

1 Introduction
In order to design a drug delivery system (DDS), various kinds of high-performance carrier materials are being developed to deliver the necessary amount of drug to the targeted site for a necessary period of time, both efficiently and precisely. Cyclodextrins (CDs) are potential candidates for such a role because of their ability to alter physical, chemical and biological properties of guest molecules through the formation of inclusion complexes in both solution and solid state [1,2].
Cyclodextrins (CDs) [3] are cyclic oligosaccharides made up of glucopyranose units bonded together via a (1, 4)-linkages. The most common CDs are α, β and γ CD, containing six, seven and eight units, respectively. In particular, β-cyclodextrin (β-CD) has an internal cavity shaped like a truncated cone of about 8 Å deep and 6.0–6.4 Å in diameter. This cavity possesses a relatively low polarity that can accommodate guest organic molecules inside. Due to this configuration the CDs are able to form inclusion complexes with various molecules and ions [4].
The main applications of natural CDs are to enhance solubility, stability and bioavailability of the drug molecules; furthermore, the enhancement of drug activity, selective transfer and/or reduction of side effects can be achieved by means of host-guest complexation [5–7]. Drugs used in the fight against cancer can be divided into four categories according to their mechanism of action:
- • alkylating agents;
- • RNA/DNA antimetabolites;
- • antimitotic agents;
- • inhibitors of topoisomerases I and II.
2-Chloroethylnitrosoureas (CENU) are an important family of alkylating agents with a broad spectrum of activity [8,9]. However, their contribution to cancer chemotherapy is limited by their toxic side effects, which are related to the formation of carbamoylating species (isocyanate) during their decomposition [10,11]. 2-Chloroethylnitrososulfamides (CENS) constitute a new family of oncostatic agents structurally related to 2-chloroethylnitrosoureas, but devoid of any carbamoylating activity. Promising agents prepared on this basis have demonstrated interesting cytotoxic activity, and among them, some proved to be considerably more potent than the parent nitrosourea [12–19]. To progress to clinical evaluation, additional data on the stability and mechanism of decomposition of CENS are needed. In order to gain a better understanding of the stability of this family of compounds, the first experimental investigation was carried out on the fragmentation of CENS in aqueous media and in serum using HPLC on-line solid phase extraction, LC–MS and conventional UV spectrophotometry [20,21].
Different investigations of the complexation between CENS and β-CD and modified β-CD were carried out following two different procedures, in both the liquid phase and the solid state. These complexes can increase the stability and improve the solubility and bioavailability of the substance and modify the pharmacokinetics of the resulting drugs with a subsequent reduction in adverse effects [22,23].
Moreover, when the guest is an aromatic compound, which has a strong absorption band in the UV-Vis region, spectroscopic methods can be used to determine the stability constants in the solution [24,25]. The changes in the absorbance of the guest (employed so far) in the presence of increasing concentrations of the host have been elaborated using the Benesi-Hildebrand linear model for a 1:1 complex [22].
The aim of the present work is to make a qualitative study of the interaction of N-nitroso, N-(2-chloroethyl), N′, N′-dibenzylsulfamid (CENS-Dibenz) with β-CD using spectroscopic as well as theoretical techniques. In the experimental part, the stoichiometry and stability constant of the inclusion complex are investigated by the fluorescence method. 1H NMR spectroscopy was also used to study inclusion complex. In the second part, we performed a detailed theoretical study focused on the modeling of the molecular interactions of β-CD molecule with this anticancer drug and the evaluation of the interaction energies, the free energies of binding, and the analysis of the thermodynamic parameters in the host-guest inclusion complex. An intensive investigation of the 3D geometry of this complex in vacuum is carried out using molecular mechanics and semi-empirical quantum chemical calculations.
2 Experimental
Bidistilled water was used in all of the experiments. The investigated compound was synthesised according to the procedure described in the literature [12]. All chemicals used were of high purity (analytical grade). Fluorescence spectra were obtained by means of a Shimadzu spectrofluorophotometer (type RF-5301 PC) equipped with a xenon arc lamp. The fluorescence analysis was carried out at the ambient temperature.
The slit width for excitation and emission were both set to 5 nm. Each spectrum is an average of three consecutively acquired spectra. For the complexation, we employed a standard solution of CENS-Dibenz (1 × 10−6 mol.L−1) which was obtained by dilution of 10−2 mol.L−1 of synthesised N-nitroso, N-(2-chloroethyl), N′, N′-dibenzylsulfamid. For spectral analysis, all measurements of fluorescence were carried out at the maximum excitation wavelength of 265 nm and the maximum emission wavelength of 362 nm.
The β-CD solution was obtained by dilution of a suitable quantity of β-CD in bidistilled water; the concentration was in the range [10−2–10−4 M]. Solutions of the β-CD complexes were prepared by adding 5.0 mL of the stock solutions of CENS-Dibenz and 5.0 mL of the stock solutions of β-CD with various concentrations in a graduated flask of 10 ml then mixed by using an ultrasonic processor (type Geprufte Sicherheit UP 50H).
1H NMR (400 MHz) spectra were recorded on a Brucker DRX-400 spectrometer using DMSO-d6 as solvent.
3 Molecular modeling studies
Molecular modeling of the inclusion complex was carried out with the use of Hyperchem 7.51 [26] and Gaussian 03 W version 6.0 [27] package for molecular mechanics and quantum mechanics methods, respectively. The CENS-Dibenz structure is shown in Fig. 1.

Molecular structure of CENS-Dibenz: a: planar representation; b: optimized by PM3 with numbering of the atoms used in the present work.
The initial geometry of the CENS-Dibenz structure was built with the use the Hyperchem software. The geometry of the β-CD was obtained from its crystallographic structure as determined by X-ray diffraction, which is archived in the Cambridge databank.
In order to simulate the inclusion process, the glycosidic oxygen atoms of the β-CD were placed on the YZ plane and their center was defined as the center of the coordination system. The β-CD was then maintained in this position while the guest molecule was introduced along the X-axis into the cavity of the β-CD.
The bond 2526 of the guest coincides with X-axis and the relative position between the host and the guest molecule was measured by the distance along the X-axis (Fig. 2) of the CENS-Dibenz with the sulfur atom to the coordinate center. Inclusion was emulated by manual displacement of the guest molecule from −7 to +7 Å, with 1 Å steps. At each step the geometry of the complex was entirely optimized by using the Allinger MM+ (with the Polak-Ribiere conjugate gradient algorithm) force field, which is well-known to minimize the energy of structures with a gradient of 0.01 Kcal/mol root mean square (RMS).
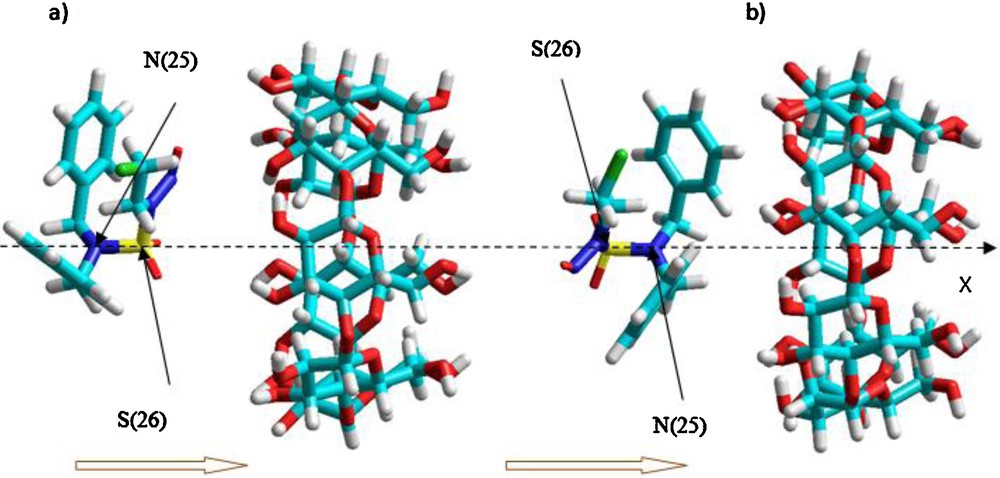
Procedure of inclusion of CENS-Dibenz into β-CD according to both orientations: a: orientation ‘A’; b: orientation ‘B’. The horizontal arrows indicate the direction of displacement of the guest molecule during the complexation process. The bond 2526 coincide with X-axis and sulfur atom labeled 26 represents the atom of reference.
The following nomenclature will be used to interpret the computational results obtained in the present work. The two different orientations in which the CENS-Dibenz can be introduced into β-CD, i.e., the one in which the NO group is oriented towards the wider rim of the β-CD and the other in which both phenylic cycles are oriented towards the largest rim of the β-CD will be referred to as complex A and complex B, respectively (Fig. 2). The method that was used to locate the structure with the lowest energy was reported in references [28,29].
The structure generated at each step is then optimized by allowing the initial conformations to change while keeping movement of the reference atom and of the β-CD completely restricted. Once the preliminary minimum level of energy was determined for each orientation, the system was re-optimized without the constraints.
In order to explore more conformational space and to find a more stable structure of the complex, the CENS-Dibenz was rotated in the cavity around the X-axis with 20 degree intervals from 0 to 360 degrees and the system was re-optimized at each position without imposing any restrictions.
After this initial procedure, the energy minimization was carried out without constraints by using the PM3 semi-empirical method. Thus, the PM3 semi-empirical method proved to be a powerful tool in the study of the conformation of cyclodextrin complexes and its performance is better than the Austin (AM1) method for biochemical systems due to its ability to better describe the interactions between non-bonded atoms, i.e. hydrogen bonds and steric effects [30].
This method has a high computational efficiency in calculating the CDs systems and its precision is comparable to that of ab-initio calculations with medium-sized basis sets [31,32]. It was selected for the study of the inclusion process of the β-CD with CENS-Dibenz in the present article.
Frequency calculations, using PM3, were also performed to confirm the completeness of the optimization, and no negative eigenvalues (eigenvalues of the Hessian matrix) were found for the final structures. Thus, for the equilibrium geometries optimized with the PM3 method in the case of our complexes, DFT single point calculations at the B3LYP/6-31G(d), MPW1PW91/6-31G(d) levels and Hartree-fock HF/6-31G(d) were carried out in the vacuum.
Finally, and in order to carry out an approach on the ideal geometry of the molecule with a superior level of theory, the system was subjected to a full optimization by using a hybrid approach developed by Morokuma and others [33], called the ONIOM method, which means “Our own N-layer Integrated molecular Orbital and molecular Mechanics”. This hybrid method allows a partition of the molecular system into two, three or more layers, and distributes the computational methods (MO or MM) among these layers. In the terminology of Morokuma [34,35], the full system is called “real” and is treated with the two low and high levels given by the theory. Total energy EONIOM is then expressed by the equation below:
| (1) |
4 Results and discussion
4.1 Fluorescence response of the inclusion complex
When the organic molecules are included within a β-CD cavity in aqueous solution, the water is expelled. This strong hydrophobic interaction produces the stable inclusion complexes. The stability derives from the polar character of the guest molecule and the functional group that attaches to it. The molecules that are included in the host cavity may have an increase in fluorescence and modified spectra. This type of interaction occurs with the molecules, which have a greater affinity for the hydrophobic interior of the β-CD molecule than for the aqueous phase [36].
Fluorescence spectra were performed to measure the stoichiometry and the stability constant between the CENS-Dibenz and the β-CD. Fig. 3 shows the effect of the β-CD on the fluorescence spectra of the CENS-Dibenz in aqueous solution. A remarkable growth of the intensities of emission was observed with the increasing concentration of the β-CD. Such increase in the emission yields point to a penetration of the CENS-Dibenz in the host cavity and leading to the formation of the inclusion complex.
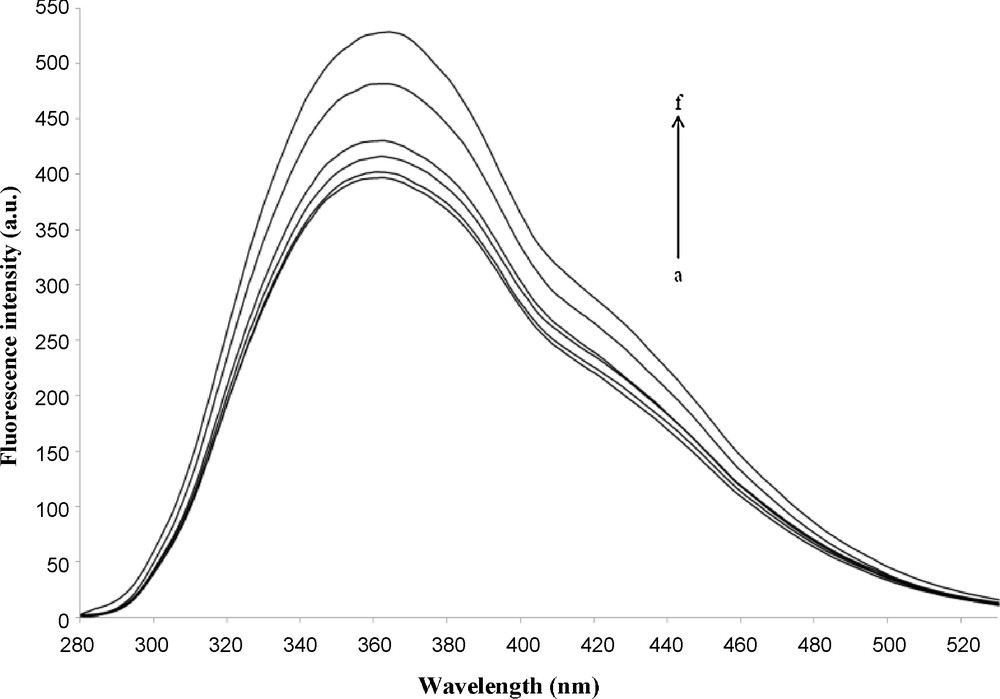
Fluorescence spectrum of CENS-Dibenz at different β-CD concentrations. [β-CD] (mol.L−1): a−f: 0, 10−4, 5 × 10−4, 10−3, 5 × 10−3, 10−2, [CENS-Dibenz] = 1 × 10−6 mol.L−1, λex = 265 nm.
The stoichiometry and the stability constant of the complex were established by means of the Benesi-Hildebrand method [24]. If we suppose that the β-CD forms an inclusion complex of 1:1 with CENS-Dibenz, Eq. (2) applies:
| (2) |
In this approach, a plot of 1/(F − F0) versus 1/[β-CD] is presented, where F is the fluorescence observed at each tested concentration, F0 is the fluorescence intensity of the analyte in the absence of the β-CD, and [β-CD] is the concentration of the β-CD. A linear plot is required for this double reciprocal plot in order to conclude a 1:1 stoichiometry. In the case where a 2:1 stoichiometry is predominant, the applicable equation is:
| (3) |
For a complex of 2:1, a straight line would be obtained when 1/(F − F0) is plotted versus 1/[β-CD]2.
Thus, the plot of 1/(F − F0) versus 1/[β-CD] displayed in Fig. 4 shows a good linearity (R = 0.99). This indicates the formation of the inclusion complex between the host and the guest with a stoichiometry of 1:1. The K constant for the guest-host inclusion complex obtained from the fluorescence spectroscopy was calculated using data of the Fig. 4 and proved to be equal to 417.63 M−1 at 25 °C. On the contrary, a downward concave curvature is obtained when the same data are fitted to a 2:1 complex, with the use of Eq. (3). This observation suggests that the stoichiometry of the complex is not 2:1.

Benesi-Hildebrand plot of 1/(F-F0) vs. 1/[β-CD] of CENS-Dibenz in the presence of β-Cyclodextrin at 25 °C. λex = 265 nm, λem = 362 nm.
4.2 1H NMR measurements
4.2.1 Formation of solid state inclusion complex
NMR studies are an important means for characterizing inclusion complex formed between cyclodextrin species and guest molecule [37] This spectrometry allows us to locate precisely the location of interaction between the host and the guest molecule (Fig. 5a). This is confirmed without ambiguity by comparing the changes in chemical shifts between free compound and compound accommodated in the cavity. Significant differences were observed in the spectra of the CENS-Dibenz/β-CD inclusion complex (Fig. 5b). We noticed, the appearance of a large band between 3.25–3.75 ppm due to the overlap of H3, H5 and the two H6 protons multiplets and the interactions between protons H3 and H5. In addition, the methylene protons CH2NNO and CH2Cl of CENS associated with cyclodextrin undergo a slight shift to low fields. Chemical shifts of the protons of CENS, free and after complexation with β-CD are:
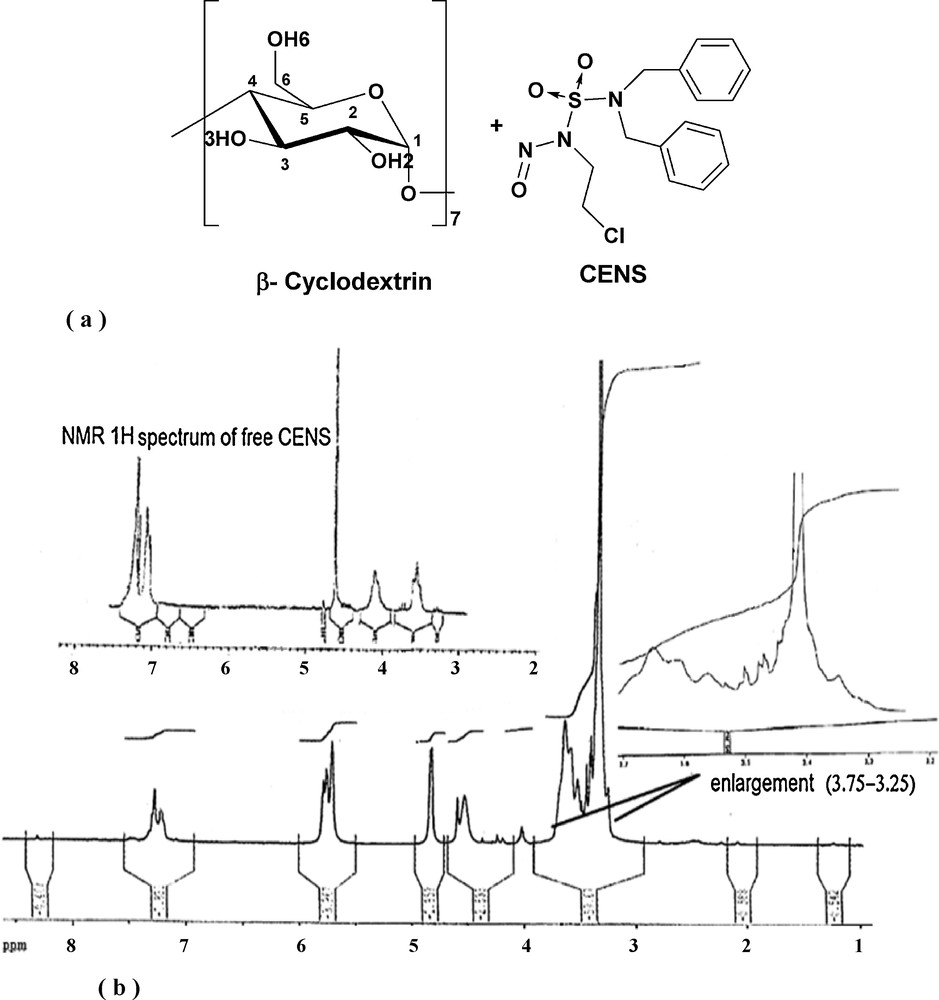
a: chemical structures of β-cyclodxtrin and CENS; b: 1H NMR spectrum of the CENS-Dibenz/β-CD inclusion complex.
CENS-Dibenz:1H NMR (400 MHz, DMSO-d6)
δ in ppm: 7.05–7.30 (m, 10H, 2ArH), 4.65 (s, 4H, 2CH2 Bn), 4.10 (t, 2H, CH2NNO), 3.55 (t, 2H, CH2Cl).β-Cyclodextrin 1H NMR (400 MHz, DMSO-d6) was taken from the reference [37].
CENS-Dibenz/β-CD inclusion complex: 1H NMR (400 MHz, DMSO-d6)
δ in ppm: 7.25–7.15 (m, 10H, 2Ar–H), 5.80–5.65 (m, 14H, OH3 + OH2 of β-CD), 4.80 (d, 7H, H1 of β-CD), 4.55 (s, 4H, CH2Bn), 4.50 (t, 7H, OH6 of β-CD), 4.05 (t, 2H, CH2NNO), 3.70–3.55 (m, 21H, (H6 (a, b) +H3) of β-CD), 3.50 (t, 2H, CH2Cl), 3.45–3.28 (m, 21H, (H5+ H4 + H2) of β-CD).
4.3 Molecular mechanical analysis
The energy change occurring with the formation of the CENS-Dibenz/β-CD complex defines the complexation energy Ecomplexation during the inclusion process and makes it possible to find the most stable structure between all the configurations under study. It can be calculated using Eq. (4):
| (4) |
Fig. 6 represents the variation of the complexation energy and its energetic contribution (van der Waals and electrostatic) at different positions x for both orientations ‘A’ and ‘B’ (Fig. 6 a and b). As can be observed, the complexation energy of the two orientations is negative at each distance ongoing from −7 Å to +7 Å, which indicates that the inclusion process of the CENS-Dibenz in the β-CD is thermodynamically favorable.

Complexation energy, van der Waals and electrostatic interactions for the CENS-Dibenz/β-CD complex versus coordinate X (Å) in vacuum, calculated by MM+ method: a: orientation ‘A’; b: orientation ‘B’.
For the ‘A’ orientation, the process of inclusion shows that at the starting point of docking (distance of −7 Å between the sulfur atom and the centre of mass of the β-CD), the complexation energy is −17.77 Kcal/mol. Then it decreases at a distance of 1 Å while one of the two aromatic rings is completely included in the cavity with a maximum value of the energy of complexation of −29.01 Kcal/mol. Beyond this point, which is defined as the minima of the Complexation, the complexation energy starts to increase and reaches a value of −15.65 Kcal/mol at the end of the docking process.
In the case of the ‘B’ orientation the minimum of energy is located between −3 Å and −2 Å. The energy values corresponding to these positions are −29.47 and −29.56 Kcal/mol, respectively. It is significant to note from the graph that the contribution of the van der Waals interactions to the total interaction energy is much larger than the contribution of the electrostatic interactions.
In order to find a much more stable structure of the complex, the two preceding structures having the minimum energy were re-optimized with the use of the circling process previously described in the computational methodology. At this stage of the MM+ optimization, all the constraints are removed from the system.
We also wanted to study the effect of solvation on the inclusion process. For this purpose studies were carried out by using the MM+ force field (with Polak-Ribiere algorithm and RMS gradient of 0.01 Kcal/mol) in a periodic box of dimensions 29.07 Å × 29.07 Å × 29.07 Å as implemented in Hyperchem 7.51.
Two other types of energy are investigated starting with the most stable structure. First, the energy of interaction between the host and the guest in the optimized geometries represented by the energetic term Einteraction and calculated by using the following formula:
| (5) |
| (6) |
| (7) |
Relevant theoretical results of the in vacuum and in water mechanic molecular MM+ of the most stable structures of CENS-Dibenz/β-CD complexes.
| Energetic terms | ‘A’ orientation | ‘B’ orientation | ||
| In vacuum | In water | In vacuum | In water | |
| Ecomplexation | −30.25 | −29.94 | −29.95 | −29.66 |
| −25.52 | −25.62 | −29.21 | −29.76 | |
| −3.02 | −2.73 | −0.98 | −0.90 | |
| Einteraction (CENS-Dibenz/β-CD) | −30.14 | −30.11 | −31.74 | −30.73 |
| Edeformation (CENS-Dibenz) | −2.73 | −3.46 | 0.97 | 0.41 |
| Edeformation (β-CD) | 2.62 | 3.63 | 0.82 | 0.65 |
For the case of vacuum, the molecular mechanics MM+ calculations show that the complex A is preferred by 0.3 Kcal/mol in comparison to the complex B. For the ‘B’ orientation, the two values of the total energy of complexation and of van der Waals (VDW) interaction are almost similar; this means that the forces responsible for the stability of the guest in the complex B are the van der Waals interactions.
On the other hand, for the complex A, the dominant driving forces for complexation are obviously van der Waals, with a weak electrostatic contribution. The corresponding values are, respectively, −25.52 and −3.02 Kcal/mol. Another observation of note for the complex B is that the energy of interaction between the guest molecule and the host is larger by 0.60 Kcal/mol compared to that obtained for the complex A.
In addition, the results of the investigation of the deformation energy demonstrate that the guest molecule in the ‘B’ orientation requires more energy than in the ‘A’ orientation for the adaptation of conformation inside the β-CD cavity. The corresponding values are respectively 0.97 and −2.73 Kcal/mol. The deformation energy of the β-CD during complexation is higher in the ‘A’ orientation than in the ‘B’ orientation.
Graphical output for calculations of the geometries of complexes A and B shows that the preferred orientation of these complexes is that in which the A aromatic cycle (Fig. 1) of the guest is inside the cavity of the β-CD leaving the nitroso, chloroethylic moieties and the B aromatic cycle in an external position (Fig. 7).
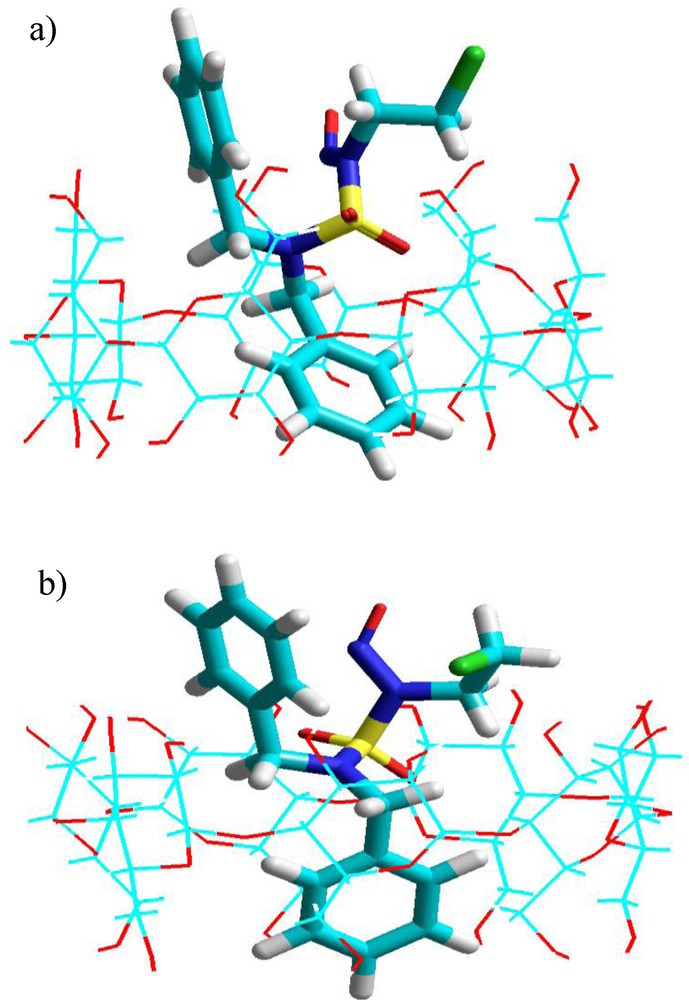
Energy-minimized (MM +) structures of the CENS-Dibenz/β-CD complex in the two distinct orientations studied in vacuum: a: ‘A’ orientation; b: ‘B’ orientation.
It is of note that for the complex A, the B aromatic cycle as well as the active groupings (the alkylating agent and the nitroso group) are directed outward through the narrow rim of the β-CD. The reverse is true for the complex B; these groupings are directed towards the wider rim of the host. Consequently, two modes of inclusion were considered according to the orientation of the components: nitrosochloroethyl-aromatic cycle B parts towards the primary alcohols groups (pag) (complex A) or towards the secondary alcohol groups (sag) (complex B). These results were corroborated by chemical analysis of analogous systems [38].
Regarding now the effect of solvation on the two complexes (Table 1); nearly the same results were obtained as those for the case of vacuum. Complexation in the presence of water is also thermodynamically favorable. It was noted that the sulfonyls groupings in the ‘B’ orientation form hydrogen bonds (O34OH) with the hydroxyls groupings of the water molecules. Thus, the main effect consists in the relative contribution of the electrostatic terms and van der Waals terms to the energy of complexation. For the ‘A’ orientation no bond with the solvent was detected.
Thus, although the structures were slightly destabilized in the aqueous solution, the energy of complexation was favored in the ‘A’ orientation and the energy difference of the complex in both orientations was 0.28 Kcal/mol. The same situation was reported in other inclusion complex cases [28,29,39]. The experimental thermodynamic values will be useful for rigorous numerical investigations, which will help draw conclusions concerning the effect of solvent on the binding of the complexation.
As was mentioned earlier, a significant characteristic of the geometry of the 1:1 complexes is that part of the guest is outside the host cavity. This suggests the possibility of the approaching of a second cyclodextrin to the previously formed 1:1 complex.
The process of 2:1 complexation was, however, emulated starting from the 1:1 stoichiometry structure of minimum binding energy (MBE) by approaching a second cyclodextrin. In this step, the center of the second cyclodextrin is always defined as the center of coordination while the guest molecule (complex A and complex B previously obtained from the 1:1 complexation) was introduced along the X-axis into the cavity of the second host. The secondary hydroxyl groupings of the β-cyclodextrin corresponding to each complex are oriented towards the wider rim of the second β-cyclodextrin (Head–Head).
The results indicate that the 2:1 complex (β-CD:CENS-Dibenz/β-CD) resulting from the complex A (‘A’ orientation) acquires a minimum of energy of complexation of −29.60 Kcal/mol which is slightly lower than that of the 1:1 complex (−30.25 Kcal/mol; Table 1). While the 2:1 complex resulting from the complex B (‘B’ orientation) acquires a minimal energy of complexation of −24.52 Kcal/mol. This value is far from that obtained for a stoichiometry of 1:1 (corresponding stoichiometry). Fig. 8 depicts the most stable structure for the β-CD:CENS-Dibenz/β-CD complex, showing intra- and inter- molecular hydrogen bonding between the two β-CDs.
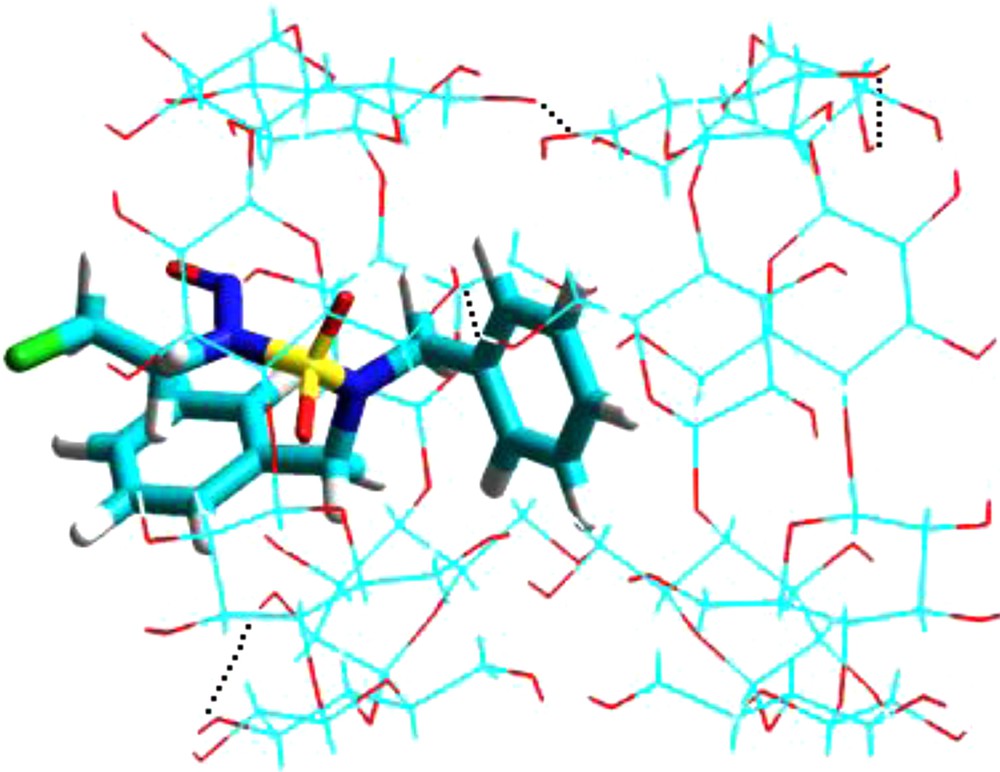
Energy-minimized (MM +) structure of 2:1 (β-CD: CENS-Dibenz/β-CD) complex studied in vacuum. Dotted lines are the hydrogen bonds.
This is in agreement with our experimental results, which confirm the presence of the majority of the complex with a stoichiometry of 1:1. However, when the CENS-Dibenz was used, we could not exclude the concomitant formation of other species with a 2:1 ratio, at least in small proportions. This can be attributed to the possible insertion of another group such as the aromatic moiety, which is comparable in size to the cyclodextrin cavity [22]. This suggestion is clearly confirmed by our preliminary theoretical calculations (Fig. 7).
Assuming a stoichiometry 1:1, it is thus difficult to choose between the preferential approaches (‘A’ or ‘B’ orientations) of the guest energies being almost similar. These results suggest that the MM+ calculations cannot establish the preferential ways about the guest penetrating the cavity of cyclodextrin and that these two orientations are two possible structures of the CENS-Dibenz/β-CD inclusion complex. In the present work it was decided to perform quantum mechanical calculations with more accurate nuclear and electronic energy terms describing the system. In this study, the equilibrium geometries of two complexes obtained previously by the MM+ were used as input for semi-empirical calculations using the PM3 Hamiltonian. All calculations, which follow, were carried out in vacuum.
4.4 Semi-empirical and quantum mechanical calculations
In our preliminary simulations, MM+ molecular mechanics calculations showed that there was no preference between the two orientations of the guest adapted in the complex with the β-CD in absence of solvent, i.e. in vacuum. Since, the docking energy is an approximation to obtain a more detailed energy description of the inclusion process. Consequently, we selected the PM3 semi-empirical quantum chemical method. The application of these semi-empirical calculations to the system under study represents a real computational challenge. Table 2 illustrates the results of the PM3 optimization for the ‘A’ and ‘B’ orientations.
Energies, thermodynamic parameters and HOMO-LUMO calculations using the PM3 method and single point energies evaluated at MPW1PM91, B3LYP and HF level for CENS-Dibenz/β-CD inclusion complexes.
| Energetic terms | CENS-Dibenz | β-CD | ‘A’ orientation | ‘B’ orientation |
| Ea | −5.31 | −1450.94 | −1474.82 | −1465.50 |
| Ecomplexation | −18.57 | −9.25 | ||
| Einteraction | −19.17 | −14.78 | ||
| Edeformation (CENS-Dibenz) | 2.36 | 1.78 | ||
| Edeformation (β-CD) | −1.75 | 3.75 | ||
| Eb HOMO (eV) | −9.89 | −10.53 | −9.68 | −9.78 |
| Ec LUMO (eV) | −1.27 | 1.47 | −1.16 | −1.54 |
| EHOMO-ELUMO gap (eV) | −8.62 | −12.00 | −8.52 | −8.24 |
| μd (Deby) | 4.15 | 9.35 | 11.27 | 9.08 |
| Hf | 209.30 | −658.82 | −466.85 | −459.07 |
| ΔH°e | −17.33 | −9.55 | ||
| Gf | 158.95 | −775.40 | −613.81 | −607.29 |
| ΔG°e | 2.64 | 9.15 | ||
| S (cal/mol.K) | 168.85 | 391.00 | 492.91 | 497.12 |
| ΔS°e (cal/mol.K) | −66.94 | −62.73 | ||
| HF/6-31G(d) | ||||
| Ea | −1,167,838.92 | −2,667,573.14 | −3,835,401.99 | −3,835,398.95 |
| Ecomplexation | 10.07 | 13.11 | ||
| MPW1PM91/6-31G(d) | ||||
| Ea | −1,172,465.46 | −2,682,031.77 | −3,854,499.63 | −3,854,496.26 |
| Ecomplexation | −2.40 | 0.97 | ||
| B3LYP/6-31G(d) | ||||
| Ea | −1,172,604.54 | −2,682,637.85 | −3,855,247.34 | −3,855,244.29 |
| Ecomplexation | −4.95 | −1.90 |
a E is the HF energy.
b Energy of the highest occupied molecular orbital.
c Energy of the lowest unoccupied molecular orbital.
d Dipole moment in Debye.
e ΔA°=Acomplex − (Aβ−CD + ACENS-Dibenz), A = H,G or S at P = 1 atm and T = 298.15 K.
The energy of complexation of the ‘A’ orientation is higher than that of the ‘B’ orientation of 9.32 Kcal/mol. This means that it is the structure A, which is more stable. Likewise, the interaction between the guest and the host in the ‘A’ orientation is higher than that obtained between the two partners guest-host in the ‘B’ orientation. The respective energy values are −19.17 and −14.78 Kcal/mol. With respect to deformation energy, and contrary to the results obtained by the MM+, the guest molecule requires more energy in complex A. In contrast, the deformation energy of the β-CD is more significant in the case of the B complex; this deformation of the β-CD appears to be one of the driving factors leading to a favored A complex. Similar results were obtained for the complexation of CENS-piperidine [40].
On the other hand, analysis of the molecular orbital energy diagram shows that the electronic structure of the guest in the CD is almost the same for the two different configurations of CENS-Dibenz. The value of the energy difference HOMO-LUMO turned out to be −8.52 e.V (complex A) which was almost similar to that for the complex B (−8.24 e.V). However, energy levels of the CENS inserted differently in the cavity of the β-CD were slightly shifted and this slight difference can lead to changes in the electronic properties (Table 2). In comparison to the guest and host in a free state, a significant increase in the polarity during the complexation of CENS-Dibenz in the β-CD was also noted. Fig. 9 shows the optimized molecular structures of the host-guest for the complexation of CENS-Dibenz with the β-CD in the two binding orientations obtained by PM3 calculations.
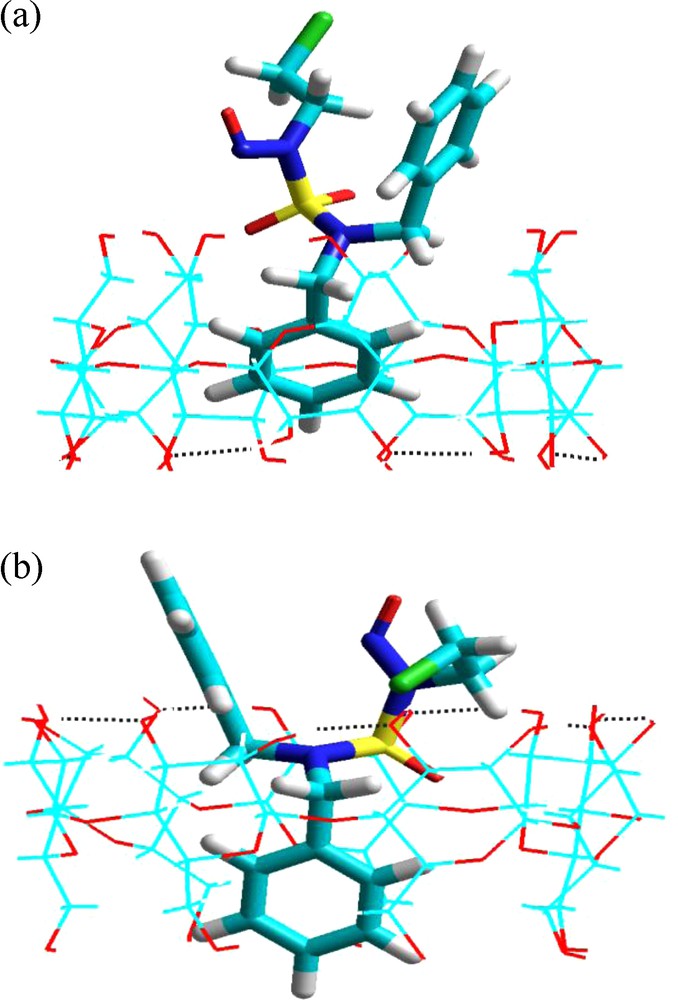
Energy-minimized (PM3) structures of CENS-Dibenz/β-CD complex in the two different orientations studied in vacuum: a: orientation ‘A’; b: orientation ‘B’. Dotted lines represent the hydrogen bonds.
It is noteworthy that the preferred mode of inclusion predicted by PM3 calculations performed in vacuum is the same as that predicted by the MM+ calculations and that one of the two aromatic cycles is inside the cavity while the other is outside in both orientations.
In Fig. 10, we summarize the computed infrared spectra of CENS-Dibenz, β-CD, complex A and complex B obtained by PM3 calculation. We can see that the IR spectra of the CENS-Dibenz/β-CD complex in the two different orientations include almost all main characteristic bands of free CENS-Dibenz and β-CD and no significant variations were observed in the absorption spectra corresponding to the both complexes which resulted from the overlapping of the simple spectra of the drug and β-CD. These results indicate that the host-guest interaction between CENS-Dibenz and β-CD is in an appropriate degree, which is enough to drive the CENS-Dibenz entering the cavity β-CD to form the stable inclusion complex, but at the same time the intrinsic properties of both the host and guest have no obvious changes.
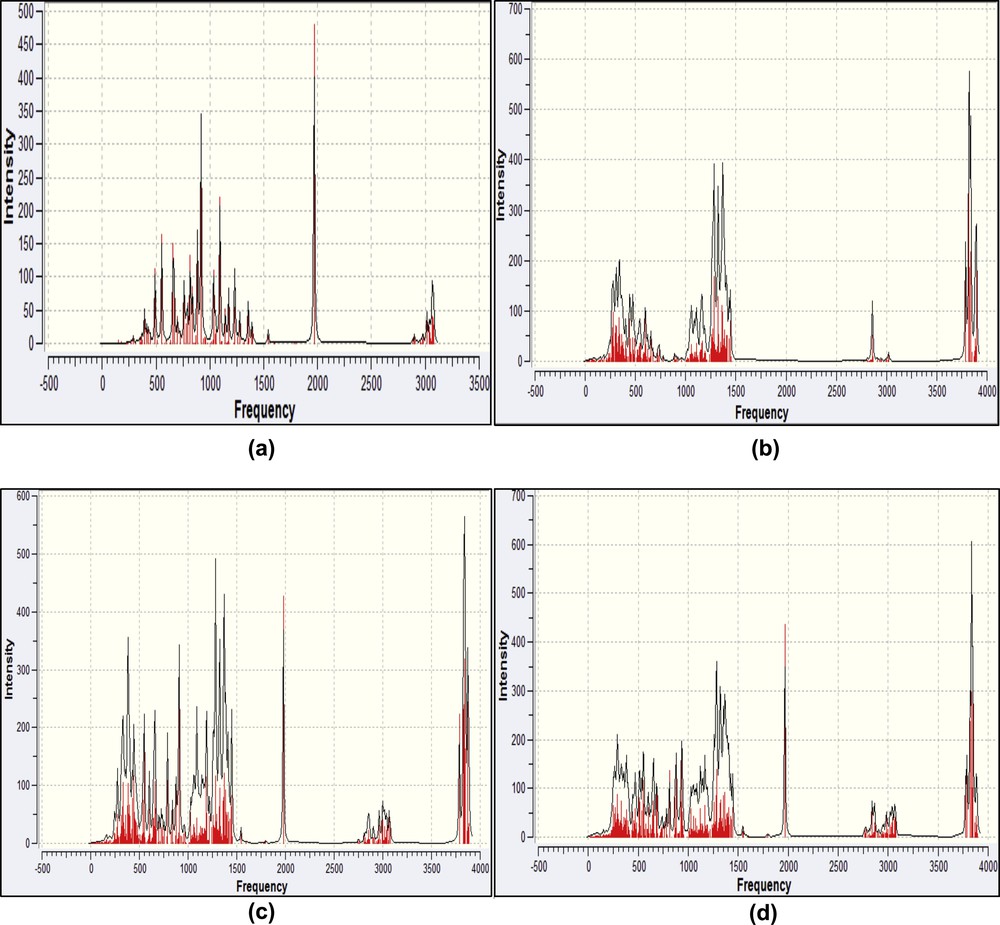
Calculated IR spectra of the isolated CENS-Dibenz (a) and β-CD (b), complex A (c) and complex B (d) by the PM3 calculation.
Most of the inclusion complexes of organic molecules with cyclodextrins for which the geometry has been investigated by NOESY/ROESY experiments and/or molecular modeling, showed unique relative orientation of the host and guest molecules in the NMR time scale.
The Specific compounds, however, can form inclusion complexes with an identical stoichiometry, but with different geometry; the phenomenon is called multimodal complexation. For instance, in a 1:1 complex of salbutamol with the β-CD either the aromatic ring of the guest molecule or its alkyl chain is located in the host cavity [41]. Lipoic acid in a complex with the β-CD also shows a bimodal complexation. One of its terminal groups, the carboxylic group, protrudes from either the wider side of the host cavity or of its narrow side. The alkyl linker is located inside the host cavity and the dithiolane ring on the opposite side to the carboxylic group [42].
Our results correspond to the results quoted previously, which are based on experiments and which confirm that it is the lipophilic part that is always inserted in the cavity of the host. The appropriate thermodynamic parameters for the complexation process, i.e. the standard free energy change (ΔG0), the standard enthalpy change (ΔH0), and the standard entropy change (ΔS0), were also performed at 298.15 k, at 1 atmosphere. The results are presented in the same Table.
From the theoretical results, it can be seen that the complexation reactions of the drug molecule with the β-CD are exothermic, as justified by the negative enthalpy changes. And the negative enthalpy changes suggest that the two processes of inclusion are enthalpically favorable in nature [32]. The enthalpy change for the ‘A’ orientation is more negative than for the ‘B’ orientation, which is surely attributed to the more tightly van der Waals interactions. The ΔS0 for the guest-host inclusion process turned out to be negative.
With regards to the difference of free energy ΔG0, this thermodynamic parameter is positive for the two complexes under study, indicating that a contribution of energy is necessary to form these complexes and that complexation hardly occurs spontaneously at ambient temperature. In addition, the stability constant K, of the complex formed at 25 °C is evaluated experimentally from the double reciprocal plot (experimental part), which allows a determination of the standard free energy change ΔG0 [43]. The free energy change can be obtained from the following formula:
This difference in results is justified by the fact that the solvation of the water molecules by the environment that takes place after inclusion is neglected in our semi-empirical calculations. Thus, we must include the solvent effect to rationalize our PM3 harmonic frequency calculations. Therefore, the obtained Gibbs free energy has no absolute significance in this case and should be considered only in a comparative sense. Similar situations were reported and models were proposed in past works [29,44].
We point out that the two thermodynamic parameters ΔH and ΔS for the formation of the inclusion complex can be easily determined from the temperature dependence of apparent formation constants by using classical equation of Van’t Hoff. We did not undertake this experimental task as our objective was to investigate only the spectrofluorimetry in order to determine the stoichiometry of the drug molecule/β-CD complex.
Finally, and starting from the single point energies which were summarized in Table 2, HF/6-31G(d), MPW1PM91/6-31G(d) and B3LYP/6-31G(d), we can see that in all cases the single point energy is lower in the complex A than that in the complex B. A very significant remark is that the energy difference between the two orientations is of the same order of magnitude for the three investigated single point calculations. These values are 3.04, 3.37 and 3.05 Kcal/mol, according to the order in which they appear in Table 2.
Besides, this suggests that the complex A is more stable than the complex B and thus predicts that the chloroethyl-nitrososulfonyl moiety has a preference to enter the cavity of the β-CD from its wide side rather than the two benzenic cycles. These results obtained by the PM3 semi-empirical optimization method agree with those obtained by the Density Functional Theory optimization (DFT: B3LYP and PMW1PM91) applied on CENS-piperidine/β-CD inclusion complex [40]. Following all these results, we can say that our method of choice (PM3 in our case) was judicious and we agree completely with authors who have deemed this method as a powerful technique that can be applied in biochemical systems.
4.5 ONIOM2 calculations
In order to improve the accuracy of the two resulting structures previously obtained by the PM3 method, we applied the ONIOM2 hybrid method to optimize the CENS-Dibenz at a high level of calculation at a medium basis set corresponding to RHF/6-31G(d) and the cyclodextrin at low level of quantum calculations PM3. All calculations were carried out according to procedure reported in the literature [45]. As listed in Table 3, the results of the ONIOM2 method have the same tendency as those of the PM3 optimization.
Energies, thermodynamic parameters calculations using the ONIOM2 method and single point energies for CENS-Dibenz/β-CD inclusion complexes.
| Energetic terms | ‘A’ orientation | ‘B’ orientation |
| Ea | −1,169,341.44 | −1,169,330.01 |
| Ecomplexation | −19.17 | −7.73 |
| Einteraction | −17.68 | −15.21 |
| Edeformation (CENS-Dibenz) | 0.27 | 4.07 |
| Edeformation (β-CD) | −1.76 | 3.41 |
| Hf | −1,168,311.72 | −1,168,301.72 |
| ΔH°b | −17.76 | −7.77 |
| Gf | −1,168,455.03 | −1,168,447.64 |
| ΔG°b | 3.22 | 10.61 |
| S (cal/mol.K) | 480.67 | 489.41 |
| ΔS°b (cal/mol.K) | −70.41 | −61.67 |
| HF/6-311G (d):HF6-31G(d) | ||
| Ea | −3,835,553.75 | −3,835,550.79 |
| MPW1PM91/6-311G(d) | ||
| MPW1PM91/6-31G(d) | ||
| Ea | −3,855,426.93 | −3,855,423.78 |
| B3LYP/6-311G(d):B3LYP/6-31G(d) | ||
| Ea | −3,854,664.72 | −3,854,674.84 |
a E is the HF energy.
b ΔA° = Acomplex − (Aβ−CD + ACENS-Dibenz), A = H,G or S at P = 1 atm and T = 298.15 K.
Consequently, the energy of complexation for the ‘A’ orientation is more negative than for the ‘B’ orientation. Indeed, this hybrid method confirms that the ‘A’ orientation is more favorable by −11.44 Kcal/mol. We also note that the relative difference in energy is slightly more significant than that was obtained by the PM3 method. Specifically, the relative difference in energy obtained by the PM3 semi-empirical method is −9.32 Kcal/mol. Table 3 also shows the great change in deformation energy for the guest molecule in the two complexes in comparison with that was previously obtained with the PM3 method.
For the ‘A’ orientation, the deformation energy of the CENS-Dibenz obtained with the PM3 and ONIOM2 methods was 2.36 Kcal/mol and 0.27 Kcal/mol, respectively. The complete opposite is found in the ‘B’ orientation, for which the PM3 and ONIOM2 methods respectively produced 1.78 Kcal/mol and 4.07 Kcal/mol for this same energy. Thus, we observe a reduction of the deformation energy of the guest in the complex A and an increase of this same energy in the complex B. This confirms that the flexibility of the drug molecule structure plays a significant role in the increase of the energy of complexation of the whole system. As for the β-CD, the corresponding deformation energy is always the same for both investigated methods.
In addition, the two inclusion processes of CENS-Dibenz are enthalpically governed with an unfavorable entropic term. For both orientations, the values of standard enthalpy change (ΔH0) are almost similar to those obtained by PM3 calculations and the entropic term indicates that the standard entropy change (ΔS0) is also negative. The standard free energy change ΔG0 though positive but remains always smallest for the complex A.
We also found that all single point calculations summarized in Table 3 confirm our results. Fig. 11 illustrates the two structures of the guest-host complexes with the minimal energies obtained by the two-layered ONIOM2 method and their calculated IR spectra.
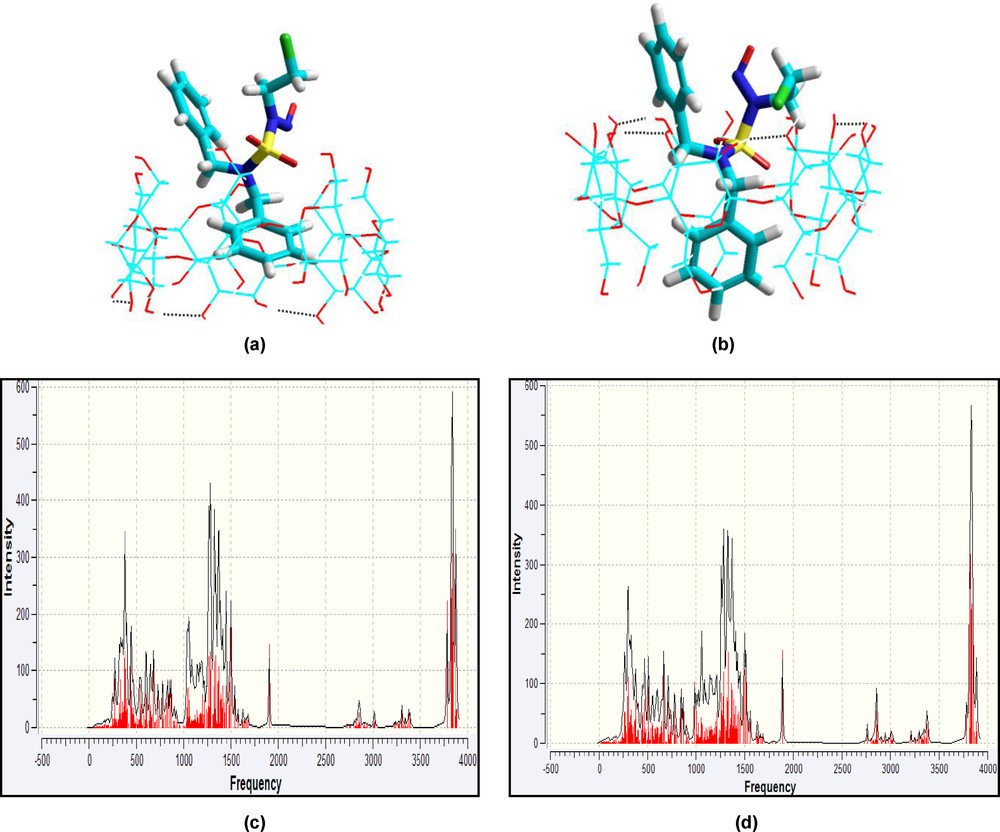
Energy-minimized (ONIOM2) structures of CENS-Dibenz/β-CD complex in the two different orientations studied in vacuum: a: orientation ‘A’; b: orientation ‘B’; c and d: simulated infrared spectra of both complexes A and B respectively. Dotted lines represent hydrogen bonds.
In Table 4, we present the bond lengths, bond angles and the most characteristic dihedral angles between the two phenylic rings A and B and the alkylating agent moiety (nitroso chloroethyl) of the drug molecule before and after complexation, obtained with the PM3 semi-empirical method and the two-layered ONIOM2 method. The experimental geometrical parameters of the free CENS-Dibenz [46] are also summarized in Table 4.
Geometrical parameters of CENS-Dibenz before and after inclusion in β-CD for the most stable A and B complexes as calculated by PM3 and ONIOM2 methods.
| Free CENS-Dibenz experimental [46] | Free CENS-Dibenz PM3/RHF6-1G(d) | CENS-Dibenz in Complex A: PM3/ONIOM2 | CENS-Dibenz in Complex B: PM3/ONIOM2 | |
| Bond lengths (Å) | ||||
| C31C132 | 1.88 | 1.77/1.79 | 1.77/1.79074 | 1.78/1.79645 |
| S26O34 | 1.401 | 1.43/1.41 | 1.44/1.42075 | 1.44/1.41979 |
| S26O33 | 1.405 | 1.44/1.42 | 1.44/1.42361 | 1.45/1.42372 |
| S26N27 | 1.707 | 1.78/1.68 | 1.78/1.67727 | 1.79/1.69417 |
| S26N25 | 1.591 | 1.50/1.64 | 1.76/1.6237 | 1.75/1.61985 |
| O29N28 | 1.20 | 1.17/1.17 | 1.17/1.1752 | 1.17/1.17799 |
| N27N28 | 1.26 | 1.39/1.34 | 1.40/1.33935 | 1.39/1.34125 |
| N27C30 | 1.68 | 1.50/1.46 | 1.50/1.46739 | 1.50/1.46641 |
| N25C8 | 1.45 | 1.50/1.48 | 1.49/1.4777 | 1.49/1.48481 |
| N25C22 | 1.47 | 1.50/1.48 | 1.50/1.47542 | 1.50/1.48001 |
| C31C30 | 1.48 | 1.51/1.52 | 1.51/1.52221 | 1.51/1.51919 |
| C31H35 | 1.02 | 1.10/1.07 | 1.10/1.07843 | 1.10/1.0799 |
| C22C16 | 1.53 | 1.49/1.51 | 1.49/1.51552 | 1.49/1.51546 |
| C8C2 | 1.50 | 1.49/1.51 | 1.50/1.52056 | 1.50/1.5167 |
| C2C1 | 1.35 | 1.39/1.39 | 1.39/1.38799 | 1.39/1.39102 |
| C3C4 | 1.37 | 1.38/1.38 | 1.39/1.3862 | 1.39/1.38607 |
| Bond angles (°) | ||||
| O34S26O33 | 122.0 | 119.77/121.01 | 118.61/120.711 | 118.64/121.645 |
| O34S26N27 | 103.8 | 109.12/106.34 | 108.05/107.613 | 108.35/105.798 |
| O34S26N25 | 108.4 | 109.11/110.807 | 109.87/107.534 | 110.33/109.599 |
| O33S26N27 | 106.2 | 106.77/105.685 | 108.01/103.951 | 105.34/102.934 |
| O33S26N25 | 110.2 | 108.96/107.72 | 108.99/110.679 | 110.00/107.961 |
| N27S26N25 | 104.9 | 101.19/103.82 | 101.98/105.679 | 102.86/108.041 |
| S26N27N28 | 119.1 | 117.40/116.44 | 117.12/113.559 | 117.15/115.398 |
| S26N27C30 | 116.1 | 120.92/121.001 | 121.35/123.807 | 121.20/122.531 |
| N28N27C30 | 122.3 | 120.99/121.26 | 121.50/122.553 | 120.77/120.233 |
| O29N28N27 | 115.6 | 118.25/114.04 | 118.14//115.139 | 118.31/114.409 |
| S26N25C8 | 120.1 | 119.93/116.628 | 121.28/118.492 | 119.11/117.593 |
| S26N25C22 | 117.9 | 120.24/115.89 | 120.24/117.826 | 120.34/117.958 |
| C8N25C22 | 118.4 | 112.63/114.265 | 113.19/119.925 | 115.31//119.196 |
| CL32C31C30 | 94.1 | 107.53/109.208 | 107.36/109.530 | 110.89/112.899 |
| N27C30C31 | 97.0 | 112.55/111.377 | 112.27/110.705 | 113.70/114.185 |
| C3C2C1 | 117.5 | 119.54/118.732 | 112.28/118.549 | 119.20/118.332 |
| C2C3C4 | 122.0 | 120.23/120.762 | 120.37/120.877 | 120.15/120.911 |
| C3C4C5 | 120.0 | 120.06/120.021 | 120.01/119.978 | 120.34/120.206 |
| C4C5C6 | 119.1 | 119.89/119.69 | 119.77/119.508 | 119.70/119.422 |
| C4C5H11 | 120.3 | 120.03/120.217 | 120.84/120.781 | 120.25/120.378 |
| C5C6C1 | 120.1 | 120.22/120.097 | 120.27/120.256 | 120.07/120.148 |
| C2C1C6 | 121.1 | 120.03/120.693 | 120.18/120.784 | 120.51/120.967 |
| N25C22C16 | 113.7 | 112.50/115.284 | 112.47/114.817 | 111.90/115.086 |
| N25C8C2 | 112.7 | 112.68/111.844 | 114.34/115.603 | 114.36/115.614 |
| Dihedral angles (°) | ||||
| C30N27S26N25 | / | 63.12/73.5413 | 110.77/124.315 | 106.24/96.2875 |
| S26N25C8C2 | / | 92.02/149.75 | 62.88/68.6089 | −77.78/−73.2656 |
| S26N25C22C16 | / | −89.48/−77.9671 | 69.60/65.5138 | −92.41/−72.2669 |
| C31C30N27S26 | / | −113.69/−113.54 | 91.42/92.8244 | −118.33/−119.616 |
| C30N27N28O29 | / | −2.77/−5.98478 | 4.49/1.36912 | −1.34/−7.04212 |
These results indicate that the guest in complexes A and B has completely changed its initial geometry. This change is clearly justified by the difference between the values of the dihedral angles of the drug molecule before and after the complexation. Thus, the molecule under consideration becomes deformed and adapts to a specific conformation inside the host cavity to form a more stable inclusion complex.
5 Conclusion
The experimental and theoretical study presented in this paper has demonstrated that CENS derivated from dibenzylamine forms an inclusion complex with β-cyclodextrin. The effect of β-cyclodextrin on the fluorescence spectrum of drug molecule was studied. Adding cyclodextrin to the CENS-Dibenz solution resulted in a significant enhancement of the luminescence. Spectrofluorimetric analysis suggests that this oncostatic drug forms a complex with β-CD in the stoichiometry of 1:1 with a stability constant of 417.63 M−1 at room temperature.
By means of theoretical methods, the present work unambiguously determined the geometrical inclusion parameters of the guest in the native cyclodextrin. The MM+ modeling in our preliminary studies indicates that all complexation energies are negative which showed that the inclusion process is thermodynamically favorable. It was also found that for the both most stable conformations, the main driving forces for complexation are dominated by non-bonded van der Waals intermolecular interactions. The values of the complexation energy of the in vacuum and in water mechanic molecular MM+ corresponding to CENS-Dibenz/β-CD complexes show that there is no preference for the most stable structures.
The semi-empirical quantum chemical calculations using the PM3 method were subsequently applied to determine the binding free energy, the most probable conformations and their thermodynamic parameters for each complex. The preferred orientation for the inclusion complexes obtained was complex A because it is the most energetically favorable structure. In this most stable conformation (i.e. complex A), the one of the two aromatic cycles is dipped in cavity of β-cyclodextrin, while the other cycle (ring) as well as the alkylating moiety protrude from the primary hydroxyl rim and are exposed to the outside of the host. The enthalpy and entropy changes suggest that the formation of the inclusion complex is an enthalpy-driven process.
The variation of free energy ΔG0 was found to be positive for both complexes in vacuum thus indicating that a contribution of energy is necessary to form these complexes. This result is completely different to that obtained from a fluorescence study. In fact, the simulation of the complexes without an explicit inclusion of the solvent can give misleading results.
Finally, we find that both PM3 and Hybrid method ONIOM2 calculations predict the same mode of inclusion of the drug molecule in the native cyclodextrin and the inclusion of the alkylating moiety via the wider (secondary) rim of the cone is still the dominant binding process.
The present findings can be expected to open the door to extensive future studies on the encapsulation of CENS and its related derivates in modified β- cyclodextrins as well as on its interaction with other suitable drug delivery vehicles like the calixarenes.
Acknowledgments
We are very grateful for financial support within the General Direction of Research Scientific and Technology Development (GDSRTD) through the PNR research of the Algerian Ministry of Superior Teaching and Scientific Research (MSTSR).