

1 Introduction
Chromones [1] and their derivatives are the naturally occurring compounds ubiquitously found in the plant kingdom, and therefore are present in representative amounts in normal human diet. These phytochemicals possess a wide spectrum of biological activities such as anti-inflammatory [2], antimicrobial [3], anticancer and antitumour [4] and antioxidant [5] properties. In fact, the chromone moiety is an important element of the pharmacophores of many biologically active molecules displaying diverse medicinal applications [2].
Intramolecular H-abstraction [6–9] in 3-alkoxy-2-arylchromones is of considerable significance. These chromones on photo-irradiation undergo cyclisation via γ-H abstraction to yield the angular tetracyclic products and the product formation depends upon the nature of 3-alkoxy group. The 3-alkoxy-2-thiophenyl-4H-chromen-4-ones on photo-irradiation produced cyclized dihydro and dehydrogenated photoproducts [10,11]. From these, the cyclized dihydro photoproducts were formed via 1,5-sigmatropic H-shift whereas the cyclized dehydrogenated products are formed by the expulsion of H2 during ketonisation directly. In the present study, we report the results of our investigation on the photoreactions of 3-alkoxy-6-chloro-2-(3-methylthiophen-2-yl)-4H-chromen-4-ones. The main objectives are:
- • how does the methyl group at 3’-position effect the product formation through its steric/inductive effect;
- • does the demethylation occurs leading to the formation of cyclized aromatic products as represented earlier or simple cyclisation occurs without its expulsion giving cyclized photoproducts analogous to dihydro-photoproduct having angular methyl group during photocyclisation;
- • to unravel the effect of different groups at C-3 on the product formation/distribution.
2 Results and discussion
The required substrates, 3-alkoxy-2-(3-methylthiophen-2-yl)-4H-chromen-4-ones 3(a–e) were synthesized by the alkylation of 3-hydroxychromen-4-one 2 that was obtained by the condensation of 5-chloro-2-hydroxyacetophenone with 3-methylthiophene-2-carboxaldehyde in the presence of NaOH/EtOH [12] to give chalcone 1, followed by the treatment with H2O2/−OH under AFO conditions [13] (Scheme 1).
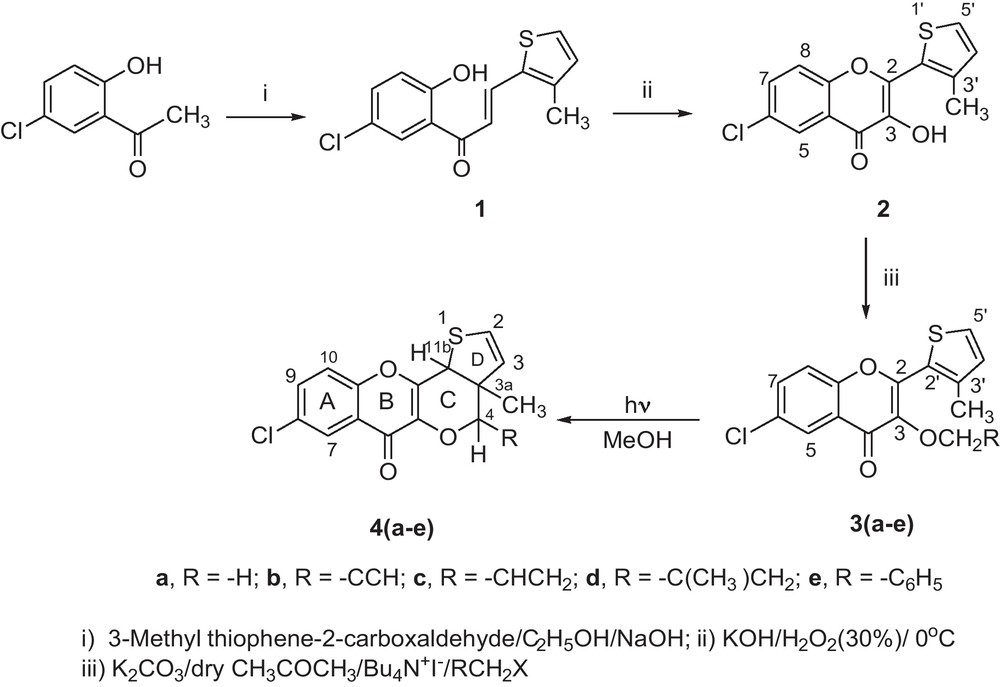
Synthesis and photolysis of chromenones 3(a–e).
The structures of compounds 3(a–e) were found to be consistent with their spectral parameters (IR, 1H/13C NMR). The yields of all these compounds were in the range of 80–98%.
The photolysis of methanolic solution of 3(a–e) with pyrex filtered UV-light, under nitrogen atmosphere produced photoproducts 4(a–e), and the structures of these photoproducts (Scheme 2) were confirmed by their spectral data (IR, 1H/13C-NMR) and elemental analysis.
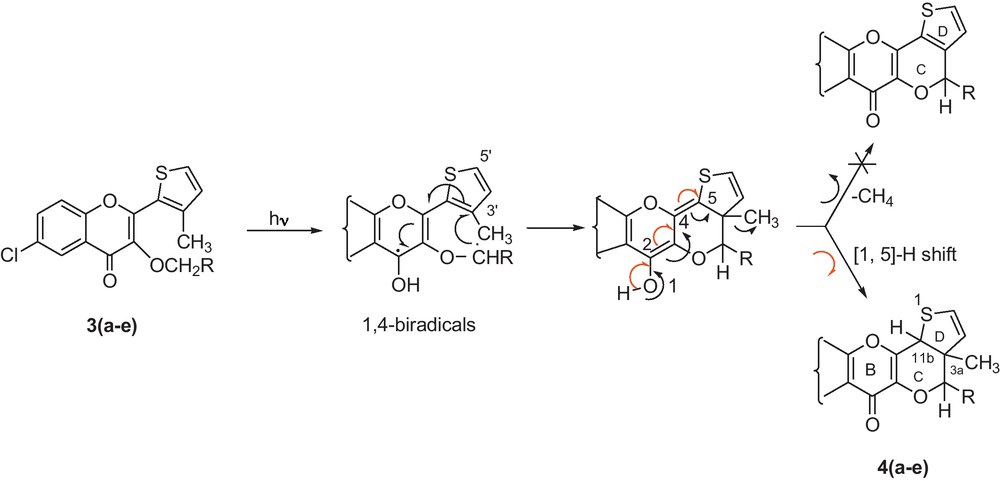
Mechanism of photocyclisation of 3(a–e).
In all these photoproducts, the ring C has half chair conformation and evidently, the C/D ring fusion is cis. For example in 4c, the trans-fusion of five-membered ring will result in higher conformational strain as calculated by MM2 energy minimizations programme [14] than the cis-fusion and this is in accordance with the earlier findings in case of the naturally occurring pterocarpans [15–18] and N-heterocycles [19]. Now assuming the C/D ring fusion as cis, the orientation of H-4 can be cis or trans with respect to –CH3 at C-3a. So, four possible 3D conformations I, II (H-4 cis to CH3-3a) and III, IV (H-4 trans to CH3-3a) were derived from MM2 energy minimizations programme which are shown in Fig. 1.
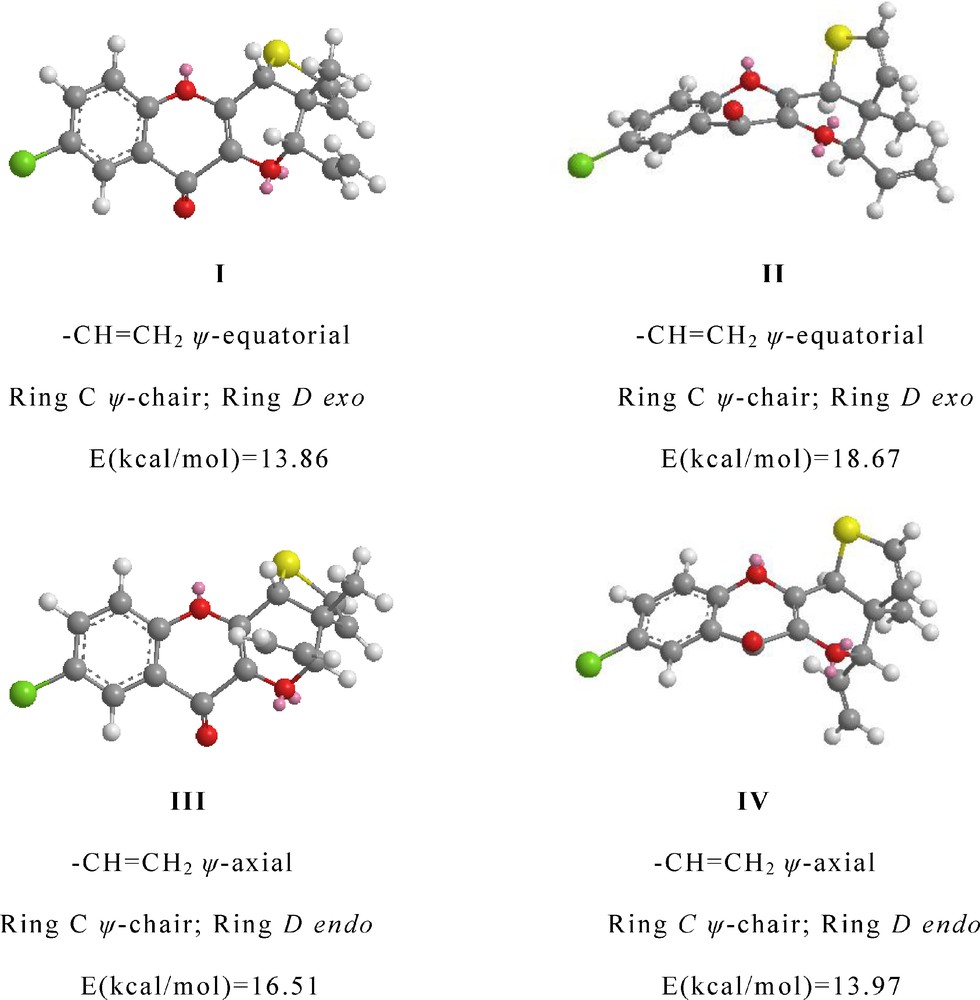
Possible energy minimized conformations of photoproduct 4c.
From the above four conformations, the one which has minimum energy i.e. conformation I, could be the possible conformation for the photoproduct 4c in which H-4 is cis to 3a-CH3 group and bulkier vinyl group is at ψ-equatorial position. Such a view is in conformity with the literature [20] where the heavier group at equatorial position in cyclohexane is always preferred.
The thiophen-2-yl chromenones having same basic skeleton similar to 3(a–e) with no 3–CH3 group at thiophenyl group furnished cyclised dihydro and cyclised dehydrogenated aromatic photoproducts [10,11]. The cyclised dihydro photoproducts were formed via 1,5-sigmatropic H-shift whereas the cyclised dehyderogenated aromatic products were formed by the expulsion of H2 during ketonisation directly. But, in the present study, the thiophen-2-yl chromenones 3(a–e) having a –CH3 group as a substituent at C-3 of thiophenyl, the formation of only cyclised dihydro type photoproducts 4(a–e) through 1,5-sigmatropic H-shift is favored. No demethylation leading to the formation of aromatic photoproducts was observed as the energy required to break C–C bond is higher than that required to break a C–H bond.
The phototransformations (Scheme 2) described above can be visualized to occur through the formation of 1,4-biradical intermediate. The products have been expected to be formed through a bond formation between –ĊH– radical and the C-3 atom of thiophene ring followed by ketonisation and H-migration to C-11b (1,5-H shift). The thiophenyl moiety at C-2 possesses only 3’-carbon available for clipping of 1,4-biradical.
Moreover, the yields of the photoproducts 4(a–e) formed depended upon the nature of the alkoxy group at C-3 position of the substrates (3a–3e). As the stability of the 1,4-biradicals generated in situ from 3a (R = –H) to 3e (R = –C6H5) increases (Scheme 2), yield of the corresponding photoproduct also increases (Table 1).
Yields of the photoproducts (4a–4e).
| Photoproduct | 4a | 4b | 4c | 4d | 4e |
| R | –H | –CCH | –CHCH2 | –C(CH3)CH2 | C6H5 |
| Yield (%) | 24 | 38 | 46 | 71 | 87 |
3 Conclusion
The 3-alkoxy-6-chloro-2-(3-methylthiophen-2-yl)-4H-chromen-4-ones upon photo-irradiation yielded angular tetracyclic products through 1,4-biradical furnished by γ-H abstraction, no demethylation occurred and therefore no cyclised aromatic photoproduct was realized.
4 Experimental
4.1 Materials and methods
Melting points were determined in open capillaries and are thus uncorrected. 1H/13C NMR spectra were recorded at 300 MHz (75.4 MHz for 13C NMR) on a Bruker spectrometer using TMS as internal standard. IR spectra were recorded on a MB3000 FT-IR with HORIZON MB™ FTIR software from ABB Bomen using KBr pellets. Mass spectra were recorded at 2500 eV (ESI-Source) using a Water's Q-TOF micro instrument. Elemental analysis was carried on Perkin Elmer 2400 instrument. TLC plates were coated with silica gel G (suspended in CHCl3–MeOH) and iodine vapors were used as visualizing agent. The columns for purification were packed with Silica gel 100–200 mesh in pet.ether and left overnight before use. The elution was carried out with increasing proportion of benzene in pet.ether-benzene mixture. The yields reported are based on the amount of isolated photoproducts and are calculated by excluding the recovered substrates.
4.2 Synthesis of chromones 3(a–e)
1-(5-Chloro-2-hydroxyphenyl)-3-(3-methylthiophen-2-yl)prop-2-en-1-one (1)
A solution of 5-chloro-2-hydroxyacetophenone (1.70 g, 1.0 eq.) and 3-methylthiophene-2-carboxaldehyde (1.38 g, 1.1 eq.) in absolute ethanol and sodium hydroxide (2.0 eq.) were stirred for 4 h. The dark red mixture was poured on ice-HCl to obtain 1 as yellow solid, crystallized from EtOH (1.90 g, 68.44%), m.p. 110–112 °C; IR νmax (cm−1): 1628.0 (C = O), 3405 (OH); 1H NMR (CDCl3, 300 MHz): δ 12.90 (1H, s, OH), 8.19 (1H, d, J3,2 = 15.0 Hz, H-3), 7.84 (1H, d, J6’,4’ = 2.4 Hz, H-6’), 7.45 (1H, dd, J4’,6’ = 2.4 Hz, J4’,3’ = 9.0 Hz, H-4’), 7.42 (1H, d, J5”,4” = 5.4 Hz, H-5”), 7.28 (1H, d, J2,3 = 15.0 Hz, H-2), 7.00 (1H, d, J3’,4’ = 9.0 Hz, H-3’), 6.97 (1H, d, J4”,5” = 5.4 Hz, H-4”), 2.46 (3H, s, 3”–CH3); 13C NMR (CDCl3): δ 192.24 (C-1), 162.02, 144.24, 137.26, 135.96, 134.25, 131.66, 128.75, 128.62, 123.47, 120.62, 120.17, 117.06, 14.38; Anal. Calcd. for C14H11ClO2S: C, 60.32; H, 3.98. Found: C, 60.30; H, 3.94.
6-Chloro-3-hydroxy-2-(3-methylthiophen-2-yl)-4H-chromen-4-one (2)
To a well stirred suspension of compound 1 (1.0 g, 0.003 mol) in MeOH was added aq. KOH (10 ml, 20%). This mixture was cooled to 0 °C. To this dark red solution was added H2O2 (30%) drop-wise till the colour changed to yellow and the stirring was continued for 4 h. The reaction mixture was neutralized with ice-HCl to give light yellow precipitates, crystallized (chloroform-ethanol) to light yellow solid (0.74 g, 70%), m.p. 190 °C; IR νmax (cm−1): 1597 (C = O), 3232 (OH); 1H NMR (CDCl3, 300 MHz): δ 8.23 (1H, d, J5,7 = 2.4 Hz, H-5), 7.66 (1H, dd, J7,5 = 2.4 Hz, J7,8 = 9.0 Hz, H-7), 7.56 (1H, d, J5’,4’ = 5.1 Hz, H-5’), 7.51 (1H, d, J8,7 = 9.0 Hz, H-8), 7.01 (1H, d, J4’,5’ = 5.1 Hz, H-4’), 2.60 (3H, s, 3’–CH3); 13C NMR (CDCl3): δ 172.23 (C-3), 153.52, 144.02, 140.84, 137.12, 133.70, 131.44, 130.57, 129.44, 125.39, 124.72, 121.94, 119.73, 16.79 (3’–CH3); Anal. Calcd. for C14H9ClO3S: C, 57.44; H, 3.10. Found: C, 57.39; H, 3.07.
6-Chloro-3-methoxy-2-(3-methylthiophen-2-yl)-4H-chromen-4-one (3a)
The compound, 2 (2.92 g, 1.0 eq.), (CH3)2SO4 (1.38 g, 1.1 eq.), dry K2CO3 (1.1 eq.) and tetra-n-butylammonium iodide (100 mg, 3.0 mol%) were refluxed in dry acetone (20 ml) for 4 h. Filtration, evaporation of solvent and crystallization of the residue (EtOH) gave 3a (80%) as a creamish solid, m.p. 136–138 °C; λmax (MeOH) 297, 264 nm; IR νmax (cm−1): 1643.0 (C = O); 1H NMR (CDCl3, 300 MHz): δ 8.24 (1H, d, J5,7 = 2.4 Hz, H-5), 7.62 (1H, dd, J7,5 = 2.4 Hz, J7,8 = 9.0 Hz, H-7), 7.53 (1H, d, J5’,4’ = 5.1 Hz, H-5’), 7.46 (1H, d, J8,7 = 9.0 Hz, H-8), 7.00 (1H, d, J4’,5’ = 5.1 Hz, H-4’), 3.99 (3H, s, –OCH3), 2.66 (3H, s, 3’–CH3); 13C NMR (CDCl3): δ 172.84 (C-4), 153.14, 141.83, 139.46, 133.46, 131.66, 130.74, 130.12, 125.33, 125.16, 124.57, 119.28, 77.19, 59.96 (3-OCH3), 17.33 (3’–CH3); Mass (m/z): 307 (M + 1, 88%); Anal. Calcd. for C15H11ClO3S: C, 58.73; H, 3.61. Found: C, 58.78; H, 3.63.
The other ethers 3(b–e) were synthesized by reacting 3-hydroxychromenone 2 with propargyl bromide, allyl bromide, methyl allyl chloride, and benzyl chloride respectively by the procedure as used for compound 3a.
6-Chloro-2-(3-methylthiophen-2-yl)-3-propargyloxy-4H-chromen-4-one (3b)
Yield 88%, light yellow solid; m.p. 156–158 °C; λmax (MeOH) 294, 263 nm; IR νmax (cm−1): 1636.3 (C = O); 1H NMR (CDCl3, 300 MHz): δ 8.23 (1H, d, J5,7 = 2.7 Hz, H-5), 7.63 (1H, dd, J7,5 = 2.7 Hz, J7,8 = 9.0 Hz, H-7), 7.55 (1H, d, J5’,4’ = 5.1 Hz, H-5’), 7.48 (1H, d, J8,7 = 9.0 Hz, H-8), 7.00 (1H, d, J4’,5’ = 5.1 Hz, H-4’), 5.07 (2H, d, J1”,3” = 2.4 Hz, H-1”), 2.65 (3H, s, 3’–CH3), 2.40 (1H, t, J3”,1” = 2.4 Hz, H-3”); 13C NMR (CDCl3): δ 172.50 (C-4), 153.22, 152.10, 142.10, 141.00, 133.62, 131.47, 130.87, 130.27, 129.80, 125.15, 124.78, 119.36, 76.59, 76.33, 59.03 (C-1”), 17.39 (3”–CH3); Mass (m/z): 331 (M + 1, 36%); Anal. Calcd. for C17H11ClO3S: C, 61.73; H, 3.35. Found: C, 61.78; H, 3.37.
3-Allyloxy-6-chloro-2-(3-methylthiophen-2-yl)-4H-chromen-4-one (3c)
Yield 95%, creamish solid; m.p. 130–132 °C; λmax (MeOH) 296, 266 nm; IR νmax (cm−1): 1643 (C = O), 1605 (C = C); 1H NMR (CDCl3, 300 MHz): δ 8.23 (1H, d, J5,7 = 2.4 Hz, H-5), 7.61 (1H, dd, J7,5 = 2.4 Hz, J7,8 = 9.0 Hz, H-7), 7.52 (1H, d, J5’,4’ = 5.1 Hz, H-5’), 7.46 (1H, d, J8,7 = 9.0 Hz, H-8), 7.00 (1H, d, J4’,5’= 5.1 Hz, H-4’), 6.07 (1H, ddt, J2”,1” = 6.6 Hz, J2”,3”a = 17.4 Hz, J2”,3”b = 10.8 Hz, H-2”), 5.37 (1H, dd, J3”a,1” = 1.5 Hz, J3”a,2” = 17.4 Hz, H-3”a), 5.23 (1H, dd, J3”b,2” = 10.5 Hz, J3”b,1” = 1.2 Hz, H-3”b), 4.76 (2H, d, J1”,2” = 6.6 Hz, H-1”), 2.64 (3H, s, 3’–CH3); 13C NMR (CDCl3): δ 172.87 (C-4), 154.07, 153.12, 141.83, 137.87, 133.44, 133.21, 131.54, 130.69, 129.99, 125.20, 125.12, 124.76, 119.31, 119.18, 72.99, 17.36 (3’–CH3); Mass (m/z): 333 (M + 1, 3%); Anal. Calcd. for C17H13ClO3S: C, 61.35; H, 3.94. Found: C, 61.33; H, 3.92.
6-Chloro-2-(3-methylthiophen-2-yl)-3-(2-methylallyloxy)-4H-chromen-4-one (3d)
Yield 95%, light yellow solid; m.p. 98–100 °C; λmax (MeOH) 297, 263 nm; IR νmax (cm−1): 1636 (C = O), 1605 (C = C); 1H NMR (CDCl3, 300 MHz): δ 8.19 (1H, d, J5,7 = 2.7 Hz, H-5), 7.62 (1H, dd, J7,5 = 2.7 Hz, J7,8 = 7.8 Hz, H-7), 7.52 (1H, d, J5’,4’ = 4.8 Hz, H-5’), 7.46 (1H, d, J8,7 = 7.8 Hz, H-8), 6.98 (1H, d, J4’,5’ = 4.8 Hz, H-4’), 5.09 (1H, s, H-3”b), 5.03 (1H, s, H-3”a), 4.72 (2H, s, H-1”), 1.80 (3H, s, 2”–CH3), 1.58 (3H, s, 3’–CH3); 13C (CDCl3): δ 172.88 (C-4), 154.02, 153.21, 141.76, 141.00, 138.46, 133.45, 131.33, 130.69, 129.87, 125.30, 125.15, 124.86, 119.35, 114.08, 75.67 (C-1”), 19.67, 17.10 (3’–CH3); Mass (m/z): 347 (M + 1, 8%); Anal. Calcd. for C18H15ClO3S: C, 62.33; H, 4.36. Found: C, 62.30; H, 4.31.
3-Benzyloxy-6-chloro-2-(3-methylthiophen-2-yl)-4H-chromen-4-one (3e)
Yield 95%, light yellow solid; m.p. 132–134 °C; λmax (MeOH) 297, 263 nm; IR νmax (cm−1): 1628.0 (C = O); 1H NMR (CDCl3, 300 MHz): δ 8.26 (1H, d, J5,7 = 2.4 Hz, H-5), 7.62 (1H, dd, J7,5 = 2.4 Hz, J7,8 = 8.7 Hz, H-7), 7.42–7.46 (3H, m, H-8, 3”, 7”), 7.49 (1H, d, J5’,4’ = 5.1 Hz, H-5’), 7.39–7.28 (3H, m, H-4”, 5”, 6”), 6.93 (1H, d, J4’,5’ = 5.1 Hz, H-4’), 5.22 (2H, s, –OCH2–), 2.55 (3H, s, 3’–CH3); 13C (CDCl3): δ 172.95 (C-4), 154.14, 153.21, 141.82, 138.13, 136.40, 133.48, 131.32, 130.73, 129.91, 128.95, 128.22, 125.27, 125.12, 124.83, 119.38, 73.75 (–OCH2–), 17.11 (3’–CH3); Mass (m/z): 383 (M + 1, 4%); Anal. Calcd. for C21H15ClO3S: C, 65.88; H, 3.95. Found: C, 65.79; H, 3.88.
4.3 Photo-irradiation of chromenones 3(a–e)
4.3.1 Photolysis of compound (3a)
A methanolic solution (150 ml) of chromenone 3a (500 mg, 1.6 mmol) was irradiated with light from a 125 W Hg vapor lamp in a pyrex reactor under Nitrogen atmosphere for 90 min. The removal of solvent left a gummy solid which was chromatographed to yield 4a.
Compound (4a): yield 24%, light yellow solid; m.p. 174–176 °C; IR νmax (cm−1): 1649.7 (C = O); 1H NMR (CDCl3, 300 MHz): δ 8.25 (1H, d, J7,9 = 2.1 Hz, H-7), 7.60 (1H, dd, J9,7 = 2.4 Hz, J9,10 = 9.0 Hz, H-9), 7.42 (1H, d, J10,9 = 9.0 Hz, H-10), 6.27 (1H, d, J2,3 = 6.6 Hz, H-2), 5.43 (1H, d, J3,2 = 6.6 Hz, H-3), 4.44 (1H, s, H-11b), 4.08 (1H, d, H-4a), 3.88 (1H, d, H-4b), 1.36 (3H, s, 3a–CH3); 13C (CDCl3): δ 170.52 (C-6), 153.58, 151.94, 148.39, 138.83, 133.47, 130.57, 125.45, 124.73, 119.48, 100.60, 78.73, 65.88, 40.83, 17.90 (3a–CH3); Mass (m/z): 307 (M + 1, 3%); Anal. Calcd. for C15H11ClO3S: C, 58.73; H, 3.61. Found: C, 58.78; H, 3.64.
4.3.2 Photolysis of compound (3b)
A methanolic solution of 3b (500 mg, 1.5 mmol) on photolysis for 45 min furnished 4b.
Compound (4b): yield 38%, creamish white solid; m.p. 202–204 °C; IR νmax (cm−1): 1643 (C = O), 2114 (CC); 1H NMR (CDCl3, 300 MHz): δ 8.25 (1H, d, J7,9 = 2.7 Hz, H-7), 7.60 (1H, dd, J9,7 = 2.7 Hz, J9,10 = 9.0 Hz, H-9), 7.42 (1H, d, J10,9 = 9.0 Hz, H-10), 6.30 (1H, d, J2,3 = 6.0 Hz, H-2), 5.54 (1H, d, J3,2 = 6.0 Hz, H-3), 4.83 (1H, d, J4,2’ = 2.1 Hz, H-4), 4.54 (1H, s, H-11b), 2.58 (1H, d, J2’,4 = 2.1 Hz, H-2’), 1.46 (3H, s, 3a–CH3); 13C (CDCl3): δ 170.39 (C-6), 153.66, 148.85, 137.21, 133.81, 130.67, 127.51, 127.11, 125.43, 124.55, 119.65, 76.63, 76.52, 68.40, 53.71, 51.54, 18.93 (3a–CH3); Mass (m/z): 331 (M + 1, 100%); Anal. Calcd. for C17H11ClO3S: C, 61.73; H, 3.35. Found: C, 61.78; H, 3.31.
4.3.3 Photolysis of compound (3c)
A methanolic solution of 3c (500 mg, 1.5 mmol) on photolysis for 45 min furnished 4c.
Compound (4c): yield 46%, creamish solid; m.p. 176–178 °C; IR νmax (cm−1): 1651(C = O); 1H NMR (CDCl3, 300 MHz): δ 8.26 (1H, d, J7,9 = 2.4 Hz, H-7), 7.60 (1H, dd, J9,7 = 2.4 Hz, J9,10 = 9.0 Hz, H-9), 7.41 (1H, d, J10,9 = 9.0 Hz, H-10), 6.37 (1H, d, J2,3 = 6.0 Hz, H-2), 5.99 (1H, ddd, J1’,2’ = 6.6 Hz, J1’,2’a = 17.4 Hz, J1’,2’b = 10.8 Hz, H-1’), 5.47 (1H, d, J2’a,1’ = 17.4 Hz,H-2’a), 5.35 (1H, d, J2’b,1’ = 10.8 Hz, H-2’b), 5.40 (1H, d, J2,3 = 6.0 Hz, H-3), 4.60 (1H, s, H-11b), 4.54 (1H, d, J4,1’ = 6.6 Hz, H-4), 1.26 (3H, s, 3a–CH3); 13C (CDCl3): δ 171.00 (C-6), 153.61, 148.15, 139.57, 138.47, 133.49, 130.76, 128.59, 126.51, 125.79, 124.68, 119.67, 116.50, 78.7, 54.10, 52.06, 18.97 (3a–CH3); Mass (m/z): 333 (M + 1, 88%); Anal. Calcd. for C17H13ClO3S: C, 61.35; H, 3.94. Found: C, 61.30; H, 3.91.
4.3.4 Photolysis of compound (3d)
A methanolic solution of 3d (500 mg, 1.4 mmol) on photolysis for 45 min furnished 4d.
Compound (4d): yield 71%, creamish solid; m.p. 214 °C; IR νmax (cm−1): 1659 (C = O); 1H NMR (CDCl3, 300 MHz): δ 8.24 (1H, d, J7,9 = 2.7 Hz, H-7), 7.59 (1H, dd, J9,7 = 2.7 Hz, J9,10 = 9.0 Hz, H-9), 7.39 (1H, d, J10,9 = 9.0 Hz, H-10), 6.36 (1H, d, J2,3 = 6.0 Hz, H-2), 5.44 (1H, d, J2,3 = 6.0 Hz, H-3), 5.16 (2H, d, J4,3a = 2.1, 2’–CH2), 4.56 (1H, s, H-11b), 1.94 (3H, s, 1’–CH3), 1.28 (3H, s, 3a–CH3); 13C (CDCl3): δ 170.51 (C-6), 153.62, 147.96, 139.87, 138.35, 133.61, 130.47, 128.56, 126.42, 125.39, 124.67, 119.62, 116.57, 78.34, 54.05, 52.01, 21.42, 18.78 (3a–CH3); Mass (m/z): 347 (M + 1, 27%); Anal. Calcd. for C18H15ClO3S: C, 62.33; H, 4.36. Found: C, 62.28; H, 4.31.
4.3.5 Photolysis of compound (3e)
A methanolic solution of 3e (500 mg, 1.3 mmol) on photolysis for 45 min furnished 4e.
Compound (4e): yield 87%, creamish solid; m.p. 186–188 °C; IR νmax (cm−1): 1651 (C = O); 1H NMR (CDCl3, 300 MHz): δ 8.28 (1H, d, J7,9 = 2.4 Hz, H-7), 7.62 (1H, dd, J9,7 = 2.4 Hz, J9,10 = 8.4 Hz, H-9), 7.28–7.47 (6H, m, H-10, 2’, 3’, 4’, 5’, 6’), 6.46 (1H, d, J2,3 = 6.0 Hz, H-2), 5.16 (1H, d, J3,2 = 6.0 Hz, H-3), 5.12 (1H, s, H-4), 4.74 (1H, s, H-11b), 1.60 (3H, s, 3a–CH3); 13C (CDCl3): δ 170.51 (C-6), 153.68, 148.10, 138.72, 135.75, 133.70, 130.56, 128.23, 127.96, 127.85, 126.93, 125.48, 124.72, 119.64, 76.61, 53.57, 52.22, 17.77 (3a–CH3); Mass (m/z): 383 (M + 1, 44%); Anal. Calcd. for C21H15ClO3S: C, 65.88; H, 3.95. Found: C, 65.91; H, 3.96.
Acknowledgements
Grateful acknowledgment is made to CSIR, New Delhi for generous financial support for accomplishing this work.