hindrance between the methyl groups facing each other. The adduct is then considered to be frustrated, and the nitrogen–boron bond, if it can be formed, is abnormally long (1.817 Å when BMe3 is used versus 1.691 Å when BF3 is used) [9].
It was only 60 years later that Stephan’s team exploited a frustrated adduct for its ability to heterolytically cleave a small molecule, H2. The frustrated Lewis pair formed by a highly basic and hindered phosphine, PtBu3, and a highly acidic and hindered borane, B(C6F5)3, can indeed accommodate dihydrogen within it, forming an encounter complex, strongly polarized due to the acidity contrast (Figures 1A,C). This results in the spontaneous formation of a P–H bond and a B–H bond, a phosphonium and a borohydride, respectively (Figure 1A) [10]. Schematically, the very low activation energy required for this process, compared with a similar reaction on a traditional Lewis pair, comes from the destabilization of the initial state, that is, its frustration, as suggested in Figure 1B. Experimentally, heterolytic breaking of the H–H bond is observed under 1 bar at just 25 °C. Moreover, the two species formed can be engaged in catalytic cycles: the FLP composed of phosphine and borane, an organocatalyst of choice for hydrogenation [11].
(A) Heterolytic H–H bond cleavage at 25 °C by a molecular FLP. (B) Simplified energy diagram of the reaction pathway. (C) Encounter complex between PtBu3, H2, and B(C6F5)3: phosphorus is in pink, boron in blue, and the two hydrogen atoms in gray [12]. (D) Surface Lewis pair in which gold acts as the Lewis acid (Guo group) [13].
2.2. Towards NanoFLPs
By analogy with this pioneering work, numerous teams have undertaken the design and use of FLPs to activate other small molecules such as CO2, CO, SO2, and so forth with remarkable results. However, there are two major limitations to the design and use of molecular FLPs as homogeneous catalysts. First, the catalyst has only one isolated active site. Only a single molecule can be activated, whereas a surface is capable of activating several molecules with neighboring adsorbates recombining to form a product. Second, the key parameters of each FLP partner—its acidity and steric hindrance—are controlled by the same means, the nature of the substituents, so that it is difficult to control one independently of the other. The use of inorganic surfaces may sometimes overcome these limitations as we shall see.
2.3. FLPs on surfaces
The most natural transposition of molecular FLPs to a heterogeneous catalyst involves grafting one of the two partners, for example, borane, onto the surface of a silica colloid. The phosphine is then in solution and free to interact with this Lewis acid surface. This has been successfully achieved for H2 activation by O’Hare’s group [14]. Although elegant, this approach is conceptually identical to the one described above, as it consists in building a supported homogeneous catalyst from an known molecular catalyst.
More attractive is the design of the surface-frustrated Lewis pairs in which one of the two molecular partners is replaced by a surface. The proof of concept was provided in 2014 by Guo’s group. The gold surface is a Lewis acid, and in the presence of an imine (Lewis base) or an amine, H2 activation is possible (Figure 1D) [13]. It should be emphasized that the majority of the results in this article correspond to activation energy predictions calculated by density functional theory, which is an interesting entry point in the field and allowed for a detailed electronic description of the surface–molecule interaction. Remarkably, a single experimental proof, using micrometric gold powder, is provided with the activation of H2 at 50 °C under 20 bar. In addition, it is proposed that more abundant metals such as copper and silver could act as Lewis acids.
In order to get the most out of each metal atom, it is natural to try and reduce the particle size to just a few tens of nanometer in diameter versus several microns for a traditional powder prepared by ball milling or another top-down synthesis. This allows a greater proportion of atoms on the surface and therefore a more judicious use of this costly resource. Moreover, metal nanoparticles are easier to stabilize in colloidal suspension form with limited sedimentation over time. Lastly, well-known synthesis routes give access to a whole family of metal nanoparticles with controlled composition, morphology, and surface chemistry (nature of organic stabilizers).
This is how we proposed to take advantage of the FLP concept in colloidal catalysis. Our design involves replacing the molecular Lewis acid (or base) with a nanoparticle. The second molecular partner, of opposite basicity, is then simply introduced into the solvent. When this molecular partner approaches the surface, we expect the spontaneous formation of a frustrated Lewis pair on the nanoparticle (which we call NanoFLP). The case of a Lewis acid nanoparticle (e.g., metallic nickel) and a Lewis base partner (typically, a phosphine) for the activation of H2 under mild conditions is illustrated in Figure 2.
Schematic diagram of a NanoFLP for activating small molecules such as CO2 and H2.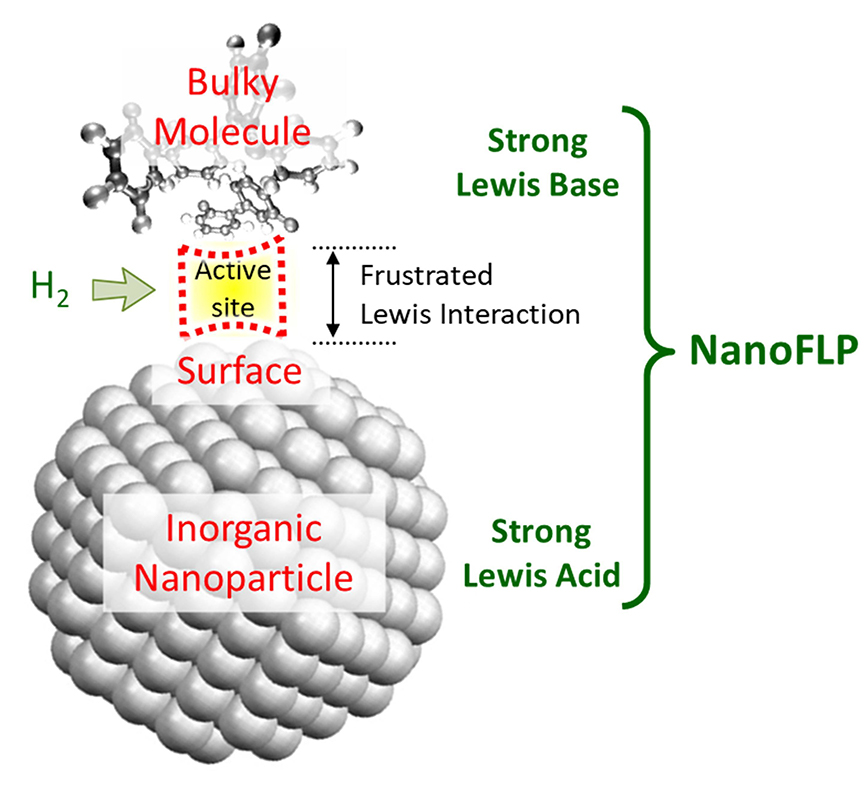
This ideal scheme is not without any obstacles. First, the surface of freshly prepared nanoparticles is often covered with organic stabilizers or ligands that ensure their colloidal stability. The molecular partner of the NanoFLP will therefore need to have sufficient affinity and be present in excess to approach the surface by displacement of the species present and/or interactions on vacant sites. Second, steric hindrance at the surface will be affected by the number of ligands present at the same time, so it is tricky to anticipate which molecule will be appropriate. Third, if the molecule–surface interaction is very strong, a classical and highly stable Lewis pair will be formed, and the corresponding site will be poisoned: the relative acidities and basicities will have to be finely tuned in order to avoid this pitfall. Finally, the chosen nanoparticle may not be stable in the presence of the Lewis base. This is what we found when exposing copper nanoparticles to tri-n-butylphosphine: a slow dissolution of the nanoparticles (leaching) occurred, creating copper–phosphine molecular complexes [8]. The Rossi group was more agile than we were, exploiting amine-coated gold nanoparticles—a combination possibly inspired by Guo’s work—to achieve catalytic hydrogenation of alkynes in colloidal suspension [15].
3. Effect of phosphines on a model reaction: Si–H bond cleavage
3.1. Nanoparticle–ligand synergy
Our primary objective was to use non-noble transition metals to create NanoFLPs. We therefore chose an easier reaction than H2 activation: the activation of phenylsilane in which the Si–H bond is quite reactive. In order to monitor the progress of the reaction, we introduced benzaldehyde so that the reaction balance corresponds to aldehyde hydrosilylation (Table 1, top). This reaction is easy to achieve with simple catalysts (such as Cu(II) complexes), but the point here was to examine the suitability of forming a NanoFLP.
Catalytic activation of the Si–H bond of phenylsilane by NiCo nanoparticles in colloidal suspension in the presence of phosphine [16]

| Entry | Lewis acid | Lewis base | Silane consumption (%) |
|---|---|---|---|
| 1 | - | - | 0 |
| 2 | NiCoNps | - | 0 |
| 3 | - | PnBu3 | 0 |
| 4 | NiCoNps | PnBu3 | 77 |
| 5 | NiCoNps | PnOct3 | 55 |
| 6 | NiCoNps | PCy3 | 7 |
| 7 | NiCoNps | PMe3 | 20 |
| 8 | NiCoNps | PPh3 | 100 |
The first step, therefore, was to identify a nanoparticle–ligand pair in which each partner alone did not catalyze the reaction, but the pair did. We had long chosen to use phosphines as our Lewis base: it is easy to vary their steric hindrance and basicity by choosing one of the many commercially available structures. Paired with them, we needed Lewis acid nanoparticles: our choice was nickel–cobalt nanoparticles that we had prepared in a previous work [17, 18].
First, we verified the absence of silane consumption when the two partners (acid and base) were not simultaneously present (Table 1, entries 1–3). Their joint introduction in catalytic quantities (10 mol%) led to a silane consumption of 7–100%, depending on the nature of the substituents on phosphine (Table 1, entries 4–8).
Following this promising result, we verified that the nanoparticles were intact at the end of the reaction, first by analyzing them “post-mortem” and second by carrying out a leaching test. This involves stopping the reaction mid-conversion, separating the nanoparticles from the supernatant by centrifugation, and then checking that the reaction does not continue to advance in the supernatant.
3.2. Major role of steric hindrance
All that remained was to interpret the differences observed between phosphines. An additional advantage of this family over other Lewis bases, such as amines, is the existence of parameters established by Tolman in the 1970s. These parameters describe, on the one hand, their donor character via the Tolman electronic parameter (TEP) and, on the other hand, their steric hindrance via the Tolman cone angle [19]. The latter is calculated according to the steric hindrance of the substituents of a phosphine complexed to a nickel atom (with a Ni–P distance of 2.28 Å by convention; see Figure 3B). We did not observe any correlation between TEP and the nature of the phosphines although it seems that the optimum Tolman angle is around 140° (Figure 3C), whether the substituents are aliphatic, aromatic, or mixed. However, this apparent correlation is not sufficient to prove the existence of NanoFLP as other effects could be involved, such as the indirect influence of the phosphine on a distant active site. Nevertheless, this first result has encouraged us to attempt H2 activation, which is a more difficult process.
(A) Proposed NanoFLP formed from a nickel–cobalt nanoparticle (Lewis acid) and triphenylphosphine (Lewis base). (B) Tolman cone angle. (C) Silane consumption as a function of the phosphine used.
4. A more challenging reaction: breaking the H–H bond
To investigate this elementary process, we choose to measure the conversion of phenylacetylene (or other alkynes) as the substrate of hydrogenation. It is possible to detect styrene and ethylbenzene by 1H NMR, which makes the reaction easy to monitor (Figure 4A). Over the past years, we investigated several types of nanoparticles in this reaction. A couple of most interesting cases are discussed in what follows.
(A) Transmission electron microscopy (TEM) of nano-urchins at low magnification. (B) High-resolution TEM of an urchin branch crystallized in the orthorhombic CoP phase. (C) Catalytic reaction and experimental conditions. (D) Color-coded stereoelectronic map. Lighter green corresponds to a higher conversion of phenylacetylene as indicated in the inset.
4.1. Cobalt phosphide nano-urchins
Because we had proven that cobalt was the active site in the hydrosilylation of benzaldehyde [20], we first turned to a cobalt-rich family of nanoparticles, cobalt phosphides, which are easier to prepare and more robust than metallic cobalt nanoparticles.
First, cobalt phosphide nano-urchins (Figure 4B) crystallized in a mixture of orthorhombic CoP and orthorhombic Co2P phases were employed as a colloidal suspension. The branches turned out to be very well crystallized in the CoP phase as observed by high-resolution transmission electron microscopy (Figure 4C). We had obtained these urchins by serendipity, by attempting to reproduce a synthesis of cobalt phosphide nanosticks as published in the literature (an article that was subsequently retracted). They have the advantage of a large number of accessible cobalt sites due to the morphology of the particles.
4.2. Phosphine effect on the catalytic reaction with cobalt phosphide nano-urchins
As in the previous study, we first checked that phosphine alone did not catalyze the hydrogenation of phenylacetylene. Cobalt phosphide nano-urchins (Figures 4B,C) in the absence of phosphine gave a conversion of 13% with 95% selectivity for styrene under the conditions detailed in Figure 4A [20]. We then tried adding a catalytic amount of phosphine, and several situations were encountered. For PPh3 and PtBu3, for example, the observed conversion was not improved by the presence of phosphine. In other cases, such as with PnBu3 or PMe3, phosphine had a positive effect.
In order to refine our interpretation of the previous study, and in particular to better visualize the joint effects of electronic and steric parameters, we have represented the results in the form of a stereoelectronic map. On the ordinate is the TEP, which quantifies the donor character of the phosphine via the wavenumber corresponding to the elongation of the carbon–oxygen bond of the Ni(PR3)(CO)3 complex. A lower wavenumber corresponds to a stronger donor phosphine. The abscissa shows a steric parameter more modern and precise than Tolman’s, calculated by destabilizing a ring of eight helium atoms positioned around the phosphine [21]. These two axes give rise to a stereoelectronic map.
To visualize the influence of the nature of the phosphine on phenylacetylene conversion, we use a color code so that it is easy to see which regions of the map are more favorable and to suggest new phosphines to optimize the catalytic conversion. Thus, in Figure 4D, the experimental optimum is obtained with PnBu3, and the most favorable region appears to be at the bottom left of the map. In contrast, the top left and bottom right regions appear unfavorable. This approach makes it possible to visualize the combined effects of steric hindrance and the donor (viz. Lewis basic) nature of the phosphine.
In this study, we have not yet been able to confirm the existence of a frustrated Lewis pair interaction on the cobalt phosphide nano-urchin surface. Here, we cannot rule out an indirect, longer-range effect of phosphines on the active site. Moreover, phosphine activation of the alkyne in solution, preceding the hydrogenation step, remains conceivable at this stage.
Because of their morphology, it was difficult to assess the specific surface of the nano-urchins. Thus, we could hardly evaluate how many phosphines per surface cobalt atom was necessary for the positive effect of the phosphine on the conversion to arise. Thus, we turned our efforts to spherical nanoparticles of controlled diameter: nickel nanoparticles.
4.3. Monodisperse nickel nanoparticles
Three samples of nanoparticles, with average diameters of 11, 18, and 23 nm were prepared following a well-established route [22, 23]. As in the previous example, their catalytic activity for the hydrogenation of phenylacetylene in the presence of an additional phosphine was evaluated and the results were displayed using a stereoelectronic map, as shown in Figure 5A for 18 nm Ni nanoparticles (10 mol%), and a reaction performed at 60 °C under H2 (7 bar) for 16 h with 20 mol% of phosphine versus phenylacetylene [24]. We also showed that some phenylacetylene derivatives, as well as a linear alkyne like 1-octyne, could also be hydrogenated under similar conditions.
(A) Stereoelectronic map obtained with 18 nm Ni nanoparticles (NPs). (B) Effect of PnBu3 on the conversion. Green diamonds: critical temperatures Tc as a function of the NP average diameter. Blue circles: onset temperature for the catalytic activity of NPs without additional phosphine. The blue area suggests the activity domain for the NPs without additional phosphine. Green arrows indicate the observed enhancement. The green area suggests the region for the enhancement brought about by the presence of PnBu3 (0.2 equiv.).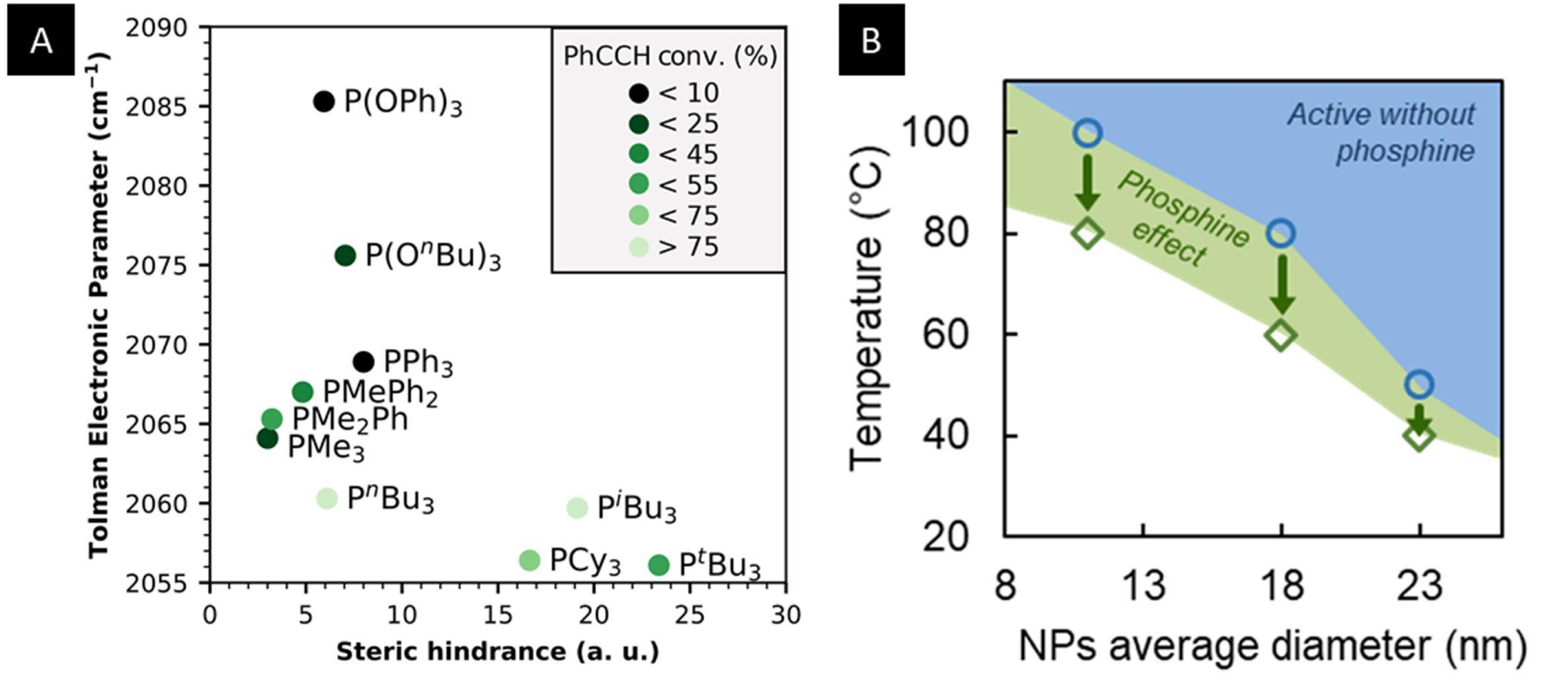
The amount of phosphine was then varied, which allowed us to demonstrate that regardless of the nanoparticle diameter, less than two phosphines per surface nickel atom were enough to enhance the conversion of phenylacetylene in a well-determined temperature domain. More specifically, we identified three regimes in terms of the effect of the phosphine. In a high-temperature regime, the phosphine did not provide an enhancement of conversion versus the reaction conducted with only the nickel nanoparticles as catalysts (blue domain in Figure 5B). In an intermediate-temperature regime, the presence of the phosphine was beneficial to the conversion (green domain in Figure 5B). Lastly, at a very low temperature, no conversion was detected. To our surprise, larger nanoparticles were comparatively more active at a lower temperature, indicating that the second regime was moved to lower temperatures. It would be interesting to assess the generality of this finding by investigating other size-tunable metal or metal phosphide nanoparticles.
At this stage, it was clear that the beneficial effect of well-chosen phosphines was to lower the temperature at which the nickel nanoparticles were able to hydrogenate the phenylacetylene. Unfortunately, an experimental measure of the activation energy associated with this reaction was out of reach at this moment. As only some phosphines, within a narrow domain of steric hindrance and Lewis basicity, were able to promote the conversion, we would be tempted to conclude that the phosphine and the nickel nanoparticles’ surface were indeed forming the desired NanoFLP in the colloidal suspension as schematized in Figure 2. This would result in a concerted H2 activation in a similar manner to that observed with molecular FLPs (Figure 1C). As depicted in Figure 6B, the phosphine would be expected to move off and on the nickel surface, as can be expected in the colloidal suspension, thus explaining why a slight excess of phosphine versus the number of Ni surface atoms is beneficial. It would once in a while form an encounter complex with H2 and a Ni surface atom, which could result in H–H bond activation with a lower activation energy than that on the bare Ni surface.
Proposed structures for the active surface. (A) Electronic effect of a nearby phosphine on H2 activation. (B) Possible NanoFLP with a nickel–phosphine active site. (C) Possible activation of the alkyne by the phosphine prior to the interaction with the surface.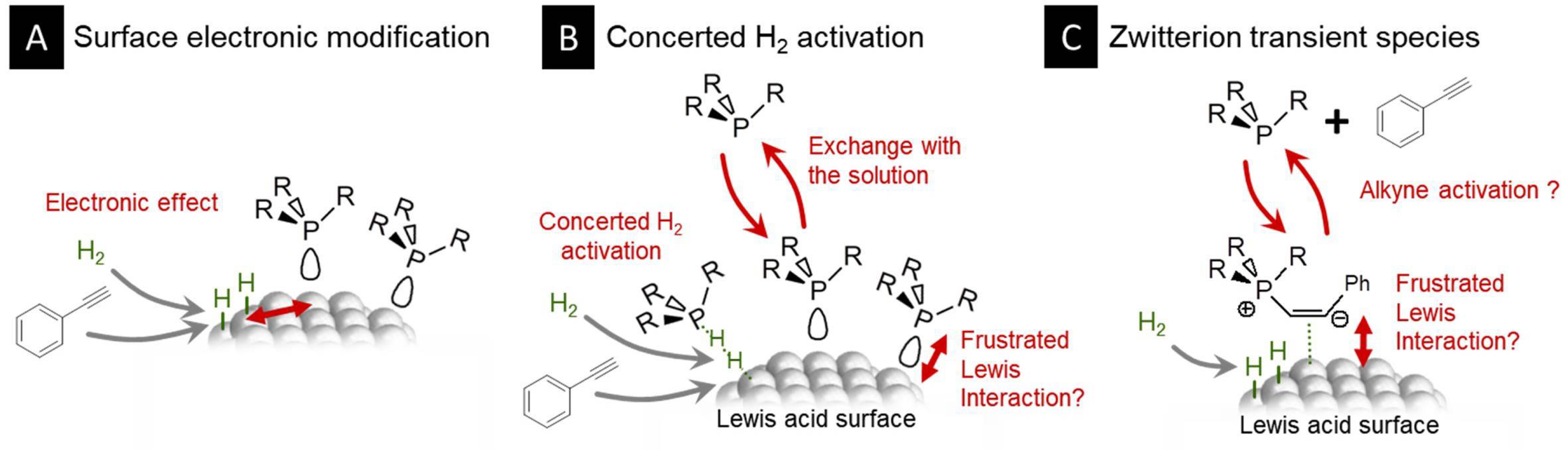
As a competitive scenario, we could propose that the phosphine may locally affect the electronic properties of the surface through its donating character. Schematically, the substrates (H2 and phenylacetylene) would be adsorbed near a phosphine-coordinated surface site, as suggested in Figure 6A, as long as the steric hindrance of the phosphine (or a very high ligand density) does not prevent this. In this case, when investigating para-substituted phenylacetylene, we might have expected some correlation of their Hammett parameter to the conversion, which was not observed. Although this is not sufficient to disqualify this mechanism, we believe that this lowers its likeliness.
A third possibility would be an activation of the alkyne by the phosphine in the solution prior to any interaction with the nickel surface as schematized in Figure 6C. The activated species, possibly a zwitterion, would then react with surface hydrides generated by H–H activation at the surface of the nickel nanoparticles. So far, our attempts to detect such intermediates were unsuccessful, but another series of experiments under harsher conditions and in the absence of nickel nanoparticles might help to identify an activated derivative of the alkyne, in line with a number of mechanistic proposals that were formulated in the context of homogeneous catalysis studies.
Our experimental data do not allow discarding the proposals made in Figures 6B and C. We believe that investigating other reaction substrates than alkynes, then pursuing the low-temperature activation of other molecules such as CO2 by potential NanoFLPs, and, last but not least, coupling a detailed kinetic study with a computational chemistry approach may shed some light on the relevance of the concept of NanoFLPs for colloidal catalysis.
5. Conclusion
The quest for NanoFLPs continues. Our aim to directly characterize the surface of the nanoparticle interacting with the phosphine during catalytic activity has come up against technical limitations that we have not yet overcome. This is why we now intend to use molecular modeling, which we recall was the cornerstone of the pioneering work by Guo’s team, to assess the activation energies associated with each of the proposed mechanisms. In any case, the research undertaken in this quest has enabled us to identify milder reaction conditions for catalytic transformations and to analyze in greater detail the contribution of phosphine ligands in colloidal catalysis.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Funding
The author would like to thank the organizations that have funded this work, and in particular the CNRS, Sorbonne Université, and the European Research Council under the H2020 program (ERC NanoFLP Grant No. 758480).
Acknowledgments
The author warmly thanks all the collaborators associated with this work, and in particular the following people who have contributed to its progress over the years: Xavier Frogneux-Plé, Antoine Pesesse, Rémi André, Cyprien Poucin, Alexy de Jesus Almeida Freitas, Alberto Palazzolo, Anthony Ropp, Karim Azouzi, Kaltoum Bakkouche, and Sebastian Adolfo Godoy Gutierrez.