

1 Introduction
For over a century, oil has been the most widely used source of energy because of its availability at low cost. After the first tensions in the oil market, other energy sources emerged, in particular for electricity generation. But oil remained predominant as energy source for transportation because of its very high power density. As energy demand is booming and oil price is rising, other fuels for transportation have to be found. Hydrogen is a good candidate because of its high power density and the possibility to produce it in a sustainable way from renewable energy sources (solar, wind…) [1–4]. It is a fuel of choice for clean transportations in polluted cities as its combustion produces only water. It can be directly used in classical thermal engines but with a low yield and emission of nitrogen oxides and ozone. Energy conversion efficiency in fuel cells is far higher (usually above 60%, as compared to 30% for thermal engines) and occurs without the exhaust of any pollutant, which places hydrogen on a parr with oil fuels if it were not for the very high price of the fuel cell.
Several hydrogen fuel cell technologies have been developed recently. Attractive applications such as transportation are very demanding though, especially in terms of engine weight and ignition delay. Only polymer electrolyte membrane fuel cells (PEMFC) can compete on this ground. However, the current technology of PEMFC involves electrodes covered with catalysts made of platinum. Platinum is precious because it is scarce (37 ppb in the Earth's crust) and consequently its price is constantly rising: currently 50 to 70 $/g, corresponding to about 2500 to 3500 $ to power a medium sized car.
Among metal catalysts, Pt is by far the best and no replacement by more common inorganic materials could be made in low temperature fuel cells so far. However, some enzymes have been well known for long as extremely efficient catalysts for the oxidation of hydrogen, although their functioning, widely different from usual inorganic catalysts, makes any comparison difficult. The need for a technological breakthrough in hydrogen fuel cell catalysts has stimulated a wide effort around hydrogenase enzymes and chemical models inspired by their astonishing performances. Biologists have screened the hydrogenases of bacteria and archaea to find new enzymes more proper for implementation in fuel cells. In some cases, hydrogenases have been genetically engineered. On the other hand, chemists have proposed dozens of biomimetic or bio-inspired synthetic compounds, a few of them showing interesting properties for hydrogen oxidation catalysis. Both approaches will be the focus of this article.
2 Structure and function of hydrogenases
Many microorganisms have developed enzymatic pathways to handle hydrogen, either for hydrogen uptake in anaerobic environments, providing the cell with low-potential electrons, or for hydrogen evolution, for the oxidative equilibrium of their electron carriers in fermentation or photosynthesis. Enzymes, called hydrogenases, catalyse the reversible oxidation of molecular hydrogen into protons and electrons, according to Eq. (1) below. Hydrogenases fall into two main classes: [FeFe] hydrogenases [5,6], present in bacteria and some unicellular eukaryotes, and [NiFe] hydrogenases [7–11] present in bacteria and archaea. The field has been reviewed recently [12]. A third class, [Fe] hydrogenases, only catalyses the first step of the reaction, i.e. the heterolytic cleavage of hydrogen, and further transfers the hydride to a cofactor [13–15]. Many studies combining different spectroscopies (EPR, IR, ENDOR…), X-ray crystallography and biochemical characterization have been reported in the past decades. However, due to the complexity of the reaction mechanism, the details of catalytic hydrogen oxidation and inhibition by oxygen are still a matter of debate [16].
| (1) |
Although [NiFe] and [FeFe] hydrogenases are not phylogenetically related, their catalytic center is built on the same scheme with two metal ions connected through thiolate bridges (Fig. 1). Thiolates are provided by cysteines in [NiFe] hydrogenases [7] while [FeFe] hydrogenases use a small organic 2-azapropanedithiolate ligand [17,18], providing a local base for proton coordination in the vicinity of the active site. The metal ions are coordinated with CO and CN−, very unusual ligands that help stabilizing the metal ion in a low redox state, and soft sulphur atoms for higher hydrogen affinity. Hydrogenases also include in their structure an electron path made of several Fe/S clusters down to the hydrogenase surface where redox partners get reduced, a hydrophobic channel tunnelling H2 to the active site [19,20] and proton transfer pathways through hydrogen bonds between amino acids from the immediate vicinity of the active site to the surface of the protein [21].

Structure of the active sites of [NiFe] hydrogenases and [FeFe] hydrogenases, both in a reduced active state (X is likely a hydride ligand).
Although hydrogenases constitute exceptional catalysts for hydrogen oxidation with turnover frequency of up to 21,000 s−1 measured on individual enzymes [22] and no overpotential [23] compared to Pt, they are studied by a handful of research groups only because of their delicate handling. Purification of native elaborate enzymes from bacterial culture is challenging, especially from anaerobic and/or extremophilic bacteria. Besides, most hydrogenases are sensitive to oxygen. Exposure to air leads to an irreversible oxygen damage of [FeFe] hydrogenases and more or less reversible inactivation of [NiFe] hydrogenases.
In a first attempt to face these problems, targeted mutations have led to engineered hydrogenases with significantly prolonged half-life in air. Dementin et al. have mutated key residues on the H2 access channel [24] in D. fructosovorans hydrogenase, an abundant and highly active enzyme in the aim of limiting O2 access to the active site. Recent results [19,25] show that O2 tolerance can be greatly increased, getting near to naturally O2-tolerant hydrogenases.
Other teams have chosen to try to overproduce very active hydrogenases in easy-to-grow bacterial systems. In most cases, the use of heterologous expression systems is at least as challenging as growing natural bacteria because of the absolute necessity to co-express the complicated protein machinery required for the complete maturation of hydrogenases. Such a machinery is in addition strain-specific for [NiFe] hydrogenases and when an organism has more than one [NiFe] hydrogenase, each of them may have its own maturation machinery [26]. Indeed, the catalytic center based on Ni and/or Fe ions involves such unusual ligands as CO and CN− and even the replacement of the sulphur atom of a cysteine by a Se in some [NiFe] hydrogenases. Only few hydrogenases can be overexpressed, such as Chlamydomonas reinhardtii [FeFe] hydrogenase in Escherichia coli [27], an enzyme irreversibly inactivated by O2. However, hydrogenases are very widespread in microorganisms, so that coupled efforts toward overexpression and mutation can lead to interesting hydrogenase enriched systems. Such systems have been developed in E. coli [28], but they are intended for fermentative H2 production [29] so far, rather than fuel cell applications.
More recently, the enzymatic arsenal of many bacteria and archaea species has been explored to discover new, less air-sensitive hydrogenases. Air compatible hydrogenases have been discovered in soil bacteria such as the Ralstonia species [30], in intestinal bacteria like E. coli [31] and extremophilic ancestral bacteria such as Aquifex aeolicus [32], leading to interesting demonstrations in fuel cells (detailed further). They typically have lower H2-oxidation rates than O2-sensitive enzymes but can be handled in air. These O2-tolerant hydrogenases show a reduced but persistent activity in the presence of oxygen up to moderately positive potentials, indicating a rapid equilibrium between enzyme fractions that are active and enzyme fractions that are oxidized to inactive but ready states. All are mostly insensitive to gases that poison Pt catalysts such as CO. However, the challenging culture of the corresponding bacteria and archaea and the low enzyme yields severely hinder the widespread use of these hydrogenases. Moreover, most of these enzymes are membrane-bound, making their purification very difficult.
Recent structural studies have shown a special [4Fe-3S] cluster in these O2-tolerant hydrogenases that replaces a [4Fe-4S] cluster in O2-sensitive enzymes [33–35]. The special redox properties of the [4Fe-3S] cluster are crucial to ensure a rapid complete reduction of reactive oxygen species at the active site to water, preventing the formation of difficult to activate partially oxidized species [35–37].
However difficult their production can be, the recent progresses in the discovery of O2-resistant hydrogenases make these enzymes attractive catalysts for hydrogen fuel cells.
3 Bio-inspired catalysts for hydrogen oxidation
Since the publication of the first crystal structure of a hydrogenase in 1995, the unusual construction of the active center has inspired chemists for the design of new molecular catalysts. Series of organometallic complexes have been synthesized following two strategies. Either the structures are built as close as possible to the geometry of the enzyme catalytic center (biomimetic approach), or the structure can be sensibly different but copy as many as possible of the surrounding elements involved in the reaction scheme (bio-inspired approach) [38]. More than 50 biomimetic Ni-Fe clusters have been described [39,40], but most have no catalytic activity. The first mimics able to activate [41–43] or produce [44–50] H2 were Ni-Ru clusters from 2006 (Fig. 2). A dinuclear Ni-Ru complex [43] proved able to catalytically oxidize H2 using Cu2+ ions in water as a source of oxidizing power. More recently various Ni-Fe [51–54] and a Ni-Mn [55,56] dinuclear mimics with either S2P2 or S4 coordination spheres on the Ni center were shown active in hydrogen evolution only. However, unfortunately none of these clusters have proven active in hydrogen oxidation so far.
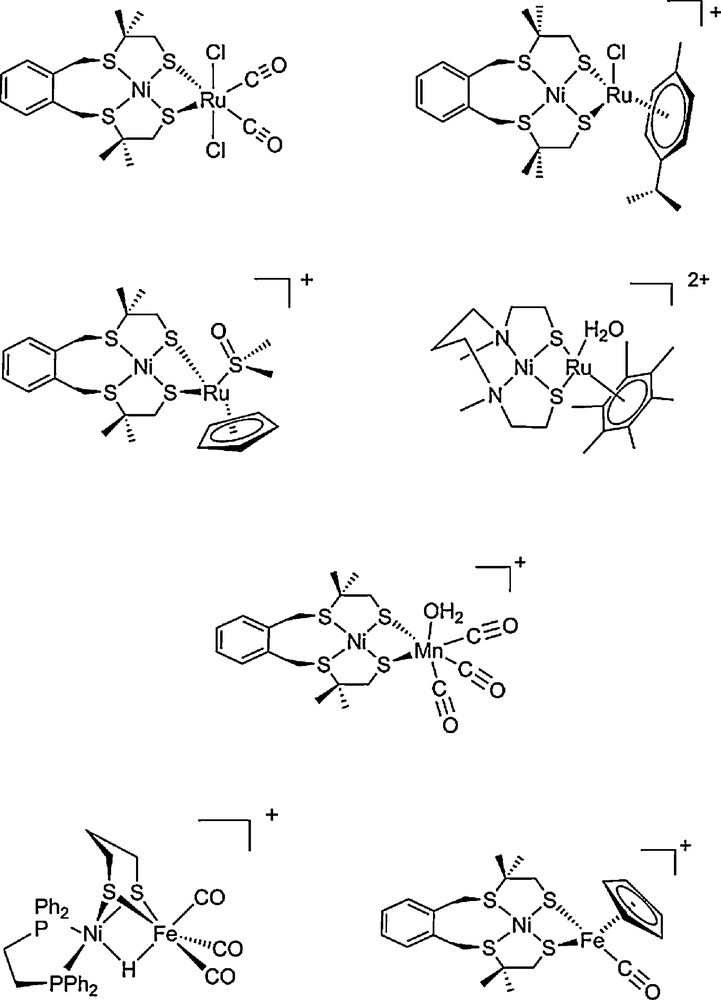
Structures of selected functional Ni-Ru [41,44,45,47–49], Ni-Mn [56] and Ni-Fe [51,52] bio-inspired models of [NiFe] hydrogenases.
In contrast, a very large number of diiron complexes have been synthesized and showed electrocatalytic activity, but mostly for proton reduction to hydrogen [40,57] and with overvoltages in the 0.5–1 V range, still quite far from the targeted 0–0.3 V range. A demonstration of hydrogen oxidation catalysis with a member of this series has been published first in 2012 (Fig. 3): the azadithiolate bridged [FeFe] cluster was bound to a Fe-cyclopentadienyl redox for electron output [58]. The compound could catalyse hydrogen oxidation in organic solvents with a handful of turnovers within a few hours.
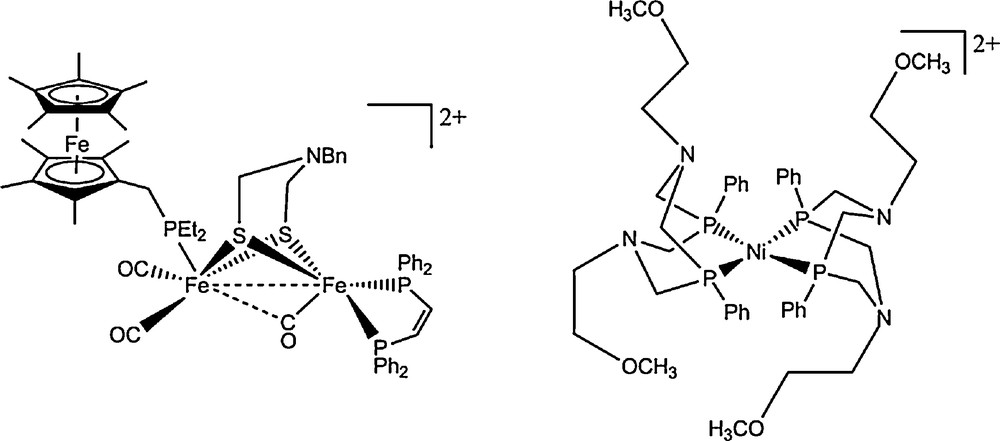
Structure of the FeFe mimic from Rauchfuss [58] able to catalyse both proton reduction and hydrogen oxidation (left) and structure of the only molecular electrocatalysts, developed by DuBois, able to reversibly mediate H2/H+ interconversion under bulk homogeneous conditions (right) [64].
The bio-inspired approach has proved more effective [3,38] in designing functional catalysts competent in hydrogen oxidation. Although many structures proposed for the hydrogen catalysis such as Co or Ni macrocyclic complexes have proved active in proton electroreduction, very few have shown a catalytic activity towards the more challenging catalytic hydrogen oxidation. A Ru porphyrin proved active in electrooxidation of hydrogen, probably as a diporphyrin sandwich complex [59]. An Ir complex [60] proved able to reduce Ag+ in organic solvent via H2 oxidation, but mechanisms are complex and slow. The DuBois’ team has developed the most competent hydrogen oxidizing clusters within their mononuclear Ni bisdiphosphine complex series. In their exquisitely complete work, they could rationalize the design of H2 active catalysts [61,62] by copying fundamental aspects of the [FeFe] hydrogenase active site:
- • an electron rich coordination sphere around the active metal ion, here two diphosphines;
- • a base in the close vicinity of the metal center, here tertiary amines in the bridges of the diphosphine ligands.
They could also give hints on how to switch from an H2 production to an H2 oxidation oriented catalyst, a stronger vicinal base being favorable to H2 oxidation. The best H2 oxidation catalyst [Ni(PCy2Nt-Bu2)2](BF4)2 catalyzes hydrogen oxidation in acetonitrile in the presence of triethylamine as a base at a turnover frequency of about 50 s−1 and a very low overvoltage (less than 0.1 V) [63]. It should be noted that the turnover frequency for the best H2 oxidizing catalyst remains small compared to turnovers for H2 reduction (130 s−1 in a similar compound). Most of these compounds require distinct conditions for catalyzing H2 production or oxidation. So far, only one compound of this series can mediate the H2/H+ interconversion in both directions and under the same conditions, thanks to a specific set of substituents at the P2N2 ligands (Fig. 3).
Most of these synthetic catalysts are active in organic solvents where they are soluble. This is somehow comparable to the hydrogenase active site embedded in the mostly hydrophobic interior of the enzyme. This however hinders their use for hydrogen oxidation into water in technological applications (fuel-cells). Catalysts have therefore to be immobilised onto a conductive substrate to be subsequently plunged into or interfaced with an aqueous electrolyte. The next section reviews different strategies developed for this aim, starting with hydrogenases for which several immobilization methods have shown successful, paving the way to organometallic catalyst grafting on electrode.
4 Electrode materials for H2 oxidation based on immobilized hydrogenases
There have been many reports dealing with the application of hydrogenases to replace Pt, since the demonstration by F.A. Armstrong that hydrogenases adsorbed on carbon electrodes [65] are comparable in activity to Pt nanoparticles [66].
4.1 Direct enzyme adsorption or binding onto the electrode
Immobilization of the catalyst on the electrode surface has been an early requirement for the electrochemical study of hydrogenases. Drop drying of a protein film onto porous carbon electrode has become a standard for voltammetry [67,68] after the extensive work of F.A. Armstrong's group and allowed for a fine mechanistic study of both hydrogen oxidation and evolution in hydrogenases, as well as the comparison of hydrogenases from different microorganisms. Depending on the surface electrostatic potential of the enzyme a favourable orientation can be spontaneously obtained, allowing direct interfacial electron transfer from the enzyme to the electrode [69]. This is because the active sites are electrically connected to the surface of the protein through the array of iron-sulfur clusters, the distal cluster lying close to the surface. Proper orientation however can be difficult to obtain, even by tuning the substrate hydrophobicity/hydrophilicity [70]. When electrostatics are not favourable, the substrate potential can be tuned using polycations to favour oriented attachment of some hydrogenases such as [NiFe] hydrogenases from A. vinosum and D. gigas and [FeFe] hydrogenase from D. desulfuricans [71]. Membrane-bound hydrogenases, for example, from A. aeolicus, can be deposited into a membrane on the electrode, taking advantage of the natural attachment of the enzyme [72]. Direct electron transfer from the enzyme to the electrode could even be obtained from membrane-bound D. vulgaris hydrogenase film on the carbon electrode when the membrane was properly oriented, i.e. the enzyme side of the membrane in contact with the electrode [73]. However, in most cases, direct electron transfer could not be obtained and a redox mediator was needed to transfer electrons to the anode as for D. fructosovorans [NiFe] hydrogenase [74].
Alternatively, hydrogenases could be covalently bound onto a carbon electrode [75]. Although the oxidation current was slightly lower than for adsorbed enzyme, covalent binding provided stability over hours of continuous operation, while control samples using adsorbed hydrogenase tended to desorb within 2 to 5 h.
The quantity of hydrogenase involved in catalysis in such systems is limited (about 10−12 mol/cm2 according to reference [71]). As hydrogenases have a considerably larger size (5 to 7 nm) than their catalytic site, the overall number of catalytically active sites per unit area is much smaller than the equivalent number of platinum active sites on a similar electrode covered with Pt nanoparticles. Therefore, although the hydrogenase catalytic efficiency is same or higher than that of Pt, the overall oxidation current density measured ranges typically in the mA/cm2 range, while current density as high as 1-10 A/cm2 can be expected from an electrode covered by a thin film of Pt [71] (in the Koutecky-Levich limit).
To catalyze a redox reaction such as hydrogen oxidation, a redox catalyst has to be in contact with a source of chemical reagent, here hydrogen gas, and also to an electrode for electron transfer. The case of hydrogen oxidation at low temperature is particularly difficult because:
- • the reaction has to proceed in water to allow for proton efflux;
- • the H2 gas is only slightly soluble in water (about 1 mM), so that the catalyst has to be maintained close to the gas/liquid interface to provide constant H2 influx;
- • the catalyst must be in contact with an electrode, usually solid.
Catalysis takes place in this gas/liquid/solid contact area. In most experimental cases, the catalytic current is therefore limited by H2 transport to the catalyst. The intrinsic catalytic rate of the catalyst can be estimated using a rotating anode to fasten H2 transport: in this case, the Koutecky-Levich model allows extrapolating the intrinsic catalytic rate at infinite rotation speed, i.e. in the absence of any mass-transport limitation. Such limit currents are not accessible on all systems (and definitely not in real fuel cells), so that anode currents from different works should be compared with caution.
In the aim of applications, the efficient wiring up of hydrogenases to a collecting electrode has quickly come into question since hydrogenase anodes obtained by protein adsorption are not stable in time: leaching of the enzyme is observed and the electrodes are stable at best for some hours when dropped in pure electrolyte solutions [76]. Two strategies have been developed: hydrogenase trapping into a porous, conductive matrix polymerized onto the collecting anode, or hydrogenase grafting onto a microporous electrode such as a carbon nanotube film. Reports are scarce but instructive.
4.2 Entrapment of hydrogenases in polymers
A number of reports describe methods to adsorb hydrogenases onto carbon electrodes modified with polymers. For example, Morozov et al. describe entrapment of [NiFe] hydrogenases from Thiocapsa roseopersicina [23,77,78], D. baculatum [23] or Lamprobacter modestogalofillum [23] in polypyrrole films. To provide proper contact to the electrode, the film also entraps viologen as a redox mediator. These modified electrodes are active in both hydrogen evolution and uptake. High H2 oxidation currents (1.2–1.7 mA cm−2) are recorded at an overpotential of 200 mV [79]. The hydrogenase density is very high in the films (45 pmol/cm2), and the redox mediator helps connecting all enzyme molecules for electrocatalysis as shown by the high turnover frequencies observed (150–220 s−1), in the same range as for hydrogenases in solution. Activities have been shown to be comparable to those of Pt-based electrodes, meaning that electrocatalysis occurs at the mass-transport limit in the film. This strategy results in very stable electrode materials, which can be functional over a period of several months [80]. The strategy has been extended recently for the entrapment of hydrogenases into a carbon nanotube network thanks to in situ polymerization of a matrix [81], resulting in a higher catalytic current thanks to an improved electron transfer to the electrode through the carbon nanotube network.
Hydrogenases, such as the [FeFe] hydrogenase from D. vulgaris can also be incorporated with methyl viologen (MV) into clay films at the surface of carbon electrodes [82]. Alternatively, the electron mediator can be incorporated within an electropolymerized film deposited together with the clay [83].
4.3 Immobilizing hydrogenases on carbon nanotubes
Carbon nanotubes (CNTs) are an interesting support for hydrogenases because they can be assembled in largely porous, conductive networks and because hydrogenases spontaneously assemble with CNTs forming biohybrids. The preparation of such biohybrids has been reported for Clostridium acetobutylicum CaHydI [FeFe] hydrogenase [84] and for [NiFe] hydrogenases from T. roseopersicina [85], D. fructosovorans [74] and A. aeolicus [86]. In the latter case H2 oxidation was obtained by direct electron transfer (without the need for a redox mediator) with a 10-fold higher catalytic current density in the case of stationary CNT-coated electrodes as compared to bare electrodes, reaching a value of 1 mA cm−2 at 60 °C. Here again, such a current density value is clearly limited by mass transport. Unfortunately, the active film proved quite unstable with a marked decrease of the catalytic current with time (up to 50–70% after 1 h at 60 °C), due to a combination of enzyme release and denaturation.
De Lacey's group chose to covalently couple hydrogenases to their substrate to avoid such long time decay. Following their first success [87] on graphite favored by the proper orientation of D. gigas hydrogenase when coupling through the enzyme carboxyl moieties, they took advantage of the very high specific area of multiwalled carbon nanotube (MWCNT) networks [76] to increase the catalytic current. A 50 μm thick MWCNT brush was grown by CVD onto a gold electrode and compared to a polished graphite electrode. The carbon substrates were first covered with a polyphenylene film bearing amine moieties by electrochemically polymerizing and reducing nitrophenyldiazonium. Hydrogenase from D. gigas was then covalently bound by amide-coupling to the amino groups of the carbon substrate. The catalytic current from the MWCNT-hydrogenase anode proved 33 times higher than for the graphite-hydrogenase anode. Although a large improvement, this ratio was not in accordance with the ratio in specific area (estimated to a factor of 1000) but with the ratio in electrochemically grafted amino group density (a factor of 50). Hydrogenases are thus not covering the whole MWCNT surface but attach at scarce defective spots in the MWCNT network. H2 oxidation currents were measured at 0.7 mA/cm2 in solution and extrapolated from rotating anode measurements to 4.1 mA/cm2 at the Koutecky-Levich limit, compared to only about 0.1 mA/cm2 for hydrogenase-on-graphite anodes. The value of 0.7 mA/cm2 obtained for covalently grafted hydrogenases should be compared with the higher H2 oxidation currents (4 mA cm−2) recorded by the Armstrong group using the same adsorbed hydrogenase, indicating that the grafting procedure may damage the enzyme. Interestingly, covalent coupling also seemed to stabilize the enzyme towards denaturation: indeed, hydrogenase-graphite electrodes were found stable for over a week while the catalytic activity of hydrogenase-MWCNT electrodes was stable over one month of continuous operation.
King's group proposed the use of C. acetobutylicum [FeFe] hydrogenase on CNT networks because it has shown a high affinity for the surface of CNTs and a special ability to direct electron transfer to the CNT [84]. The CNTs used here are single-walled nanotubes (SWCNTs), which are synthesized as a mixture of semi-conducting and highly conductive (called metallic) SWCNTs. Their recent paper [88] shows that H2 oxidation currents can be largely increased when using mixtures enriched in metallic SWCNTs with record currents up to 14 mA/cm2 in the Koutecky-Levich limit (0.5 mA/cm2 without rotation) for 84% metallic SWCNTs on carbon cloth. However, the [FeFe] hydrogenase from C. acetobutylicum has to be handled in strictly anaerobic conditions.
Our group has recently developed a coupling chemistry for the covalent attachment of hydrogenases onto CNT. Two hydrogenases were chosen in this work: the [FeFe] hydrogenase from Desulfovibrio desulfuricans, because it can be prepared in relatively large quantity and its catalytic activity is very high, but it has to be handled in anaerobic conditions; and the oxygen-resistant [NiFeSe] hydrogenase from Desulfomicrobium baculatum because it can be handled to some extent in aerobic conditions. The hydrogenases have been prepared and purified as previously described [89,90].
D. desulfuricans [FeFe] hydrogenase and D. baculatum [NiFeSe] hydrogenase did not show a high affinity for CNT compared to what has been reported for C. acetobutylicum [FeFe] hydrogenase [84], as shown by adsorption tests by atomic force microscopy (AFM). Based on previous work on other hydrogenases showing a good affinity for positively charged surface [71], we also covered CNTs with a cationic pyrene derivative synthesized on purpose (1-trimethylammonio acetyl pyrene). However, although the charge surface map (not shown) calculated for D. baculatum hydrogenase showed important acidic patches, the enzyme showed only low affinity for the cationic CNTs as shown by AFM imaging of cationic CNTs before and after incubation with hydrogenase (Fig. 4).
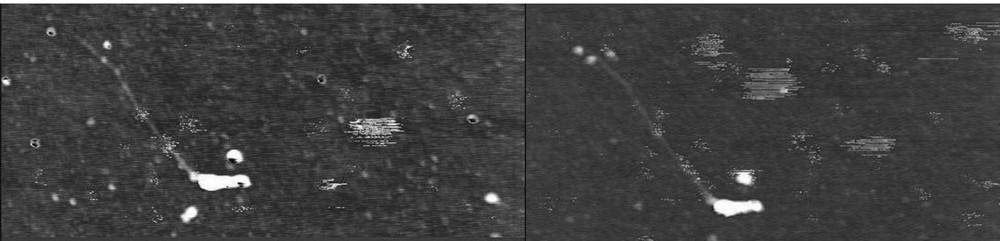
AFM images of a HiPco CNT covered with trimethylammonio acetyl pyrene before (left) and after (right) incubation with [NiFeSe] hydrogenase on a silicon wafer. Two additional dots can be seen on the top left end of the CNT, corresponding to two bound hydrogenases. The width of the images is 1.2 μm.
Following our previous work on protein to CNT grafting [91], covalent coupling of hydrogenase to CNT was then chosen to insure biohybrid stability. Biohybrids were prepared on single-walled carbon nanotubes (SWCNT) by coupling COOH groups on hydrogenases onto amine modified SWCNTs. Indeed, several hydrogenases show a crown of carboxylic groups around the electron outlet. By coupling the enzyme's carboxylic groups to the CNT's amine groups, we expected a better orientation of the enzyme on the CNT and therefore a higher yield of direct electron transfer than observed by Lojou's team while coupling enzyme's amine groups to CNT's carboxylic groups [74].
Biohybrids with a hydrogenase content as high as 40% were prepared and characterised by AFM imaging (Fig. 5) and XPS analysis (Fig. 6). SWCNTs were chosen rather than MWCNTs because they are smaller in diameter and contain no inner carbon, so that AFM and XPS detection of hydrogenases in the biohybrids were possible. SWCNTs (arc discharge, 1.2 to 1.4 nm in diameter) were purified and solubilized in an aqueous solution of 1% F127, a non-ionic surfactant. The SWCNTs were functionalized with a Boc-protected 4-aminoethylbenzenediazonium salt synthesized on purpose. SWCNTs were separated from by-products and rinsed by ultracentrifugation. Boc deprotection of Boc-aminoethylbenzene-SWCNTs into amino-SWCNTs was performed in TFA 95%. Hydrogenase was first incubated with EDC and sulfo-NHS in a MES buffer for carboxylate activation, and then amino-SWCNTs were added and reacted overnight. Biohybrids were separated from the reagents by ultracentrifugation.

AFM images of pristine SWCNT (left) and SWCNT-hydrogenase biohybrids (right). Biohybrids were prepared either in situ on a thin film of SWCNT casted on a silicon wafer (upper images) or in solution (lower images). By in situ coupling, the same SWCNTs can be imaged and compared accurately before and after coupling: coupled hydrogenases appear as additional dots on the SWCNT network.
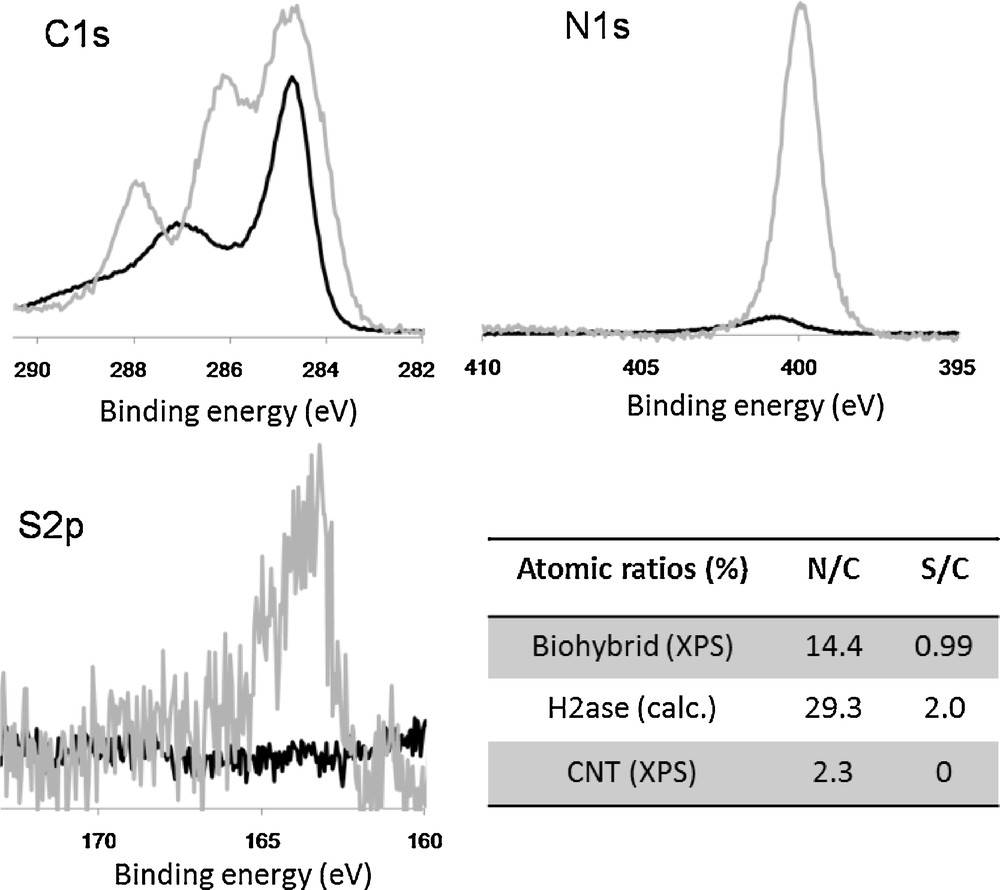
XPS analysis of [FeFe] hydrogenase-SWCNT biohybrids. C1s, N1s and S2p spectra show the XPS spectra of amino-SWCNTs (black) and biohybrids (grey) renormalized to the area of the carbon peak at 284.5 eV (aromatic carbon). The table reports XPS peak area ratios N/C and S/C from XPS data for SWCNTs and biohybrids and calculated from molecular composition for [FeFe] hydrogenase. The ratios correspond to a hydrogenase/SWCNT composition of 40/60 in the biohybrid.
AFM analysis of the biohybrid product (Fig. 5 lower part) shows star-shaped aggregates with a bumpy surface and a thick diameter as compared to amino-SWCNTs. To try and evaluate the enzyme density on SWCNTs, a similar coupling procedure was used on SWCNTs deposited on a silicon wafer (Fig. 5 upper part). This way, the very same SWCNTs could be imaged by AFM before (left) and after (right) hydrogenase coupling. Individual hydrogenases appear as bumps of 5 to 7 nm height on the SWCNTs. We could estimate to enzyme coverage to about one hydrogenase per 100 nm of SWCNT length corresponding to a SWCNT/hydrogenase mass ratio of 50/50.
The composition of D. desulfuricans [FeFe] hydrogenase/SWCNT biohybrids was established by XPS analysis of a dried sample. The C1s XPS spectra (Fig. 6) shows the appearance of two new peaks corresponding to the CN (286 eV) and CO (288 eV) carbons of the protein back-bone. The N1s and S2p spectra show the nitrogen and sulphur contribution from the protein. Comparison of the measured N/C and S/C ratios in SWCNTs and biohybrids with the calculated atomic ratios in the hydrogenase gave a mass ratio hydrogenase/SWCNT of 40/60, in coherence with the estimate from AFM images.
The catalytic activity of biohybrids was measured by the methylviologen (MV) test: [NiFeSe] hydrogenase had a catalytic activity of 1170 U/mg of protein, while [NiFeSe] hydrogenase-SWCNT biohybrids showed a catalytic activity of 10 U/mg of protein. This drop of a factor of 120 in enzyme activity can be explained by three opposite effects. On one hand the enzyme might be damaged due to covalent coupling and consequent protein denaturation. On the other hand, the steric hindrance due to the presence of a SWCNT coupled in the close vicinity to the electron outlet of the enzyme reduces electron transfer to MV, which would artefactually drop the measured catalytic activity. Finally, let us note that SWCNTs have been demonstrated as good electron acceptors for hydrogenases [84] and might compete with MV. As the MV test is a kinetic measurement, a competition for reduction might produce a large error in the concentration estimate.
Biohybrids were then deposited on carbon cloth (GDL support from fuel cell design) for electrocatalytic activity measurement in an electrochemical cell and compared to a hydrogenase protein film. A specific hydrogen oxidation activity was recorded with similar currents for biohybrid films and hydrogenase only films with the same enzyme concentration (Fig. 7). The H2 oxidation curve showed a bell shape at 0.1–0.3 V vs SCE characteristic of hydrogenase inactivation/reactivation at high potential. The catalytic current measured on biohybrid (Fig. 7B) was half of the current from pure hydrogenase (Fig. 7A) while the overall hydrogenase concentration was 4 times lower, showing that the catalytic activity was not reduced by covalent coupling. This is in deep contrast with the loss of activity estimated from the MV test, the latter being probably distorted by the presence of SWNT. The addition of a redox mediator in the solution did not enhance the current.
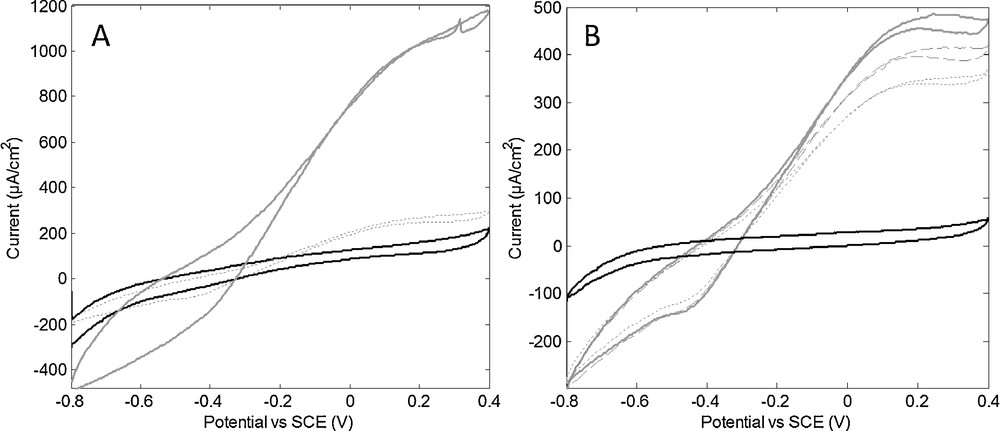
Electrochemical response of a film of D. baculatum hydrogenase (A) and a film of D. b. hydrogenase-SWCNT biohybrid (B) on carbon cloth flushed with N2 (black) or H2 (grey). The films were obtained by drying 20 μL of aqueous solutions of hydrogenase (0.25 mg/mL) and biohybrid (containing 0.06 mg/mL enzyme). Current stabilized after a reactivation period of 15 min (A) and 1 h (B) cycling (10 mV/s, electrolyte 20 mM phosphate buffer at pH 8 NaCl 0.1 M, 25 °C). Cycles at maximum currents (continuous line) are shown with cycles after 3 h (dashed line) or 18 h (dotted line) of continuous operation.
We note that the stability of the enzyme was greatly enhanced in the biohybrid: more than 50% of the enzyme activity in the biohybrid was maintained after 18 h of continuous operation while only 12% activity remained in enzyme only films.
These new results showed that hydrogenases can be covalently coupled to CNT into a well-defined biohybrid adduct at very high concentration (up to 40% enzyme in mass) while preserving the hydrogenase activity. However, deposition of biohybrids or enzymes from an aqueous solution on a carbon cloth proved very difficult to control because of the high hydrophobicity of the carbon cloth. The implementation of our biohybrid catalysts has still to be improved to allow for tests in a complete fuel cell.
4.4 Entrapment of hydrogenases in aqueous gels
An alternative immobilisation strategy was experimented recently in our team. To help in controlling deposition and manipulation of the hydrogenase while maintaining contact with CNT for electron collection, hydrogenase and CNTs were entrapped into a nanostructured aqueous gel.
The non-ionic polymer surfactant F127 (a PolyEthylene Oxide-PolyPropylene Oxide PEO-PPO-PEO triblock industrial copolymer) was chosen because of its being a non-denaturing surfactant (used for drug delivery), and its ability to make solid aqueous gels at relatively low concentration. Moreover, CNTs can be dispersed in F127 aqueous solution at high nanotube concentration [92]. F127 aqueous solutions make solid gels if oil is added [93,94], the gel having either a hexagonal linear micelle structure or a spherical micelle packing structure or a cubic bicontinuous double structure. As a support for a conductive CNT network, we targeted the cubic bicontinuous phase the structure of which is expected to help in making a percolating network through the gel.
The chosen composition was 25/20/55 water-triacetin-F127 mass composition following the work of Alexandridis et al. [94]. Gels containing either D. baculatum hydrogenase only, or hydrogenase and SWCNTs were compared to control gels without hydrogenase. The anode was made of a 3 mm thick layer of gel connected by a fine gold grid at the back through which H2 or N2 could infuse, and was separated from the electrolyte and counter electrode by a Nafion membrane. Data are recorded at room temperature.
Results are shown in Fig. 8. A voltammogram of a SWCNT-hydrogenase gel (A) shows a 18 μA/cm2 hydrogen oxidation current at 0.25 V vs SCE. Oxidation currents for a SWCNT-hydrogenase gel were compared with control gels (B). H2 oxidation was obtained from hydrogenase containing gels only, as expected. A strong increase in H2 oxidation current could be obtained in the presence of SWCNT. Let us note that hydrogenases were not covalently linked to SWCNT in this case, but confinement by the gel nanostructure (< 10 nm pores [94]) might force contact between the enzyme and the CNT.
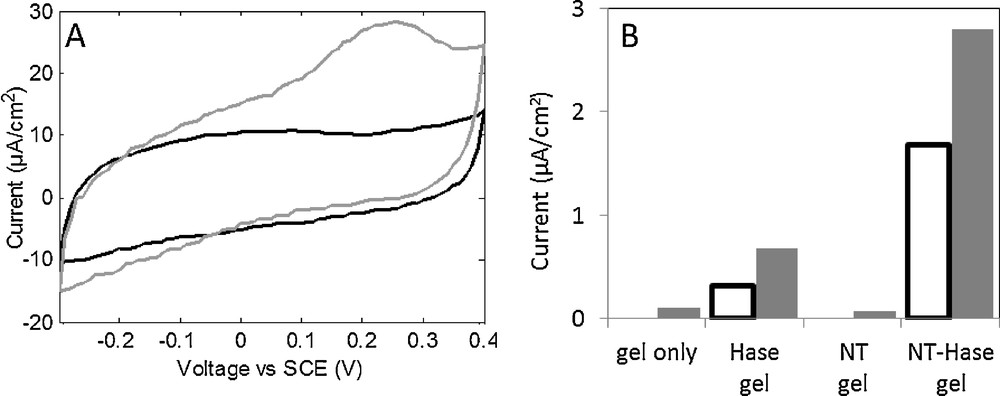
A. Cyclic voltammograms of a gel containing 0.03% SWNT and 60 nM D. baculatum hydrogenase under N2 (black) and H2 (grey) flow at 25 °C. Currents were stable from the third cycle, cycle 4 is shown (10 m V/s). B. Oxidation current at 192 mV vs SCE under N2 (white bars) and H2 (grey bars) for gels with or without SWNT (0.1%) and with or without D. baculatum hydrogenase (60 nM). Current was stabilized for 90 s.
As a conclusion, it appears that CNT aqueous gels represent an interesting support for hydrogenases in a fuel cell anode. Hydrogen oxidation currents are obtained in the 2 to 20 μA/cm2 range, even at room temperature, in a convenient paste-like gel compatible with fuel cell fabrication. The SWCNT content in the gel and of the gel thickness remain to be optimized, but oxidation currents are already quite high compared to previous work, considering that the measurements were made on static electrodes and at room temperature.
4.5 Hydrogenase-based biofuel cells
Armstrong and coworkers early used the drop-drying method to construct the first fully enzymatic hydrogen fuel cell, able to power a watch [95,96]. The fuel cell was constituted of a graphite anode modified with an O2-tolerant hydrogenase (R. metallidurans) and a graphite cathode modified by a fungal laccase (catalyzing O2 reduction) in a single chamber containing a mixture of oxygen and hydrogen (3% H2 in air, a non-explosive level). The fuel cell developed a Voc of 950 mV and a maximal power of 5.2 μW/cm2. Although extremely elegant, the whole system is far from optimum because of the low accessible H2 level, the lower catalytic activity of O2-tolerant hydrogenases under O2 atmosphere and the low catalyst density on flat electrodes.
Recently, Armstrong's group proposed an enhanced version [97] of their well-known membrane-less enzymatic hydrogen fuel cell [95,96] described above. First, a MWCNT network was used to increase the electrode area. Second, the gas mixture in the unique chamber was switched from H2-poor to H2-rich non-explosive H2/air composition (80/20), forcing the fuel cell from the H2 oxidation limiting regime into the O2 reduction limiting regime. Third, the enzymes: the O2-tolerant E. coli Hyd1 hydrogenase on the anode and bilirubin oxidase BOD on the cathode, were covalently-grafted to pyrene moieties, pyrene having a very high affinity for the CNT surface. The three improvements brought a strong jump in overall fuel cell performance, producing more than 0.1 mW/cm2 at 1 V at room temperature with a relatively good stability over 24 h continuous operation.
Another H2/O2 biofuel cell based on a hyperthermophilic O2-tolerant hydrogenase and bilirubin oxidase was reported this year by Innocent and Lojou [98]. Both electrodes were based on SWCNTs bearing carboxylic functions. Covalent attachment of the enzymes onto nanotubes was achieved through peptidic linkage between the carboxylic functions of the nanotubes and amino residues at the surface of the protein, in the presence of N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) and N-Hydroxysuccinimide (NHS). Under pure H2 and O2 saturated buffer solutions and in the absence of any other redox mediator, the biofuel cell delivers power densities up to 300 μW cm−2 at 0.6 V with an open circuit voltage of 1.1 V. These performances, which are demonstrated to be dependent on hydrogenase characteristics at high potentials, are the best ever obtained. Promising stability of the biofuel cell during 24 h of continuous use has been obtained.
5 Immobilizing bio-inspired catalysts on electrodes: Ni-based electrode materials with exceptional activity for H2 oxidation/production
Synthetic molecular catalysts active in H2 oxidation have been developed later than hydrogenases, so that the need to immobilize them on electrodes arose only very recently. Very few works are, as yet, devoted to this key point, and a major breakthrough was reported by some of the present authors in 2009 regarding novel Ni-based CNT-coated electrodes with remarkable electrocatalytic activities both for H2 evolution and uptake [99,100]. Immobilization strategies have been inspired by methods already proven on hydrogenases, such as covalent grafting on CNT through diazonium functionalization of CNTs or π- π association using pyrene derivatives.
We exploited the outstanding catalytic properties and synthetic versatility of DuBois’ complexes to evaluate the potential of hydrogenase bio-inspired catalysts as heterogeneized electrode materials for applications in hydrogen fuel cells. This led us to synthesize an analogue of DuBois complexes, namely 1(BF4)2 (Fig. 9), in which activated ester functionalities were introduced. An amidation reaction then afforded covalent attachment of the catalyst (Fig. 10) with carbon nanotubes previously decorated with amine groups at their surface through electrochemical reduction of the 4-(2-aminoethyl)phenyldiazonium salt (Fig. 9) [101]. The performances (overpotential requirement and turnover frequency) for hydrogen evolution of the grafted catalyst proved very similar to those measured for the soluble precursor 1(BF4)2 when measured in the presence of p-cyanoanilinium as the proton source and acetonitrile as the solvent.
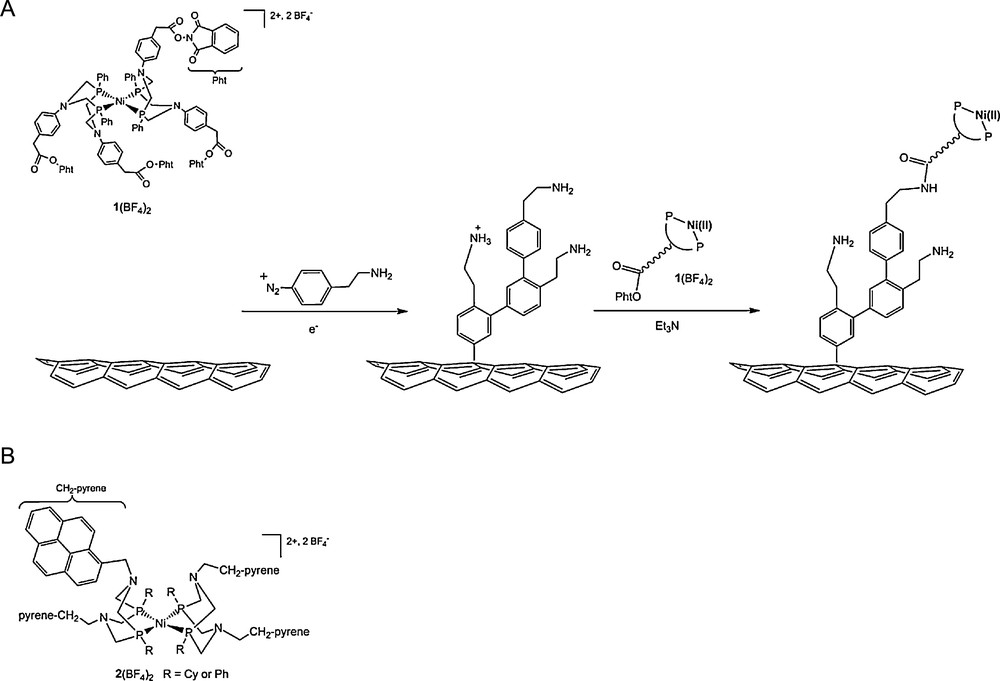
A. Electrografting of amine groups on MWCNTs and post-functionalization with complex 1(BF4)2 through amide links [99]. This is a simplified representation of the structure of the material for sake of clarity: the number of phenylene residues is indeed arbitrary and attachment of the nickel complex to two or more surface amine groups not excluded. B. Schematic structure of the nickel(II) bisdiphosphine complex 2(BF4)2 containing four pyrene π- stacking anchor sites.
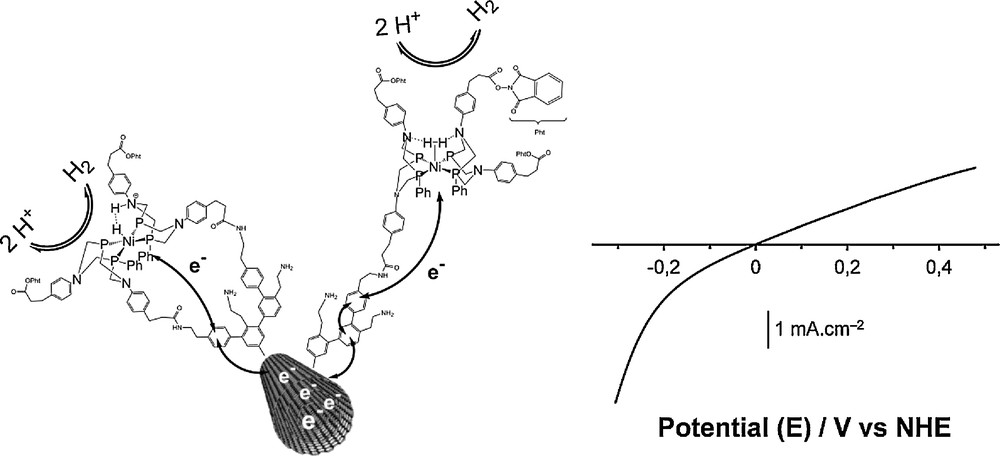
Left: schematic representation of the structure and reactivity of the bio-inspired H2-evolving nickel catalyst grafted on a carbon nanotube [99]. Electrons are exchanged between the carbon nanotube and the Ni centres where H+ is reduced to H2 or H2 oxidized to H+; Right: typical linear sweep voltammogram recorded with the bio-inspired material incorporated in a membrane-electrode assembly with Nafion and assayed in the presence of H2 (1 bar) in an aqueous electrolyte at pH 0.
Deposition of these electroactive Ni-functionalized carbon nanotubes onto a gas diffusion layer, developed for proton-exchange membrane (PEM) applications and made of carbon fibers, and coating with Nafion generated a cheap, stable, air-resistant electrocatalytic material with unique performances especially under the strongly acidic conditions required in the expanding PEM technology. This Pt-free catalyst proved very efficient for hydrogen oxidation from aqueous sulphuric acid solution at the thermodynamic equilibrium with current densities similar to those observed for hydrogenase-based materials (1–2 mA cm−2) under mass-transport limitation. The maximum current density that can be measured in the absence of such limitation (i.e. using a rotating disk electrode on which the catalytic material has been deposited) reach 40 mA cm−2. The material displays exceptional stability (> 30,000 turnovers for H2 oxidation where achieved during a 10-h experiment) and mediates H2 evolution at potentials cathodic to the H+/H2 equilibrium.
The molecular catalyst in this material is located at the crossroads of the three interpenetrated networks allowing percolation of protons (the Nafion membrane), hydrogen (the pores in the gas diffusion layer) and electrons (the carbon fibers of the gas diffusion layer relayed by the conducting carbon nanotubes). In a way and even if it is not as well defined as in the protein, the catalyst environment in this membrane-electrode assembly reproduces that found in the active sites of hydrogenases buried into the polypeptidic framework, but connected to the surface of the protein via a gas diffusion channel, a network of hydrogen-bonded amino-acids for proton transport and the array of electron-transferring iron-sulfur clusters. We should however recognise that this design emerged from the PEM technology and was not bio-inspired, even though it would be tempting to pretend for. As a result of this study and to the best of our knowledge, this has been the first report of a molecular-engineered and noble-metal free electrode material that is capable of achieving hydrogen evolution/oxidation with no overvoltage (Fig. 10). This implies that the grafting on the surface and the coating with the Nafion membrane slightly modifies the local environment of the catalytic metal center, thus improving the catalytic performances. A similar effect has been recently described for Ni-based catalysts in solution through fine-tuning of the diphosphine ligands [64].
We also prepared pyrene derivatives of such nickel bisdiphosphine complexes (Fig. 9B) displaying similar electrocatalytic activities. Immobilisation of these complexes onto CNTs, deposited on gas diffusion layers, is practically a much easier method than the covalent grafting as described above. Interestingly, surface catalyst loading can be easily tailored by varying the amount of CNTs deposited on the electrode. For example, a catalyst surface concentration as high as 8 × 10−9 mol cm−2 was obtained with a CNTs deposit of 0.2 mg/cm2. For comparison the covalent method relying on diazonium electroreduction followed by an amidation post-functionalisation step affords a limited surface coverage of 1.5 × 10−9 mol cm−2 [100]. Furthermore, when stacked onto CNTs the nickel bisdiphosphine complexes show catalytic performances comparable with those displayed by the complexes covalently attached to CNTs. We thus believe that this methodology will be useful to elaborate more efficient electrocatalytic materials.
A very important limitation in the use of platinum nanoparticles as electrocatalysts for H2 oxidation in fuel-cells comes from the poisoning effect of CO [102]. The Cherry on the Cake came from H2-oxidation catalytic studies performed for this class of Ni-functionalized materials performed in the presence of carbon monoxide (CO), a major impurity in H2 fuels derived from reformed hydrocarbons or biomass. We observed that catalytic activity for H2 uptake is sustained in the presence of carbon monoxide (50 ppm) (Fig. 11). Indeed solution studies have shown that binding of CO to nickel(II) bisdiphosphine complexes is either ineffective or reversible without inhibiting H2-oxidation catalysis [103,104]. We found that this property is retained in the materials after grafting. For comparison, commercial MEAs containing highly dispersed platinum (0.5 mgPt cm−2) are rapidly and completely deactivated over minutes under the same conditions (Fig. 11). This constitutes a major breakthrough for Nafion-based proton-exchange membrane (PEM) fuel cell technology, since CO poisoning limits the commercialization of devices based on Pt electrocatalysts.
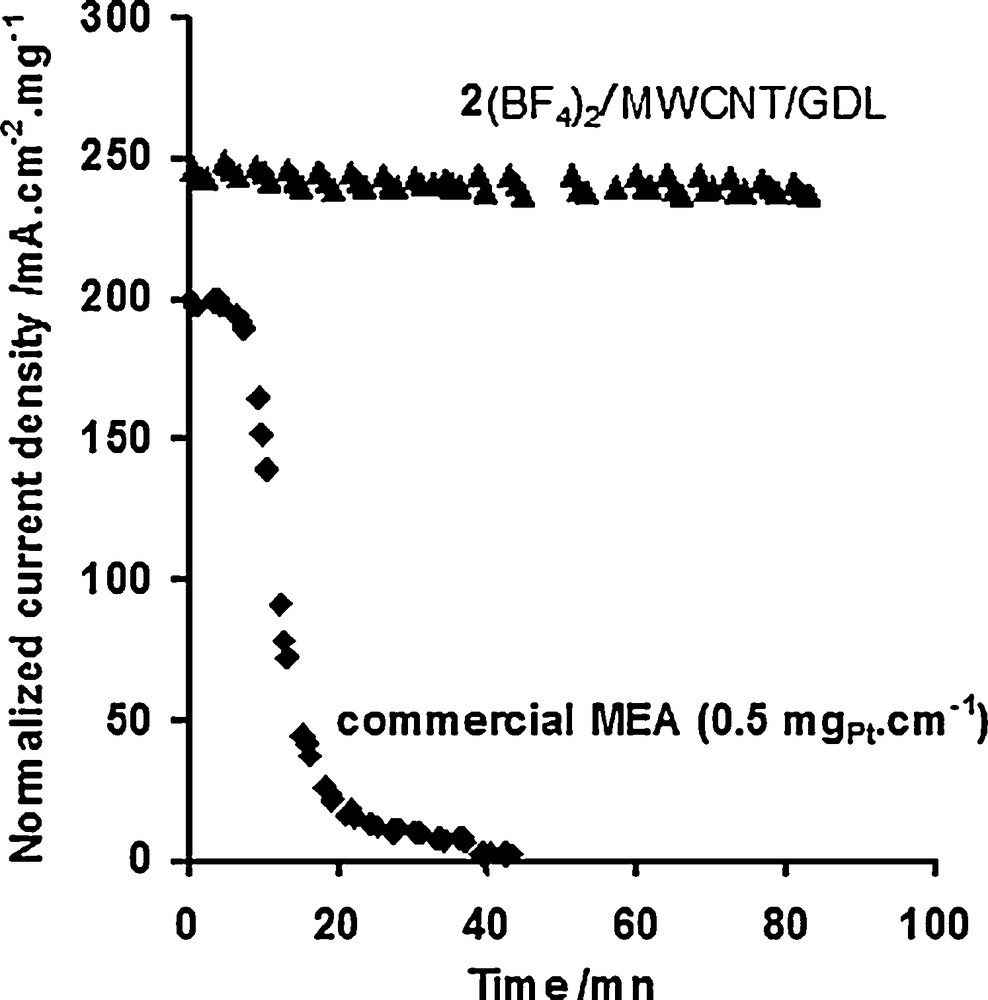
Evolution upon repeated cycling (2 mV s−1) of the H2 oxidation current density per mg of deposited catalyst recorded at 0.25 V vs NHE for a nickel-functionalized MWCNT/GDL electrode (prepared from 2(BF4)2), surface catalyst concentration: 2 ± 0.5 × 10−9 mol cm−2) and a commercial Pt-based active layer (Pt nanoparticles deposited on carbon black, 0.5 mgPt cm−2) under an atmosphere of H2 polluted with 50 ppm CO (flux rate: 20 sccm).
Reproduced with permission from [100].
6 Conclusion
Based on the above results, the assembly of a platinum free hydrogen fuel cell seems within reach. In low temperature hydrogen fuel cells, platinum is also used at the cathode as oxygen reducing catalyst. Oxygen reduction reaction (ORR) is a second important challenge toward a platinum free fuel cell. Indeed, reducing oxygen to water implies the capture of four electrons and several protons (depending on the pH) at each turnover. If partial reduction is performed, the evolved species such as hydrogen peroxide can damage catalysts and electrolyte. This reaction is often described as the most demanding one since, even in the presence of platinum, it does not proceed at the thermodynamic equilibrium (thus resulting in a loss in the energy conversion yield for the entire fuel cell) and because it requires four times more platinum as a catalyst to proceed at a rate comparable with the anode one. Nevertheless, the field is considerably more open and developed than for hydrogen oxidation probably because the materials are not sensitive to air degradation. Many precious metal-free materials have been demonstrated in the past few years with catalytic efficiency approaching that of platinum. A comprehensive review can be found elsewhere [105].
One point thus strikes one when considering the assembly of a platinum free hydrogen fuel cell: air-resistant hydrogen oxidation catalysts are rare. The functionalization of CNTs with bio-inspired molecular complexes thus appears as a straightforward methodology to prepare highly robust, CO-tolerant, noble-metal-free and bidirectional electrocatalytic nanomaterials for H2 evolution and uptake, compatible with the conditions encountered in classical proton-exchange membrane devices. The materials reported here can be prepared in one or two steps from CNTs and functionalized complexes and then deposited onto any electrode support. H2 oxidation current densities as high as those measured when native hydrogenase enzymes are immobilized onto carbon-based electrodes could be measured and specific current densities (i.e. referred to the mass of immobilized catalyst) are comparable to commercial active layers containing highly dispersed platinum (compare initial values in Fig. 11). The bio-inspired nanomaterials proved stable in the air for months with full retention of their activity. Although aging tests have been limited to few hours of continuous catalysis, this methodology thus paves the way for the implementation of noble-metal free electrocatalytic materials in operative PEM devices, the remaining challenge consisting in significantly improving the catalytic loading so as to reach the power performances displayed by commercial Pt-based active layers.
7 Experimental
7.1 Hydrogenase-SWCNT biohybrids
Biohybrids were prepared with HiPco or arc discharge SWCNTS. Arc discharge SWCNTs were purchased from the Carbon Solutions Inc. company (1.2 to 1.4 nm in diameter). HiPco SWCNT were from NT@Rice (0.9 to 1.1 nm in diameter). HiPCO SWCNT were used as received. Ultrasound sonication was used to suspend CNT powder in a surfactant solution of 2.5% sodium cholate in H2O. CS SWCNT were purified by 30 min of ultrasonication in a bath at 100% with HNO3 followed by refluxing in a mixture of 70 mL HNO3 and 40 mL H2O for 60 h. The suspension was filtered three times with Pall Life Sciences GH Polypro 0.45 μm hydrophilic filters. A washing step with 50 mL of H2O followed each filtration. The filtrate was resuspended in 200 mL of 12.5 mM NaOH aqueous solution twice and in 2% F127 in H2O after the third filtration using an ultrasound sonication bath. The CNT were heated to 70 °C and ultrasonicated at 100% in a bath for 3 to 4 h. The suspension was then run through a GE Healthcare Sephacryl S-500 High Resolution size-exclusion column while still warm. CNT-containing fractions were identified through UV-Vis spectra and pooled. Typical SWCNT concentrations were in the range 10 to 50 mg L−1.
The SWCNTs were functionalized with a Boc-protected 4-aminoethylbenzenediazonium salt synthesized on purpose.
7.2 Boc-protection of 4-(2-aminoethyl) aniline
An amount of 138 mg of 4-(2-aminoethyl) aniline was mixed with 1 mL H2O and 1 eq. (Boc)2O at room temperature and agitated on a rotating support for 30 min. A yellow powder (114 mg, 48%) was recovered by filtration with a glass fiber filter and dried under vacuum. The product was stored out of light at ambient temperature. IR spectra showed a carbonyl band at 1700 cm−1.
7.3 Synthesis of Boc-aminoethylbenzenediazonium tetrafluoroborate
An amount of 200 mg of Boc-protected aniline was dissolved in distilled acetonitrile, agitated by magnetic stirrer and brought to –48 °C by a bath of iced acetonitrile. 1 eq. NOBF4 dissolved in distilled acetonitrile was added drop by drop. The reaction was carried out for 90 min and allowed to come to room temperature. The resulting oil was triturated with ether and dried using a rotatory evaporator kept under argon followed by a recrystallization in acetonitrile/ether to obtain a light brown solid (82 mg, 27%).
7.4 Amino-SWCNTs
An amount of 3.3 mg (0.01 mmol) of Boc-aminoethylbenzenediazonium tetrafluoroborate was dissolved in 0.5 mL of a water/ethanol 1:1 mixture and added to 4 mL of SWCNTs (35 mg L−1) in F127 1% aqueous solution and allowed to react overnight. SWCNTs were separated from by-products and rinsed by ultracentrifugation (200,000 g, 1 h 20 min twice). Boc deprotection of Boc-aminoethylbenzene-SWCNTs was performed in TFA 95% for 4 h. After dilution in 10 volumes of water, amino-SWCNTs were recovered by ultracentrifugation (same conditions).
7.5 Hydrogenase-SWCNT biohybrids
A 90 μL aqueous reaction mixture with final concentration of 2 μM EDC, 5 μM sulfo-NHS, 150 mM NaCl, 1 μM [NiFeSe] hydrogenase or 1 μM [FeFe] hydrogenase, and 50 mM MES pH 6 was agitated for 20 min at 800 rpm and 25 °C. The coupling reagents were dissolved just before the reaction was started. An amount of 110 μL of amino-SWCNT solution buffered to pH 8 by a 0.15 M phosphate solution was added to the enzyme activation reaction mixture and left under agitation for 16 h. The [NiFeSe] biohybrid was ultracentrifuged at 150,000 g for 80 min at 10 °C. The supernatant liquid was removed and remaining viscous liquid was used for AFM imaging and electrochemical tests. The reaction mixture for [FeFe] was filtered using a 0.2 μm PFTE filter and rinsed with five volumes of the mixture of buffer and salt solution and H2O. The filter was dried in an oven at 50 °C before undergoing XPS analysis.
Acknowledgments
The authors acknowledge the financial support of the French National Research Agency (EnzHyd project, Grant ANR-08-PANH-008) and the Nanoscience program of CEA.