

1 Introduction
An art to develop operationally simple and environmentally benign routes for organic compounds having high synthetic as well as biological potential is one of the fundamental objectives in synthetic organic chemistry [1]. In this context, the possibility of performing one-pot condensation of three or more components, multicomponent reactions (MCRs), is of relevance both from an economical as well as ecological point of view [2]. Although the main attraction of multicomponent reactions is their ability to construct two or more C-C or C-heteroatom bonds in single step, a typical challenge in performing them is to obtain only the desired product when competitive two component reactions between the selected substrates are equally probable. Two main approaches to address this challenge rely in the selection of an appropriate reaction path as well as catalyst.
Phosphonates are important intermediates in organic synthesis and their utility in Wittig and related reactions is well documented [3]. Currently they have also been the focus of intensive studies due to their usefulness as enzyme inhibitors [4], metabolic probes [5], peptide mimetics [6], antibiotics, pharmacologic agents [7], etc. Many natural products containing C–P bond are also known to exhibit important biological activities [8]. Owing to such a wide range of applications, development of efficient protocols for the synthesis of phosphonates, phosphonic acids and related compounds via C–P bond formation is enjoying growing interest. It is worth mentioning that, apart from the Kabaschnik-Field, Michaelis-Beker, and Michaelis-Arbuzov reaction [9], addition of phosphite nucleophile across carbon-carbon double bond (phospha-Michael reaction) is an ever green and widely used method in C–P bond formation [10,11]. The reaction is usually catalyzed by alkaline earth metal oxides [12], transition metal catalysts [13], tetramethylguanidine [14], Lewis/Bronsted acids [15], MWs [16], etc. In the recent past, the phospha-Michael reaction has been explored in the synthesis of β-phosphonomalononitriles using HClO4.SiO2 [17a], 3-aminopropylated silica gel [17b], nanoflake-ZnO [17c], sodium stearate [17d], etc. as catalysts. Many of these two component protocols essentially involve addition of phosphite nucleophile to benzylidine malononitrile; however, reports on one-pot synthesis of β-phosphonomalononitriles are scanty [18]. Our interest in this class of compounds stems from the ongoing studies in our laboratory on the development of new synthetic methodologies [19] including a recent report on expeditious synthesis of α-hydroxy phosphonates using potassium phosphate as well as diethylamine as catalyst [20] (Scheme 1a). Based upon these results, we further expand this chemistry towards the synthesis of β-phosphonomalononitriles using diethylamine as a novel organocatalyst.
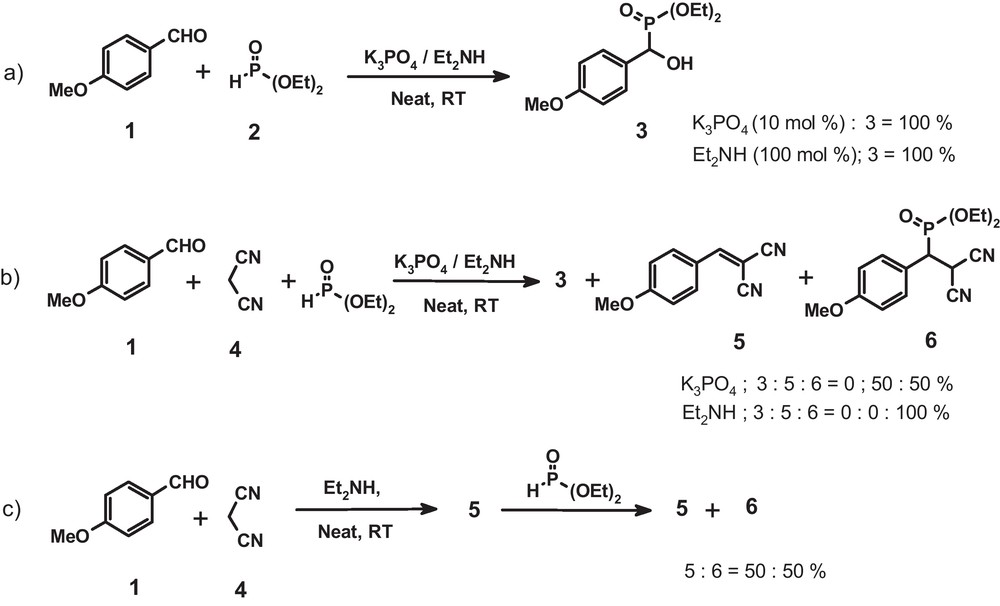
Screening of reaction path/catalyst for one-pot synthesis of β-phosphonomalononitrile.
2 Results and discussion
For one-pot synthesis of β-phosphonomalononitriles, one of the cleaner routes involves a base catalyzed, multicomponent condensation between an aldehyde, malononitrile and di/tri alkyl phosphite. On the other hand, a typical challenge in execution of this route relies in the choice of a base catalyst which would furnish the targeted β-phosphonomalononitrile, 6, in excellent yields with total avoidance of the formation of α-hydroxy phosphonate, 3, via the competitive Pudovik Reaction [21]. Thus, we planned to develop a base catalyzed one-pot protocol for the synthesis of β-phosphonomalononitriles.
Initial exploratory reaction was carried out between anisaldehyde, malononitrile and diethyl phosphite using potassium phosphate as a catalyst (10 mol %). The crude product which resulted after stirring the reaction mixture over night was identified to be a mixture of Knoevenagel condensation product, 5 and the desired β-phosphonomalononitrile, 6, in nearly 1:1 proportion (1H-NMR). Most strikingly, formation of α-hydroxy phosphonate, 3, was not noticed (Scheme 1b). In short, although potassium phosphate has been demonstrated by us as a highly efficient catalyst to effect two component condensations between an aldehyde and diethyl phosphite [20b] as well as between an aldehyde and active methylene compounds [19f], during a three component condensation using these substrates, the reaction exclusively followed the tandem Knoevenagel-phospha-Michael pathway. This break-through result directed us to test the feasibility in using other base catalysts in one-pot synthesis of β-phosphonomalononitriles. Accordingly, various homogeneous and heterogeneous base catalysts were screened for this three component condensation reaction (Table 1). It was noticed that, under solvent-free condition, compared to inorganic salts (entry 2, 3, 4 & 5, Table 1), organic bases such as pyrrolidine, piperidine and diethylamine (entry 11, 12, 13, Table 1), with very close pKa values, were equally efficient to furnish the desired β-phosphonomalononitrile in excellent yield. On the other hand, with the choice of heterogeneous catalysts like PVP, DBU, DMAP as well as DABCO, the reactions furnished a mixture of α-hydroxy phosphonate and β-phosphonomalononitrile in different proportions. From the view point of operational simplicity, another cleaner route for the synthesis of β-phosphonomalononitriles involves the sequential, one-pot, Knoevenagel-phospha-Michael addition reaction (Scheme 1c). During exploration of this route, Knoevenagel condensation between anisaldehyde and malononitrile was initially effected using diethylamine as catalyst. To the resultant product, 5, diethyl phosphite (1 eqv) was added and stirring was continued over night. The resultant crude product was identified (1H- NMR) to be a mixture of Knoevenagel condensation product, 5 and the desired phospha-Michael addition product, 6, in nearly 1:1 proportion. Finally, a control experiment was carried out using the same substrate combination. In absence of a catalyst, the reaction furnished a mixture of products (Entry 1, Table 1). Based upon the results summarized in Table 1 and on the basis of parameters of commercial availability, low toxicity, cost, ease in handling as well as its easy removal from the reaction mixture, diethylamine was recognized to be the best suited organocatalyst in the one-pot synthesis of B-phosphonomalononitriles. A careful literature survey very interestingly revealed that there are no reports on the use of diethylamine in any multicomponent reaction. Thus, it was then planned to explore the generality of the reaction conditions towards the synthesis of various β-phosphonomalononitriles. Initially, the reactions were performed between malononitrile, diethyl phosphite and various aromatic aldehydes bearing electron withdrawing (6h, Table 2) as well as electron donating groups (6e, g, Table 2) and having obtained the satisfactory results, the scope of the protocol was extended towards heteroaromatic (6n, Table 2) as well as non-aromatic aldehyde (6o, Table 2). Except aliphatic aldehyde, (6p, Table 2), in all cases, respective β-phosphonomalononitrile was obtained in a short time, in excellent yield as well as purity. (Table 2) We then investigated the possibility of replacing diethyl phosphite with dimethyl and di-iso-propyl phosphite. (Table 2, entry b, d). In all the cases the reactions were equally smooth and furnished corresponding β-phosphonomalononitrile in excellent yield. In an attempt to broaden the scope of this protocol, studies were also performed using ethyl cyanoacetate, diethyl malonate and phenylsulfonyl acetonitrile as other active methylene components. Interestingly, with the choice of ethyl cyanoacetate alone, the reaction proceeded smoothly to furnish diastereoisomeric mixture of desired phosphonocyanomalonate (6q, Table 2, dr = 60:40, 1H NMR) while the reactions using diethyl malonate or phenylsulfonyl acetonitrile yielded a mixture of products.
Effect of catalyst in one-pot synthesis of β-phosphonomalononitrile, 6e.
| No. | Catalyst | Time | Yield (%)a | ||
| (mol %/mgm) | (h) | 3 | 5 | 6 | |
| 1 | – | 24 | 40 | 40 | 20 |
| 2 | K3PO4 (10) | 12 | – | 50 | 50 |
| 3 | K3PO4 (20) | 12 | – | 50 | 50 |
| 4 | Borax (200 mg) | 12 | 20 | – | 70 |
| 5 | MgO (200) | 10 | 20 | 20 | 60 |
| 6 | Amberlite (OH−, 200 mg) | 16 | 20 | 80 | – |
| 7 | PVP (100 mg) | 16 | 20 | 70 | – |
| 8 | DBU (10) | 30b | 80 | 20 | – |
| 9 | DABCO (10) | 30b | 80 | 20 | – |
| 10 | DMAPc | 2 | 40 | 20 | 40 |
| 11 | Pyrrolidine (10) | 30b | – | – | 90 |
| 12 | Piperidine (10) | 30b | – | – | 90 |
| 13 | Diethylamine (10) | 15b | – | – | 95 |
a Yields based upon TLC comparison with authentic samples.
b Time in minutes.
c Dimethylamino pyridine.
Diethyl amine catalyzed one-pot synthesis of β-phosphonomalononitriles.
| Entry | Product (6) | Time (min) | Yield (%) | Ref. |
| a | 10 | 96 | [18c] | |
| b | R = CH3 | 10 | 94 | – |
| c | R = C2H5 | 15 | 90 | [17d] |
| d | R = HC(CH3)2 | 15 | 93 | – |
| e | 15 | 95 | [18a,18c] | |
| f | 10 | 95 | [17d,18c] | |
| g | 10 | 93 | – | |
| h | 20 | 94 | – | |
| i | 15 | 96 | – | |
| j | 15 | 90 | [17f] | |
| k | 20 | 95 | [17b] | |
| l | 15 | 93 | [17e] | |
| m | 30 | 84a | – | |
| n | 20 | 90 | [17a] | |
| o | 30 | 87 | [17a,b] | |
| p | 3b | 30 | [17a] | |
| q | 15 | 95c | [17b] |
a Mixture of products.
b Time in hours.
c dr = 60:40 (1H NMR).
As regards the one-pot synthesis of β-phosphonomalononitriles, an interesting observation is noteworthy. The catalyst, diethylamine, which was found to be highly efficient to furnish β-phosphonomalononitrile following the multicomponent pathway (Scheme 1b), was unable to drive sequential, Knoevenagel-phospha-Michael reaction to completion (Scheme 1c). At this stage of our work, we do not have any concrete answer to explain this anomaly. However, with reference to our recent report on catalyst as well as solvent-free protocol for Michael addition of anilines to enones [22] and our current experiences in the synthesis of β-phosphonomalononitriles, we believe that this anomaly is possibly concerned with the difference in the amount of heat liberated in following the two different pathways. It could be shown very easily that, under solvent-free condition, the heat released upon addition of diethylamine to a mixture of aldehyde, malononitrile and diethyl phosphite is much larger than that released upon addition of diethyl phosphite to a stirred mixture of Knoevenagel condensation product and diethylamine. In short, the heat released during this MCC reaction might be sufficient enough to drive this tandem Knoevenagel-phospha-Michael reaction to completion.
Following this initial success in the development of a one-pot protocol for the synthesis of β-phosphonomalononitriles, it was planned to explore the applicability of the developed protocol towards designing of molecular frame work in a few biologically active compounds. In this context, our attention became focused on the synthesis of 2-amino-4H-chromenes (Fig. 1).

Structures of a few biologically active 2-amino-4H-chromene derivatives.
The chromene moiety often appears as an important structural motif in many natural products while chromene pharmacophore can serve as a useful building block in the generation of a variety of natural products with important biological activities [23]. Among different types of chromenes, 2-amino-4H-chromene derivatives constitute an important class of heterocycles customarily used in cosmetics, pigments and biodegradable agrochemicals [24]. They are also known to exhibit important biological activities such as antimicrobial, antiviral, antitumor, antihypertensive anti-ischemic behavior, etc. [25–27]. With the rapid progress both in chemistry as well as biology of 2-amino-4H-chromenes, development of a protocol meeting to the demands of an “ideal synthesis” [28] is highly desirable. It is worth mentioning that, amongst various 2-amino-4H-chromene derivatives, synthesis of (2-amino-3-cyano-4H-chromen-4-yl) phosphonic acid diethyl ester derivatives, 11, has been reported earlier using InCl3 [29a], potassium phosphate [29b], β-cyclodextrin [29c] as well as ethylenediaminediacetate [18c] as catalysts. In light of the developed protocol for the expeditious synthesis of β-phosphonomalononitriles, we envisaged that, diethyl amine catalyzed three component condensation between ortho-hydroxy benzaldehyde, malononitrile and diethyl phosphite would easily furnish diethyl (2-amino-3-cyano-4H-chromen-4-yl) phosphonate, 11a, following one of the paths depicted in Scheme 2.
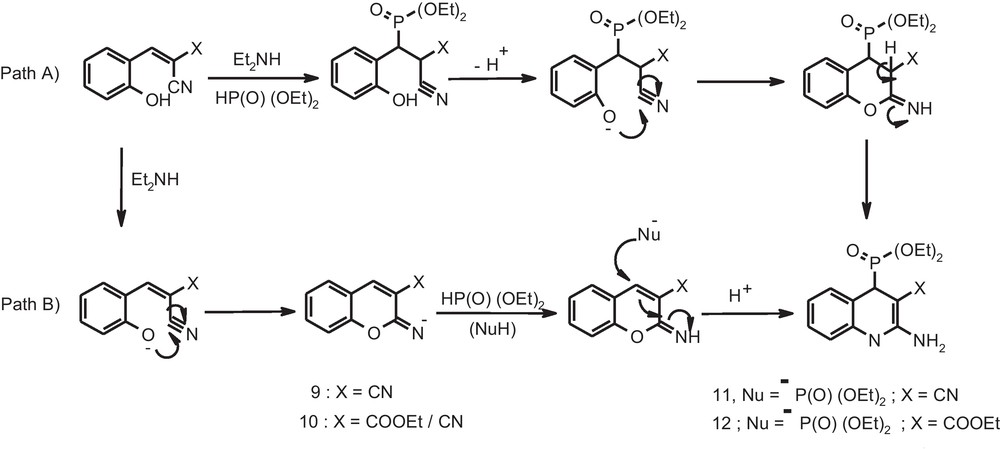
Plausible mechanism for the formation of (2-amino-3-cyano-4H-chromen-4-yl) phosphonic acid esters.
In a template reaction, salicylaldehyde, malononitrile and diethyl phosphite were stirred together at ambient temperature in presence of diethylamine (10 mol %) as catalyst. It was truly gratifying to notice that the resultant product in excellent yield was the desired chromene derivative, 11a (Table 3, Scheme 3).
Diethyl amine catalyzed synthesis of (2-amino-3- cyanochromen-4-yl) phosphonic acid diethyl esters, 11a-h.
| Entry | Product (11) | Time (h) | Yield (%)a | M.P (° C) (Rep) ref./Ods. |
| a | 2 | 95 | (140–142) [29a]/139–140 | |
| b | 6 | 92 | 150–152 | |
| c | 2.5 | 94 | (172–174) [29b]/172–174 | |
| d | 2 | 95 | (180–82) [29a]/180–182 | |
| e | 2.5 | 93 | (198–200) [18c]/194–197 | |
| f | 2 | 95 | (213–215) [18c]/216–218 | |
| g | 2 | 90 | (160–162) [18c]/161–164 | |
| h | 2 | 92 | (180–182) [18c]/178–182 |
a Salicylaldehyde, malononitrile and diethyl phosphite (2 mmol, each) ethanol (3 mL), diethylamine (10 mol %), rt.

Diethylamine catalyzed synthesis of (2-amino-3-cyanochromen-4-yl) phosphonic acid diethyl esters, 11.
This initial success prompted us to explore the potential of this protocol in the synthesis of various 2-amino-4H-chromene derivatives. Accordingly, the reactions were performed between various substituted salicylaldehydes, malononitrile and diethyl phosphite. The attempted reactions under solvent-free conditions were slightly exothermic and failed to furnish corresponding 2-amino-4H-chromene derivatives in high yields as well as purity. However, with the choice of ethanol as an ecofriendly reaction medium, the reactions with all the substituted salicylaldehydes proceeded smoothly (Table 3). To expand the scope of the developed protocol, malononitrile component in above reaction was then replaced by ethyl cyanoacetate. However, the reaction product was identified to be a mixture (TLC) and modification of the reaction conditions as regards change in the reaction temperature as well as solvent was not found to be beneficial to obtain the desired phosphonate ester, 12, (Scheme 2) as a sole product. Interestingly, when the diethyl phosphite component in above reaction was replaced with triethyl phosphite, the reaction did not go to completion.
3 Conclusion
In conclusion, we have described for the first time, the use of diethylamine as a highly efficient organocatalyst in a one-pot, multicomponent synthesis of β-phosphonomalononitriles at ambient temperature. Usability of the same catalyst in synthesis of diethyl (2-amino-3-cyano-4H-chromen-4-yl) phosphonic acid esters has also been described. Easy commercial availability of non-toxic catalyst at extremely low cost, ambient temperature, excellent yields, operational simplicity, easy work-up procedures have made this greener protocol a user friendly alternative to the exiting ones.
In continuation of these studies, diethylamine catalyzed synthesis of other chromene derivatives B-F (Fig. 1) has also been accomplished and those results will be reported separately.
4 Experimental
Dialkyl phosphites, aldehydes (Aldrich), potassium phosphate (Lancaster), diethylamine and malononitrile (SD Fine Chemicals, Mumbai) were used as received. Melting points were recorded in open capillaries using Kumar melting point apparatus. IR spectra were recorded as KBr discs or as neat, using Perkin Elmer Spectrum-1 IR spectrometer. 1H (300 MHz) and 13C (75.4 MHz) NMR spectra were recorded as CDCl3 solutions using TMS as internal standard on Bruker Avance II spectrometer. GCMS spectra were recorded using Shimatzu-QP 2010 analyzer.
4.1 Typical experimental procedure
4.1.1 Synthesis of β-phosphonomalononitriles, 6a–o
To an equimolar and well stirred mixture of aldehyde, diethyl phosphite and malononitrile (2 mmol, each) was added diethyl amine (10 mol %) and stirring continued. Upon completion of reaction, (tlc) water (10 mL) was added and the reaction mixture was extracted using ethyl acetate. The organic extract was washed with water, dried over anhydrous sodium sulphate and solvent removed. The resultant oil was chromatographed over silica-gel. Elution with hexane-ethyl acetate (90:10) furnished the desired β-phosphonomalononitrile in excellent yield.
4.1.2 Synthesis of (2-amino-3-cyano-chromene-4-yl) phosphonic acid diethyl ester derivatives, 11a–g
To a well stirred solution of salicylaldehyde, diethyl phosphite and malononitrile (2 mmol, each) in ethanol (3 mL) was added diethyl amine (10 mol %) and stirring continued. Upon completion of the reaction, (tlc) water (10 mL) was added and stirring continued till a free flowing colourless solid was obtained. It was filtered, washed successively with water, n-hexane and dried in air.
4.2 Spectral data of the representative compounds
[1-(4-Methylphenyl)-2,2-dicyanoethyl] phosphonic acid dimethyl ester; (6b): IR (CHCl3): 2987, 2197, 1648, 1256, 1233, 1043 cm−1; 1H NMR (300 MHz, CDCl3): δ = 2.4 (s, 3H), 3.53 (d, 3H, 2JHP = 10.5 Hz), 3.82 (d, 3H, 2JHP = 10.8 Hz), 3.64 (d, 1H, J = 10.8 Hz), 4.46 (t, 1H, J = 10.5 Hz), 7.23-7.34 (m, 2H), 7.36 (m, 2H); 13C NMR (75.4 MHz, CDCl3): δ = 21.26, 25.08, 44.12 (d, 1JCP = 145.14 Hz, CH), 53.30 (d, 2JCP = 7.38 Hz, OCH3), 54.55 (d, 2JCP = 6.93 Hz, OCH3),110.88, 111.06, 111.20, 126.97, 127.05, 129.09, 129.17, 129.91, 130.14, 139.45; GCMS: m/z = 294, 278, 213, 167, 115, 110, 93, 79, 63.
[1-(4-Methylphenyl)-2,2-dicyanoethyl] phosphonic acid di-iso-propyl ester; (6d): IR (CHCl3): 2984, 2256, 2206, 1632, 1381, 1233, 1027 cm−1; 1H NMR (300 MHz, CDCl3): δ = 0.87 (d, 3H, J = 6.6 Hz), 1.29 (d, 3H, J = 6.3 Hz), 1.36 (dd, 6H, J = 6.1 & 2.7 Hz), 2.39 (s, 3H), 3.44 (dd, 1H, 2JHP = 18.3 Hz, 3JHH = 8.4 Hz), 4.38-4.50 (m, 2H), 4.8-4.84 (m, 1H), 7.19–7.26 (m, 2H), 7.31–7.37 (m, 2H); 13C NMR (75.4 MHz, CDCl3): δ = 21.25, 22.90 (d, 3JCP = 6.5 Hz, CH3), 23.80 (d, 3JCP = 5.4 Hz, CH3), 23.98 (d, 3JCP = 3.7 Hz, CH3), 24.36 (d, 3JCP = 3.6 Hz, CH3), 25.70 (CH), 44.78 (d, 1JCP = 145.2 Hz, CH), 72.29 (d, 2JCP = 7.5 Hz, CH), 73.41 (2JCP = 7.5 Hz, CH), 111.04 (d, 3JCP = 12.8 Hz, CN), 111.32 (d, 3JCP = 9.8 Hz, CN), 127.56, 127.63, 129.29, 129.37, 129.95, 129.97, 139.19, 139.22 ppm; GCMS: m/z = 334, 277, 250, 187, 143.
[1-(3,4-Dimethoxyphenyl)-2,2-dicyanoethyl] phosphonic acid diethyl ester; (6g): IR (CHCl3): 2945, 2833, 2221, 1267, 1032 cm−1; 1H NMR (300 MHz, CDCl3): δ = 1.17 (t, 3H, J = 7.2 Hz), 1.38 (t, 3H, J = 7.2 Hz), 3.48 (dd, 1H, 2JHP = 21.6 Hz, 3JHH = 7.8 Hz), 3.74–3.82 (m, 1H), 3.90 (s, 3H), 3.91 (s, 3H), 3.97–4.07 (m, 1H), 4.12–4.24 (m, 2H), 4.44 (dd, 1H, 3JHP = 9.0 Hz and 3JHH = 7.8 Hz), 6.90 (d, 1H, J = 8.4 Hz and 2.1 Hz), 6.97–7.04 (m, 2H); 13C NMR (75.4 MHz, CDCl3): δ = 16.19 (d, 3JCP = 3.8 Hz, CH3), 16.28 (d, 3JCP = 3.8 Hz, CH3), 25.80 (CH), 44.23 (d, 1JCP = 144.7 Hz, CH), 55.72, 55.90, 63.14 (d, 2JCP = 6.7 Hz, CH2), 64.24 (d, 2JCP = 6.7 Hz, CH2), 111.12 (d, 3JCP = 10.5 Hz, CN), 111.40 (d, 3JCP = 10.5 Hz, CN), 111.97, 112.04, 122.10, 122.15, 122.20, 149.46, 149.97, 149.99 ppm; GCMS: m/z = 352, 287, 259, 214, 177, 151, 138, 121.
[1-(3-Nitrophenyl)-2,2-dicyanoethyl] phosphonic acid diethyl ester, (6 h): IR (CHCl3): 2944, 2833, 2205, 1537, 1354, 1251, 1024 cm−1; 1H NMR (300 MHz, CDCl3): δ = 1.26 (t, 3H, J = 7.2 Hz), 1.37 (t, 3H, J = 6.9 Hz), 3.71 (dd, 1H, 2JHP = 21.6 Hz, 3JHH = 7.5 Hz), 3.95–4.27 (m, 4H), 4.58 (t, 1H, 3JHP = 3JHH = 7.8 Hz), 7.69 (t, 1H, J = 7.8 Hz), 7.87 (dd, 1H, J = 7.5 and 0.9 Hz), 8.32–8.37 (m, 2H); 13C NMR (75.4 MHz, CDCl3): δ = 16.09 (d, 3JCP = 5.2 Hz, CH3), 16.18 (d, 3JCP = 6.0 Hz, CH3), 25.07 (CH), 43.45 (d, 1JCP = 143.1 Hz, CH), 63.80 (d, 2JCP = 6.7 Hz, CH2), 64.35 (d, 2JCP = 6.7 Hz, CH2), 111.20 (d, 3JCP = 9.8 Hz, CN), 111.30 (d, 3JCP = 11.3 Hz, CN), 124.21, 124.58, 124.67, 130.33, 133.09, 133.17, 135.37, 135.45, 148.46 ppm; GCMS: m/z = 337, 291, 183, 174, 153, 128, 109, 91.
[1-(4-iso-propylphenyl)-2,2-dicyanoethyl] phosphonic acid diethyl ester, (6i): IR (CHCl3): 2944, 2833, 2220, 1246,1028 cm−1, 1H NMR (300 MHz, CDCl3): δ = 1.11 (t, 3H, J = 6.9 Hz), 1.28 (d, 6H, J = 6.9 Hz), 1.37 (t, 3H, J = 7.2 Hz), 2.94 (septet, 1H, J = 7.2 Hz), 3.53 (dd, 1H, 2JHP = 21.8 Hz, 3JHH = 8.4 Hz), 3.70- 3.78 (m, 1H), 3.95–4.04 (m, 1H), 4.10–4.23 (m, 2H), 4.48 (t, 1H, 3JHP = 8.4 Hz, 3JHH = 8.1 Hz), 7.27 (d, 2H, J = 8.4 Hz), 7.37 (dd, 2H, J = 8.4 & 1.4 Hz); 13C NMR(CDCl3, 75.4 MHz): δ = 16.04 (d, 3JCP = 5.4 Hz, CH3), 16.23 (d, 3JCP = 5.8 Hz, CH3), 23.84 (CH3) 25. 46 (CH), 44.44 (d, 1JCP = 144.7 Hz), 63.08 (d, 2JCP = 6 Hz, CH2), 64.14 (d, 2JCP = 6.7 Hz, CH2), 110.95 (d, 3JCP = 12.7 Hz, CN), 111. 14 (d, 3JCP = 10.6 Hz, CN), 127.38, 127.40, 127.49, 127.57, 129.18, 129.28, 150.16, 150.20 ppm; GCMS: m/z = 334, 319, 291, 269, 241, 197,171, 156, 138, 111, 91.
[1-(2-Thiophenyl)-2,2-dicyanoethyl] phosphonic acid diethyl ester, (6 n): IR (CHCl3): 2944, 2216, 1247, 1026, 713 cm−1; 1H NMR (300 MHz, CDCl3): δ = 1.26 (t, 3H, J = 7.2 Hz), 1.38 (t, 3H, J = 7.2 Hz), 3.87 (dd, 1H, 2JHP = 21.9 Hz, 3JHH = 6.6 Hz), 3.92–4.25 (m, 4H), 4.46 (dd, 1H, 3JHP = 8.4 Hz, 3JHH = 6.9 Hz), 7.10 (dd, 1H, J = 4.8 & 3.9 Hz), 7.33 (t, 1H, J = 2.4 Hz), 7.39 (d, 1H, J = 5.1 Hz); 13C NMR (75.4 MHz, CDCl3): δ = 16.18 (d, 3JCP = 3.7 Hz, CH3), 16.25 (d, 3JCP = 3.7 Hz, CH3), 26.53 (CH), 39.98 (d, 1JCP = 146 Hz, CH), 63.60 (d, 2JCP = 6.7 Hz, CH2), 64.44 (d, 2JCP = 6.7 Hz, CH2), 110.79 (d, 3JCP = 10.5 Hz, CN), 110.91 (d, 3JCP = 10.5 Hz, CN), 125.67, 127.23, 127.26, 129.29, 129.39, 130.61, 130.70 ppm; GCMS: m/z = 298, 233, 205, 190, 162, 138, 123, 111.
[1-(Cyclohexyl)-2,2-dicyanoethyl] phosphonic acid diethyl ester, (6o): IR (CHCl3): 2933, 2255, 1650, 1450, 1240, 1022, 971 cm−1; 1H NMR (300 MHz, CDCl3): δ = 1.25-1.42 (m, 11H), 1.70–2.24 (m, 7H), 4.15–4.26 (m, 4H), 4.37 (dd, 1H, 2JHP = 14.7 Hz, 3JHH = 3.7 Hz); 13C NMR (75.4 MHz, CDCl3): δ = 16.30 (d, 3JCP = 6.0 Hz, CH3), 16.36 (d, 3JCP = 6.0 Hz, CH3), 21.81, 25.68, 26.19, 26.28 (CH2), 31.14, 31.21, 31.37, 31.47, 37.59, 37.63 (CH), 44.19 (d, 1JCP = 140.2 Hz, CH), 62.83 (d, 2JCP = 7.5 Hz, CH2), 63.05 (d, 2JCP = 7.5 Hz, CH2), 111.27, 111.32, 112.08, 112.28 ppm; GCMS: m/z = 298, 258, 233, 190, 177, 162, 138, 123, 111, 97.
(2-Amino-6-chloro-3-cyano-4H-chromene-4-yl) phosphonic acid diethyl ester, (11b): Colourless solid; mp = 150–152 °C; IR (KBr) = 3349, 3153, 2189, 1654, 1419, 1244, 1037, 973 cm−1; 1H NMR (300 MHz, CDCl3): δ = 1.27 (t, 3H, J = 7.2 Hz), 1.36 (t, 3H, J = 7.2 Hz), 3.83 (d, 1H, 1JHP = 18.5 Hz), 4.03-4.18 (m, 4H), 5.00 (brs, 2H), 6.94 (d, 1H, J = 8.7 Hz), 7.24 (dt, 1H, J = 8.7 Hz & 2.4 Hz), 7.32 (t, 1H, J = 2.4 Hz); 13C NMR (75.4 MHz, CDCl3): δ = 16.41 (d, 3JHP = 5.2 Hz, CH3), 16.48 (d, 3JHP = 5.5 Hz, CH3), 35.35 (d, 1JCP = 147.7 Hz, CH), 63.12 (d, 2JCP = 7.5 Hz, CH2), 63.38 (d, 2JCP = 7.5 Hz, CH2), 117.70, 117.74, 118.43, 128.99, 129.03, 129.23, 129.29, 148.38, 148.45, 161.64, 164.68 ppm; HRMS: Mass Calcd for (C14H16O4N2PCl + Na): 365.0434; Obs. mass: 365.0465.
Acknowledgement
The authors (UVD and MAK) are grateful to UGC, New Delhi for financial assistance (F. No. 34-362/2008) as well as for a Junior Research Fellowship, respectively. We are also thankful to DST and UGC, New Delhi for financial assistance under DST-FIST and UGC-SAP programme, respectively. We are sincerely grateful to Prof. A. Srikrishna (IISc, Bangalore) for fruitful discussions and timely help.