

1 Introduction
During the past three decades, a number of phosphates belonging to the A2O-M2O3-P2O5 (A = monovalent and M = Al, Ga, Cr,Fe) system were showed to exhibit fast A+ ion transport properties: among them are the Nasicon-type monophosphates A3Cr2(PO)4 and A3Fe2(PO)4 [1–4] the monodiphosphata Na7(MP2O7)4PO4 [5] and the Na7M3(P2O7)4 [6,7]. All these compounds have a 3D framework made up of corner sharing MO6 octahedra and PO4 tetrahedra, with the A+ ions located in the interstitial space. During an attempt to find new materials with optimal properties and enrich this family of compounds, we successfully synthesized the solid-state compound AgCr2(PO4)(P2O7).
This study reports its synthesis by the high temperature solid-state reactions and the single crystal structural determination are reported for the title compound, and the electrical conductivity has been measured as a function of temperature. Furthermore, the infrared spectrum of AgCr2(PO4)(P2O7) is reported and band assignments are made.
2 Experimental
2.1 Materials and measurements
All reagents were purchased commercially and used without further purification. A qualitative energy dispersive spectroscopy (EDX) analysis, performed on JEOL-JSM 5400 scanning electron microscope, revealed the presence of only Ag, Cr, P and O elements. The infrared spectrum for AgCr2(PO4)(P2O7) was recorded at room temperature using single crystals on a Nicolet 470 FTIR spectrophotometer (resolution: 0.125 cm−1), over the range 1400–400 cm−1 using the KBr disk method. The impedance measurements were carried out in a Hewlett-Packard 4192-A automatic bridge monitored by a HP microcomputer. The frequency range was 5 Hz–13 MHz. Pellets of 13 mm diameter and 0.6 mm thickness were prepared by pressing the powder sample at 10 tones. Then the pellets were sintered at 600 °C for 24 h. Silver electrodes were painted on the two faces of the pellets with a silver paste, and then the painted pellets were heated at 300 °C/1 h. The impedance measurements were carried out at steady-state temperatures on the pellets in still air.
2.2 Synthesis
Single crystals of AgCr2(PO4)(P2O7) were prepared by crystallization in a flux of molybdate MoO3(Acros, 99%), in an atomic ratio P: Mo = 4: 1. Appropriate amounts of AgNO3 (Fluka, 99%), Cr(NO3)3 9H2O (Fisher, 98.6%) and (NH4)2HPO4 (Merck, 99%) were mixed by dissolving in aqueous nitric acid and the obtained solution was dried at 353 K. The resulting dry residue was ground in an agate mortar to ensure its best homogeneity, and then gradually heated up to 873 K in a platinum crucible. After being reground, the mixture was melted for 1 h at 1123 K and subsequently cooled at a rate of 10 K.h−1 down to 673 K, after which the furnace was turned off. The crystals obtained by washing the final product with warm water, in order to dissolve the flux, are essentially composed by green crystals of AgCr2(PO4)(P2O7).
2.3 Structure determination
The structure was determined from single X-ray data, collected at room temperature by an Enraf-Nonius CAD4 [8] diffractometer using monochromated Mo-Kα radiation (λ = 0.7107 Å) and ω/2θ scans. The unit cell parameters and the orientation matrix were determined from a least-squares fit of 25 reflections in the range 10.95° ≤ θ ≤ 14.98°. A total of 1401 unique reflections were measured (Rin = 0.082) but only 809 were considered as observed according to the statistic criterion [I > 2σ(I)].
The intensity data were corrected for Lorentz and polarization effects. Absorption corrections were done analytically taking into account the size and shape of the crystal (Tmin = (Tmin = 0.20; Tmax = 0.49). The structure was solved by direct methods [9], which allowed us to obtain the positions of the Cr and Ag atoms. The P and O atoms were subsequently localized by inspection of the difference electron density maps [10].
At this stage the atomic coordinates and isotropic thermal factors were refined to R = 0.131 and RW = 0.346. When all the atoms were anisotropically refined, the agreement factors R and RW converged to 0.050 and 0.159, respectively but the difference synthesis map shows strong electronic residues at 0.247, 0.187, and 0.518, which allowed us to think that the four silver atoms of the cell are spread randomly over eight positions. All these positions cannot be fully occupied at the same time because two position related by a symmetry center are distant by only 0.33 Å. Finally, when all the atoms were anisotropically refined, the agreement factors R and Rw converged to 0.043 and 0.136 and all ADPs are positive with normal standard deviations. The cationic distribution was confirmed by a Bond Valence Sums (B. V. S.) calculation using the Brown and Altermatt method [11]. Experimental conditions for intensity measurements, structure solution and refinement are reported in Table 1. Final atomic coordinates and equivalent isotropic displacement parameters are listed in Table 2.
Details of the data collection and structural refinement for AgCr2(PO4) (P2O7).
| Crystal data | |
| Chemical formula | AgCr2(PO4)(P2O7) |
| Crystal system | Monoclinic |
| Space group | C2/c |
| a (Å) | 11.493 (1) |
| b (Å) | 8.485 (1) |
| c (Å) | 8.790 (1) |
| β (°) | 114.56 (1) |
| Z | 4 |
| ρcal (g.cm–3) | 4.095 |
| Data collection | |
| Crystal dimensions | 0.1 × 0.07 × 0.07 mm |
| Diffractometer | CAD4 (Enraf-Nonius) |
| Radiation | λ (Mo Kα) = 0.7107 Å |
| Monochromator | Graphite |
| μ(mm–1) | 5.903 |
| Scan type | ω/2θ |
| Scan speed | Variable |
| 2θmax (°) | 59.9 |
| Number of unique reflections; Rint | 809; Rint = 0.082 |
| Number of observed reflections [I > 2σ(I)] | 633 |
| F (000) | 912 |
| Structural refinement | |
| Intensity corrections | Lorentz-polarisation |
| Absorption correction (Tmin, max) | Analytical (0.20, 0.49) |
| Structure solution | Direct Methods |
| Reliability factors | R1 = 0.043; wR2 = 0.135; S = 1.21 |
| Number of parameters | 86 |
| (Δρ) max, min/e. Å–3 | 1.63;-1.89 |
Atomic coordinates and displacement parameters Ueq(Å2) for AgCr2(PO4) (P2O7).
| Atom | Wyckoff | x (σ) | y (σ) | z (σ) | Ueq (σ) |
| Ag | 8f | 0.2563 (4) | 0.2343 (4) | 0.5109 (4) | 0.0230 (1) |
| Cr | 8f | –0.1258 (2) | 0.2382 (1) | 0.4593 (2) | 0.0044 (1) |
| P1 | 8f | 0.3883 (2) | 0.0636 (1) | 0.2870 (3) | 0.0043 (2) |
| P2 | 4e | 0 | 0.0434 (1) | 0.25 | 0.0052 (1) |
| O1 | 4e | 0.5 | –0.0170 (4) | 0.25 | 0.0082 (2) |
| O2 | 8f | –0.3101 (4) | 0.1597 (1) | 0.3738 (2) | 0.0057 (2) |
| O3 | 8f | 0.4512 (1) | 0.1736 (4) | 0.4365 (4) | 0.0088 (2) |
| O4 | 8f | 0.3116 (2) | –0.0683 (1) | 0.3142 (3) | 0.0074 (2) |
| O5 | 8f | 0.0999 (2) | 0.1517 (4) | 0.2258 (3) | 0.0112 (2) |
| O6 | 8f | 0.5737 (4) | 0.4482 (4) | 0.4077 (1) | 0.0129 (4) |
Further details of the crystal structure investigation can be obtained from the Fachinformationszentrum Karlsruhe, 76344 Eggenstein-Leopoldshafen, Germany, (fax: +49 7247 808 666; crysdata@fiz.karlsruhe.de on quoting the depository number CSD 422998.
3 Results
3.1 Description of the structure
The new compound AgCr2(PO4)(P2O7) is isostructural to AgV2(PO4)(P2O7 [12]. Its framework is formed by [PO4] tetrahedra, [P2O7] groups and [CrO6] octahedral. Projection of the structure along ac plan is depicted in Fig. 1.

A projection along the ac plan of AgCr2(PO4) (P2O7).
The projection of the structure along ab plan (Fig. 2) shows that it can be described as the stacking along b of octahedral layers and tetrahedral layers alternatively.
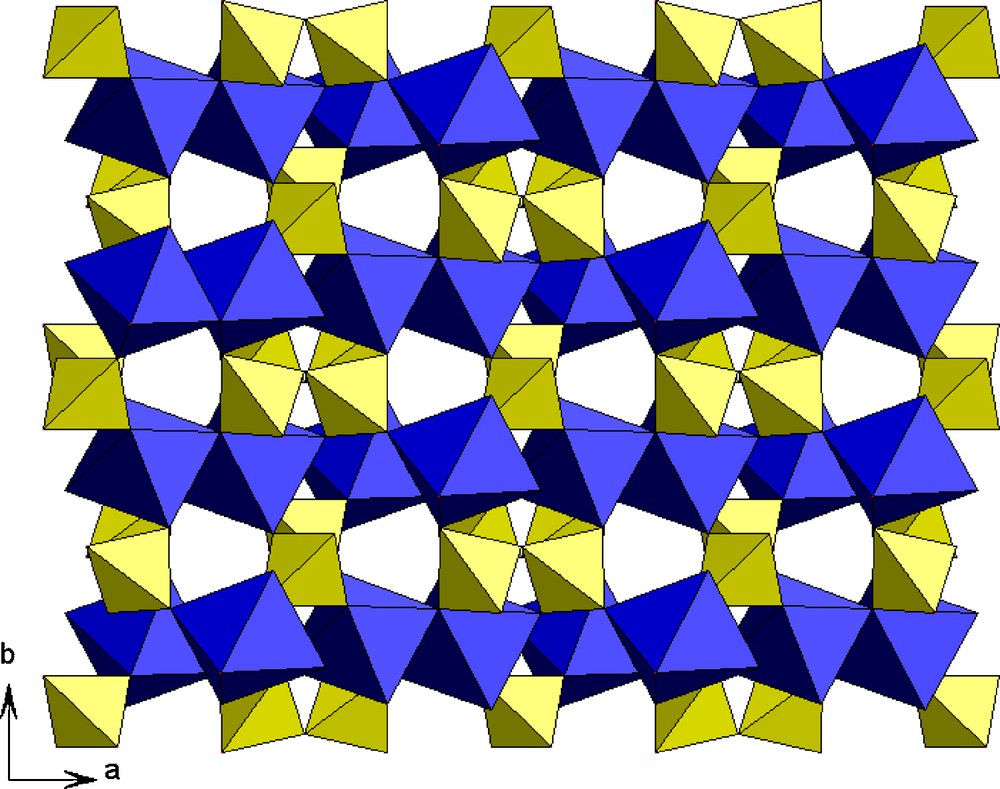
A projection along the ab plan of AgCr2(PO4) (P2O7).
The structure can be described as the stacking along c direction of [Cr2P3O16]∞ layers parallel to (001) two successive layers being eniantiomorphic with respect to each other. As it is shown in Fig. 3, each layer consists of [Cr2P2O14]∞ chains running along [110] direction and built up from Cr2O10 bioctahedral units sharing their corners with diphosphate P2O7 groups laterally. These chains are interconnected through the monophosphate groups.
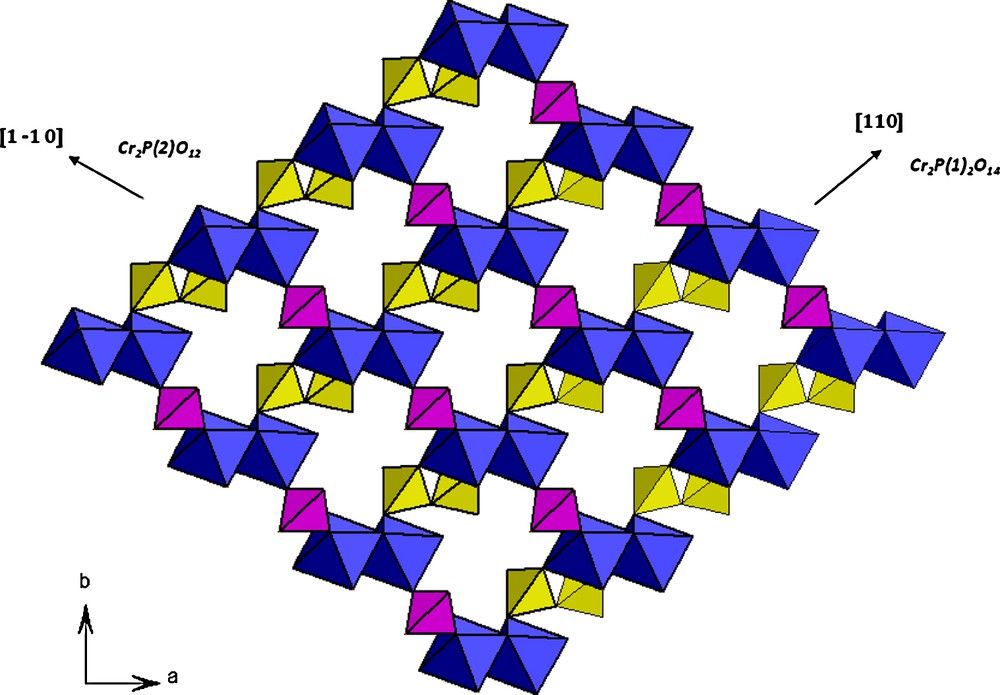
Connection between the CrO6 octahedron and its neighboring PO4 tetrahedra.
Another way to describe this structure is to consider the [Cr2PO12]∞ chains built up of corner sharing Cr2O10 and monophosphate groups running along [1–10] direction .These chains are interconnected through [P2O7] groups.
A projection along the [111] direction of AgCr2(PO4)(P2O7) shows the six-edged tunnels occupied by the Ag+ ions (Fig. 4).

A projection along the [111] direction of AgCr2(PO4)(P2O7) showing the six-edged tunnels, occupied by the Ag+ ions.
In what follows, we will describe the structural environment for each atom.
3.1.1 Chromium atoms
The CrO6 octahedron is rather irregular; with the smallest and largest distances being in the range from 1.905 (5) Ǻ to 2.082 (4) Ǻ. Average Cr–O distances is 1.989 (5) Å. This value is close to the ionic radii sum (2.02 Ǻ) of Cr3+ and O2− [13]. Each [CrO6]octahedron shares one common edge (O(2)–O(2)) with itself to forms [Cr2O10] dimers with a Cr-Cr distance of 3.225 (6)Å and all of its corners with six P(1)2O7 and four P(2)O4.
The O-Cr-O bond angle ranging from 77.2 (2)° to 96.05 (2)° and from 168.70 (2)° to 177.60 (2)° for the cis and trans angles, respectively (Table 3).
Main interatomic distances (Å) et angles (°) for AgCr2(PO4) (P2O7).
| CrO6 | |||
| Cr-O(6) | 1.905 (5) | O(6)-Cr-O(5) | 94.6 (2) |
| Cr-O(5) | 1.917 (4) | O(6)-Cr-O(3) | 91.7 (2) |
| Cr-O(3) | 1.971 (5) | O(5)-Cr-O(3) | 93.1 (2) |
| Cr-O(4) | 2.016 (3) | O(6)-Cr-O(4) | 177.60 (19) |
| Cr-O(2) | 2.044 (5) | O(5)-Cr-O(4) | 85.1 (2) |
| Cr-O(2) | 2.082 (4) | O(3)-Cr-O(4) | 90.66 (19) |
| <Cr-O> | 1.989 (4) | O(6)-Cr-O(2) | 88.1 (2) |
| O(5)-Cr-O(2) | 93.7 (2) | ||
| O(3)-Cr-O(2) | 173.20 (18) | ||
| O(4)-Cr-O(2) | 89.52 (19) | ||
| O(6)-Cr-O(2) | 91.72 (19) | ||
| O(5)-Cr-O(2) | 168.7 (2) | ||
| O(3)-Cr-O(2) | 96.05 (19) | ||
| O(4)-Cr-O(2) | 88.18 (19) | ||
| O(2)-Cr-O(2) | 77.2 (2) | ||
| P(1)O4 | |||
| P(1)-O(4) | 1.506 (5) | O(4)-P(1)-O(3) | 114.6 (3) |
| P(1)-O(3) | 1.529 (5) | O(4)-P(1)-O(2) | 112.4 (3) |
| P(1)-O(2) | 1.555 (5) | O(3)-P(1)-O(2) | 110.6 (3) |
| P(1)-O(1) | 1.602 (6) | O(4)-P(1)-O(1) | 106.6 (3) |
| <P1-O> | 1.547 (5) | O(3)-P(1)-O(1) | 107.6 (2) |
| P(2)O4 | |||
| P(2)-O(6) | 1.526 (5) | O(6)-P(2)-O(6) | 116.0(4) |
| P(2)-O(6) | 1.526 (5) | O(6)-P(2)-O(5) | 110.5 (2) |
| P(2)-O(5) | 1.556 (5) | O(6)-P(2)-O(5) | 106.1 (3) |
| P(2)-O(5) | 1.556 (5) | O(6)-P(2)-O(5) | 106.1 (3) |
| <P(2)-O> | 1.541 (5) | O(6)-P(2)-O(5) | 110.5 (2) |
| O(5)-P(2)-O(5) | 107.4 (4) | ||
| AgO7 | |||
| Ag -O(5) | 2.429 (21) | Ag -O(5) | 2.473 (22) |
| Ag -O(4) | 3.086 (19) | Ag -O(6) | 3.098 (20) |
| Ag -O(3) | 2.614 (26) | Ag -O(4) | 2.857 (19) |
| Ag -O(3) | 2.727 (20) | ||
| <Ag -O> | 2.755 (20) |
3.1.2 Phosphorus atoms
The particularity in these phosphates is the fact that [PO4] and [P2O7] groups coexist in the same compound. The [PO4] tetrahedral are slightly distorted due to the corner sharing with [CrO6] polyhedra.
The diphosphate unit P(1)2O7 also adopts a binary internal symmetry with the bridging oxygen O(1) located into a two-fold axis. The average values for the P–O distances and P–O–P angles are 1.547(5) Å, 109.45(2)° respectively for [P(1)O4] tetrahedra. All these distances and angles are of the same magnitude in accordance with previous observations of [PO4] tetrahedra involved in other diophosphate anions [14–16]. This group shares the terminal oxygen atoms with four [Cr2O10] dimers.
The P(2)O4 tetrahedron is bonded to four different [CrO6] octahedral. The P(2)-O distances range from 1.526(3) Å to 1.557(5) Å (av. 1.541 Å) and the O-P-O angles are in the 106.1°–116.0° range (av. 109.43°), which is in a good agreement with those previously observed. The calculated average values of the distortion indices [17], corresponding to the different angles and distances in both [P(2)O4] and [P(1)O4] tetrahedra [DI(O–P–O) = 0.25–0.028; DI(P–O) = 0.009–0.019 and DI(O–O) = 0.016–0.021] exhibit a pronounced distortion of the P(1)–O distances and O–P(1)–O angles; so each phosphate group can be considered as a rigid regular arrangement of oxygen atoms (Table 4).
ID for the coordination polyhedra around Cr and P.
| Cr | P1 | P2 | |
| IDd | 0.029 | 0.019 | 0.009 |
| IDa | 0.044 | 0.028 | 0.025 |
| IDo | 0.037 | 0.021 | 0.016 |
If we consider only Ag–O distances less than 3.1 Å, the Ag cations are surrounded by seven oxygen atoms, with distances ranging from 2.429(21) Å to 3.098(20) Å forming irregular coordination polyhedra. The bond valence sum [18] calculations confirm the one, three and five-fold valence of the Ag, Cr and P atoms, respectively.
3.2 IR spectroscopic investigation
3.2.1 PO43− vibrations
The internal vibrational modes of a free PO43− tetrahedron with Td symmetry are decomposed as A1 + E + 2 T2. The mode with the A1 symmetry type is the symmetric stretching mode (ν1) of the P–O bonds. The modes with the E and T2 symmetry types are bending O–P–O modes (ν2 and ν4, respectively). The second mode with the T2 symmetry is the asymmetric stretching mode (ν3). For a free PO43− tetrahedron, ν1 = 938 cm−1, ν2 = 420 cm−1, ν3 = 1017 cm−1and ν4 = 567 cm−1.
3.2.2 P2O74− vibrations
According to Corbridge [19], the P2O7 group (which can be written O3P–O–PO3) can be described for the vibration spectrum interpretation as an assembly of the vibrations of the PO3, and the P–O–P groups. The assignment of the P2O74− modes is carried out in terms of PO3 and P–O–P vibrations. In these conditions, the infrared bands for AgCr2(PO4)(P2O7) shown in Fig. 5 are distributed in four distinct wave number ranges, 400–675 cm−1, 720–800 cm−1, 850–990 cm−1, and 990–1400 cm−1. Band assignments for the fundamental modes of this compound are given in Table 5 confirm the presence of the mono and diphosphate groups in the title compound.
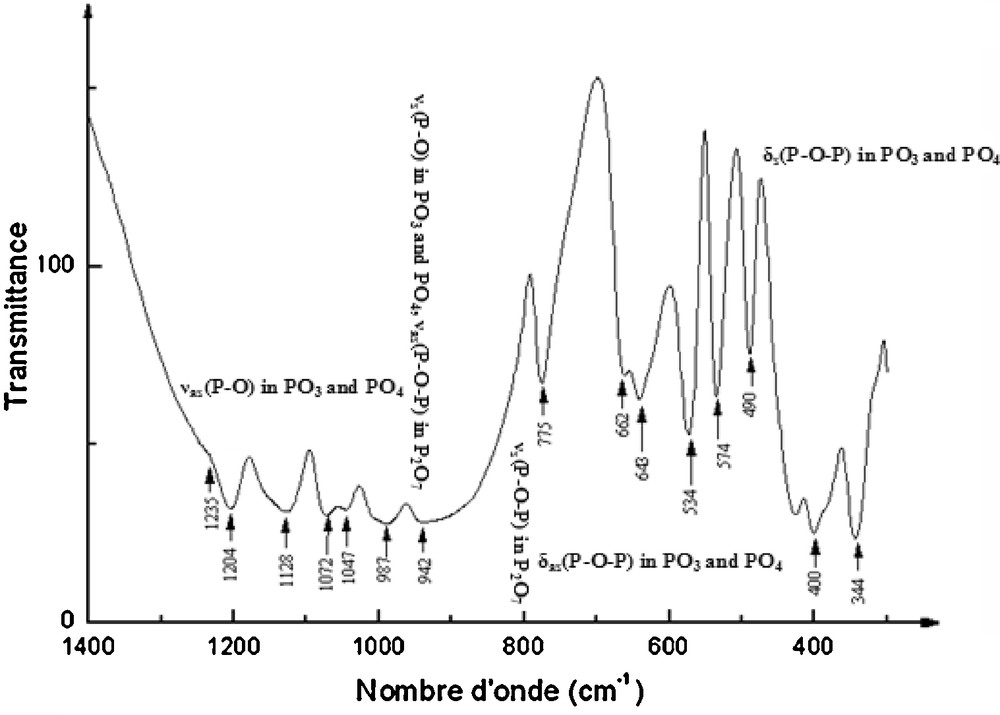
Infrared analysis spectrum ofAgCr2(PO4) (P2O7).
Band assignment (cm–1) for AgCr2(PO4) (P2O7).
| Bands (cm–1) | Assignment |
| 1235, 1204, 1128, 1072, 1047 | νas(P-O) in PO3 and PO4 |
| 987, 942 | νs(P-O) in PO3 and PO4, νas(P-O-P) in P2O7 |
| 775 | νs(P-O-P) in P2O7 |
| 662, 643, 575, 534 | δas(P-O-P) in PO3 and PO4 |
| 490, 425, 400 | δs(P-O-P) in PO3 and PO4 |
3.3 Ionic conductivity
Typical complex plots of imaginary part of impedance–Z” versus the real part Z’ at various temperatures (Z* =Z’-jZ”) are shown in Fig. 6 in which semi circles are observed. The centers of these semicircles are depressed below the baseline. The ionic conductivity as function of the temperature has been obtained from the values of intercept of the extrapolated high-frequency semicircles with the real axis. It should be noticed that conductivity in this phosphate is of ionic type; the electronic conductivity in oxides is due to overlapping of non-completely filled d or f orbitals of cations or to electron hopping from aliovalent ions, assisted by an oxygen anion. In this case, we would exclude both mechanisms so conductivity should be totally ionic, some electronic could be present in grain boundaries, but negligible.
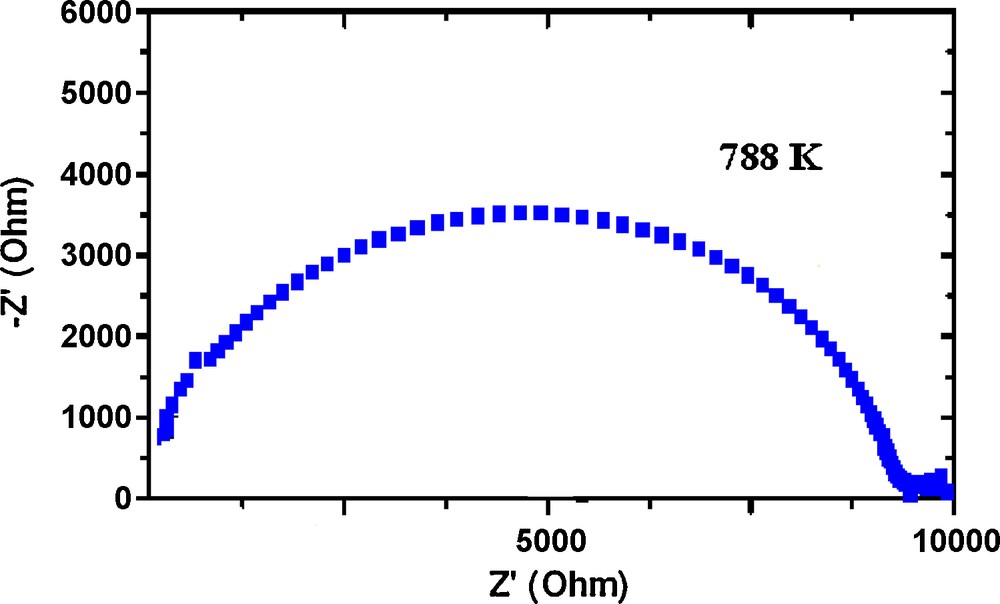
Complex impedance diagram for AgCr2(PO4)(P2O7) at 788 K.
The conductivity variation indicates an increase of conductivity with rise in temperature with a typical Arrhenius-type behavior having linear dependence of thermal conductivity logarithm log(σT) on inverse of temperature 103/T (K−1) (Fig. 7). This type of temperature dependence of the conductivity indicates that the electrical conduction in the material is a thermally activated process. It can be explained in accordance with the expression: σT = A0 exp(-Ea/kT), where A0 is the pre-exponential factor, Ea the activation energy, T the absolute temperature and k the Boltzmann constant. The conductivity value is σ = 58.27 × 10−7 Ω–1 cm–1 at 788 K (Table 6) and the ionic jump activation energy is 1.36 eV. This material show low conductivity performances, as compared to other compounds [20–24], this probably can be explained by other unfavorable structure features: ions Ag+ lie in too small polyhedral as mentioned in the discussion above and are lying with completely occupation site.
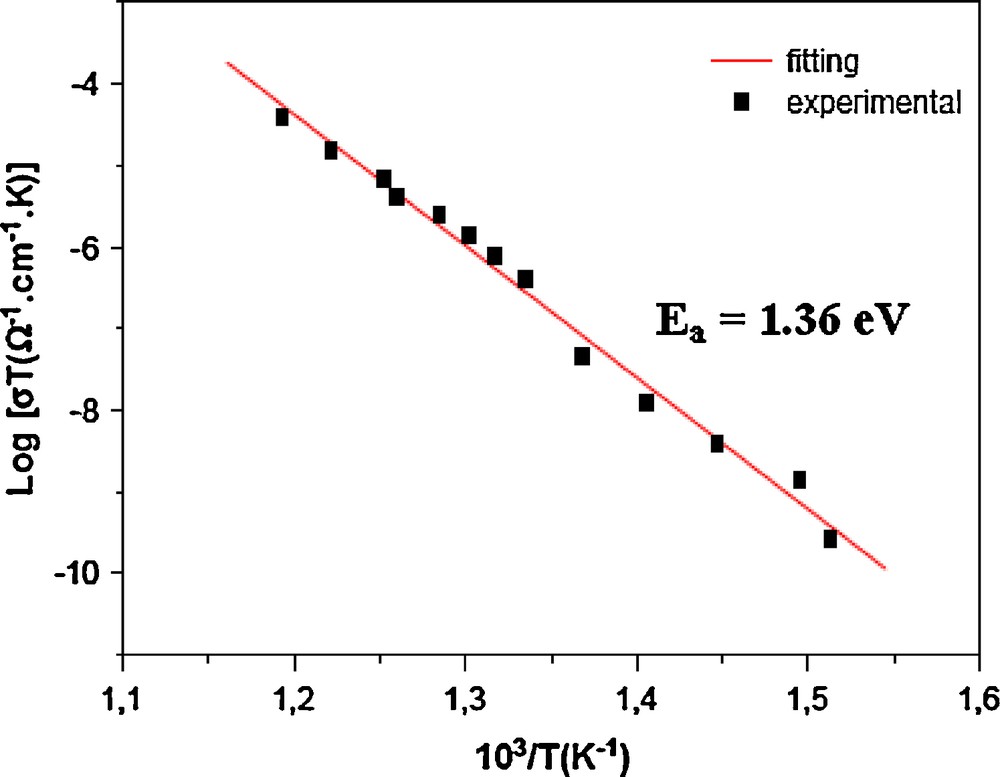
Arrhenius plots of conductivity for AgCr2(PO4)(P2O7) single crystals.
Ionic conductivity (σ) of AgCr2(PO4) (P2O7).
| T(K) | R(Ω) | σ(Ω−1Cm-1) 107 |
| 661 | 588,848 | 1.04 |
| 669 | 238,416 | 2.15 |
| 691 | 193,451 | 3.16 |
| 711 | 118,222 | 5.17 |
| 731 | 68,600 | 8.9 |
| 749 | 27,311 | 22.36 |
| 759 | 20,994 | 29.08 |
| 768 | 16,223 | 37.64 |
| 778 | 12,759 | 47.86 |
| 788 | 10,480 | 58.27 |
| 798 | 8,427 | 72.45 |
| 818 | 6,210 | 98.33 |
| 838 | 4,253 | 143.58 |
4 Conclusions
Crystal structure of AgCr2(PO4)(P2O7) has been resolved in C2/c space group. The structure was solved by direct methods. It was refined to R = 0.043 (Rw = 0.135) for 809 independent reflexions. Two kinds of [PO4] exist and the mean value of P–O is 1.547 (5) Å for one and 1.541(5) Å for the other. In P2O7, the angle P–O–P is 129.4 (5)°. The distances P–O of bridge are 1.602(4) Ǻ, the mean value of P–O in terminals PO3 is 1.530 Ǻ. The structure contains tunnels along the [101] directions in which the Ag+ ions are located. The coordination numbers of the silver ions are ten. The open framework of the title compound and the location of the silver ions in its tunnels make it a possible candidate for ionic conductivity However, this material shows low conductivity performances with a conductivity value of σ = 58.27 × 10−7 Ω−1 cm−1. The compound was also characterized by infrared spectroscopy and the results are in a good agreement with structural data.
Acknowledgements
I thank Dr. Amor Haddad (Laboratoire de matériaux et cristallochimie [LMC], institut supérieure des sciences appliquées et technologie, avenue El Mourouj, 5111 Mahdia, Tunisia).