

1 Introduction
One of the most promising solutions to the anticipated crisis in future energy production resides in the combination of two renewable resources, water and sunlight [1]. Conversion of solar energy to chemical energy through the light-driven reduction of water into hydrogen not only provides an attractive approach to store the abundant flow of sunlight falling on earth but also to generate a fuel with the highest energy output relative to molecular weight that can be used to generate electricity in fuel cells with very high energy conversion efficiencies [2]. Furthermore hydrogen is a carbon-neutral and environmentally friendly fuel since it generates only water as a by-product during reaction with oxygen in a fuel cell.
A first issue related to water splitting resides in the multielectronic nature of the reactions at work, resulting in kinetic limitations. Thus the development of photoelectrolyzers requires efficient electrocatalysts, proceeding at diffusion controlled rates without demanding large driving forces called overpotentials [3]. Practically, the overpotential, which corresponds to the extra energy required for the redox reaction as compared to that defined by its standard potential, should remain below 0.2 V for a process to be economically viable. This requirement applies both for the water oxidation and the water reduction reactions. So far, the most efficient electrocatalysts are based on noble metals, in particular platinum, in a pure form or as alloys. However, the wide and continued application of Pt-based systems is highly doubtful because of the limited amount of this metal on our planet and its high cost [4]. Intense research activity in this field has led to the development of new electrocatalytic materials based on non noble metals [5].
A second important aspect resides in the development of efficient and stable photosensitizers (PS), namely with high extinction coefficients in the visible region and long-lived excited states allowing for electron transfer from or to the photo-excited state. These components serve to absorb sunlight photons, in place of water which is unable to do so, and induce a charge separated state combining “holes” with the potential for water oxidation to oxygen and highly reducing “electrons” for water reduction to hydrogen. Current research takes advantage of the interesting photo-physical and -chemical properties of molecular compounds, such as inorganic complexes and organic dyes, as well as of semiconducting solid materials such as metal oxides.
With this combination of multielectronic catalysts and light-harvesting materials, a simple photoelectrochemical cell such as that shown in Fig. 1 can be built. It is interesting to note that by doing so, we reproduce some of the basic components at work during biological light-driven water-splitting processes in photosynthetic microorganisms. First, Photosystem II absorbs solar energy and generates a “hole” used for water oxidation catalyzed by a unique Mn4Ca complex. In the cell of Fig. 1, this process is achieved at the photoanode. Second, the electrons extracted from water are transferred through a long-range pathway, thus preventing charge recombination, to an enzyme, a hydrogenase, which catalyzes the reduction of protons to hydrogen. In the solar cell of Fig. 1, the electrons generated at the photoanode are collected at the cathode where a catalyst operates the production of H2. Proton transfers, not discussed here, in both biological and chemical devices, are crucial processes which have to be optimized.
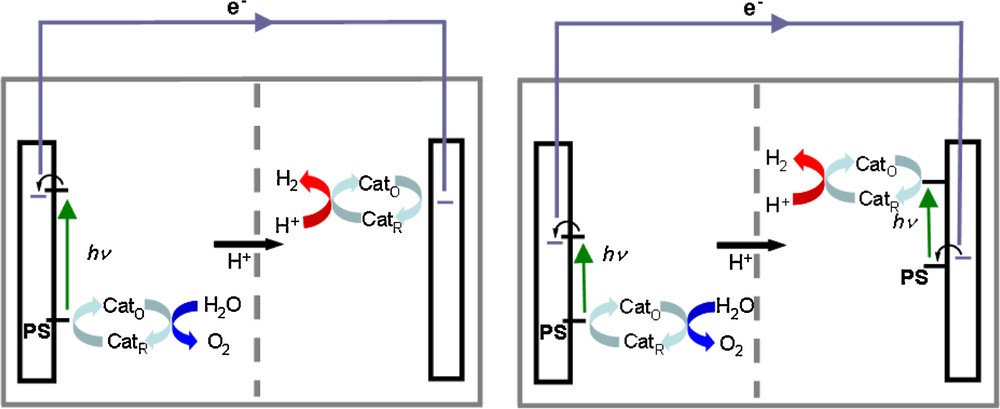
Schematic representation of photoelectrochemical cell for water splitting based on one (left) and two (right) photosensitizer (PS). Cat and Cat’ are catalysts for water oxidation and reduction, respectively, existing in the reduced (R) and oxidized (O) states.
To provide a complete view of the problem, it should be added that in frequent cases the electrons extracted from water do not have the minimal energy required for reducing the protons and thus need to be re-energized. Thus a photocathode rather than a cathode is required in the technological device (Fig. 1, right). The same occurs in living organisms which have evolved a second photosystem (Photosystem I) for that purpose [6].
In this review, we will first give an overview of the photoelectroelectrocatalytic systems based on tridimensional solid materials before describing the recent achievements for the construction of systems for water splitting based on molecular components, either enzymes or mimics.
2 Photocatalytic water splitting using semiconductor materials
The seminal work on that topic was reported by Fujishima and Honda in 1972 with a water splitting cell made from a TiO2 photoanode and a Pt cathode [7]. Utilization of simple semiconductors as PS such as TiO2, with wide band gaps and displaying both valence band edge potential competent for oxygen evolution and conduction band edge potential competent for proton reduction, is however challenging since these materials hardly absorb visible light but UV radiations. Two solutions have been developed to circumvent this limitation. The first one exploits the properties of other semi-conductor materials with smaller band gaps, such as WO3, α-Fe2O3 or GaInP2, that overlap with the solar spectrum but in that case the band-gap is not properly positioned for water splitting. In the case of WO3 and α-Fe2O3 for instance, the potential for the photoexcited electron in the semiconductor is more positive than the potential needed to reduce water to H2 (Fig. 2). By contrast, the photogenerated holes in GaInP2, Si or GaAs are not oxidizing enough to promote oxygen evolution from water at pH 1. As a result, the photoelectrochemical cell should be voltage biased with another photovoltaic device in a tandem cell configuration. The second solution consists in sensitizing TiO2 with molecules–typically ruthenium diimine complexes but first-row transition metal complexes and organic dyes have proved competent for that use–that are covalently grafted at the surface of the oxide [8]. A third alternative relies on the introduction, through hydrogenation, of disorders in a TiO2 layer which consequently becomes black and thus absorbs a large part of the visible solar spectrum [9]. Exploitation of such a material for water splitting has not been reported so far.
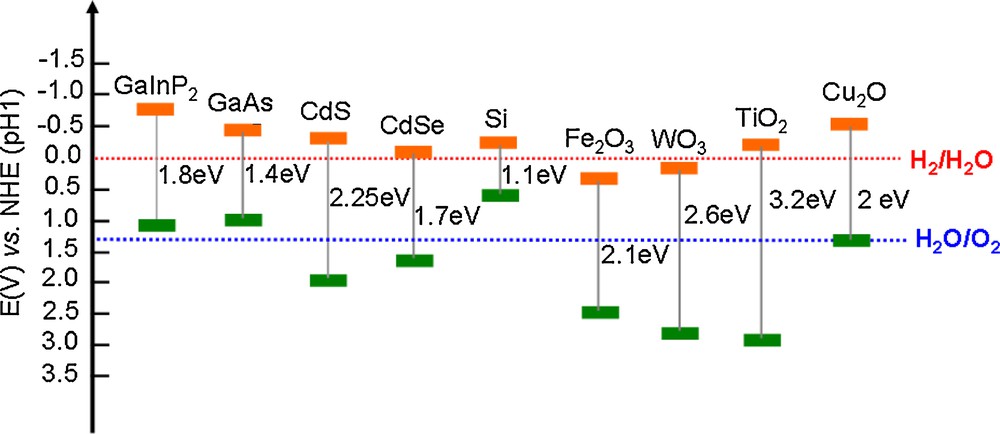
Band positions of several semiconductor materials in contact with aqueous electrolyte at pH 1 (green lines represent the upper edge of the valence band whereas the red ones represent the lower edge of the conduction band). The visible spectrum corresponds to energies from 1.56 eV (800 nm) to 3.12 eV (400 nm).
Three examples of operating cells are known in the literature. The monolithic photovoltaic-photoelectrochemical device reported by Khaselev and Turner combines a GaInP2/GaAs p/n, p/n photovoltaic cell and two Pt-based active layers (Fig. 3a) [10]. Rocheleau et al. initially designed a similar hybrid-junction photoelectrode configuration, incorporating cheap, environmentally benign and highly-stable O2-evolution photocatalysts, such as α-Fe2O3 or WO3, deposited on top of a solid-state junction to generate the bias for water reduction on a platinum electrode (Fig. 3b) [11–14]. These first examples display quite good efficiencies for energetic conversion (12.4% for the cell reported by Khaselev and Turner) but are very expensive due to the fabrication of the photovoltaic assembly and the presence of Pt as a catalyst. Platinum-free devices are also developed integrating a multi-junction photovoltaic device based on amorphous silicon, and CoMo (hydrogen evolution)/Fe:NiOx (oxygen evolution) electrocatalytic layers working under basic conditions, though with associated corrosion issues [15,16]. The last example is the so-called tandem-cell developed by M. Grätzel at EPFL (Switzerland) developing 3.5 mA cm−2 with an overall yield of ∼5% that consists of a WO3/Pt or a Fe2O3/Pt photoelectrochemical cell biased with a Dye Sensitized Solar Cell (Fig. 3c) [8]. It is important to note that, in the three examples shown in Fig. 3, hydrogen evolution is catalyzed at the surface of a platinum electrode. Platinum is also sometimes used to catalyze oxygen evolution. When WO3 or α-Fe2O3 is used at the anode, oxygen evolution is not really catalyzed and occurs thanks to the high driving force provided by the very positive potential of the valence band of the oxide material: however photogenerated valence band holes are kinetically inefficient for abstracting electrons from water because of low conductivity properties that limit their diffusion to the surface [17,18]. Recent studies have demonstrated that coating of these materials with a catalytic layer [19–23], such as the cobalt oxide deposit described by Nocera [24,25], results in a significant reduction of the overvoltage for water photo-oxidation.
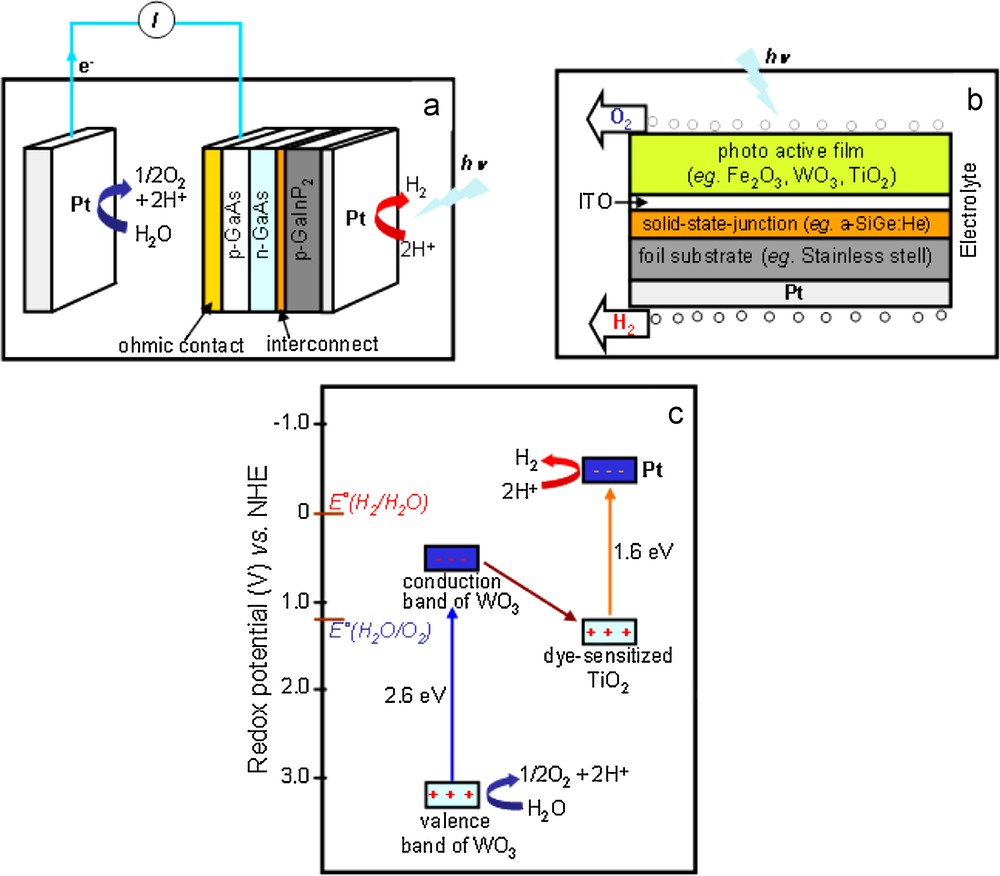
Selected devices for photocatalytic water splitting: (a) Turner's monolithic photoelectrochemical-photovoltaic design [10], (b) Hawaï hybrid planar multijunction photoelectrode [11–14] and (c) principle of Grätzel's tandem cell [8].
Current research is made toward the use of materials absorbing in the visible spectrum and being competent for both hydrogen and oxygen evolution [26]. CdS or CdSe materials are rather unstable under continuous irradiation [27]. Promising materials include nitride [28] or oxynitride [29,30] compounds and composite oxides such as In1−xNixTiO4 [31]. Further combinations of dye-sensitized nanostructured TiO2 layers with nanoparticulate catalysts are developed, namely by the group of Mallouk [32]. However in both approaches, noble metal compounds are typically used as catalysts or cocatalyts for hydrogen evolution (Pt) and water oxidation (Pt, RuO2 or IrO2).
3 Photocatalytic water splitting using molecular materials
In line with the biological solutions to photocatalytic water splitting, an emerging approach to the design of a photoelectrochemical cell is molecular electrocatalysis and photochemistry. In particular “bioinspired electrocatalysis” has recently emerged as an interesting and productive strategy in that respect.
The design of enzyme mimics has been greatly facilitated by the detailed characterization, at the atomic level, of the active sites of the natural systems, photosystem II [33–37] and hydrogenases [38]. Obviously, their active centres are attractive sources of inspiration because of their relative structural simplicity and of their utilization of first-row non-noble transition metals, manganese and calcium for water oxidation, nickel and iron for water reduction, to achieve catalysis. During the last years, this approach has provided a wealth of very interesting active compounds on which technological developments can be built up.
As discussed above, another important aspect regarding water photolysis is light collection. We mentioned above that there is still a need for an electrode material which has the ability to collect the visible portion of the sunlight. Again molecular science may provide original solutions through the development of both inorganic PS, such as diimine complexes of ruthenium, iridium, rhenium or copper, and organic dyes. Efficient combinations of electrocatalysts with PS will then yield new photocatalysts [39–42]. This field also benefits from all the knowledge in photonics accumulated in the context of dye-sensitized solar cells implying the grafting of molecular PS on metal-oxide surfaces and related light-driven electron transfers [8]. Again bio-inspiration can lead to the design of smart linkers that may favour proton-coupled electron transfers within the system [43–45].
Nevertheless, it appears clearly that the molecular electrocatalysis approach will demonstrate its practical potential, as far as solar cells are concerned, only through the development of methods for the immobilization of the best molecular electro- and photocatalysts on cheap electrode materials, either carbon or transparent metal-oxides, and the evaluation of these new original electrode surfaces. Here we describe some of the very first attempts to design such electrode materials based on molecular PS and/or catalysts that can be used for water electrolysis and photoelectrolysis. The examples discussed herein indeed show that the immobilization approach very often results into increased catalytic activities and robustness of the catalytic centers and allows the exploitation of water-insoluble complexes.
4 Photocatalytic H2-evolving systems and photocathode design
While early achievements in this field have been made with multi-component systems where PS and catalysts are free in solution, a series of supramolecular photocatalytic H2-evolving systems have been recently reported based on the assembly of either photosystem I and hydrogenase or inorganic PS and bio-inspired catalysts.
4.1 Photocatalytic bio-constructs
Goldbeck et al. have used a dithiol ligand to connect a [4Fe-4S] cluster of Photosystem I directly to the distal [4Fe-4S] cluster of the [FeFe]-hydrogenase from Chlostridium acetobutylicum. The complex is able to use ascorbate as the sacrificial electron source and produces hydrogen under illumination [46]. A similar system (Fig. 4) was constructed by Heberle et al. using the membrane-bound hydrogenase from Ralsthonia eutropha H16 fused with an extrinsic subunit (PsaE) of an histidine-tagged form of PS I [47]. This system has been immobilized at the surface of a nickel-functionalized gold electrode through a nickel His–Tag interaction. When this electrode is poised at −0.09 V vs NHE, so as to reduce the soluble electron carrier N-methylphenazonium methyl sulphate (PMS), and under visible illumination (λmax = 700 nm), photocurrents are observed (85 nA cm−2), 30% of which could be assigned to hydrogen evolution [48].
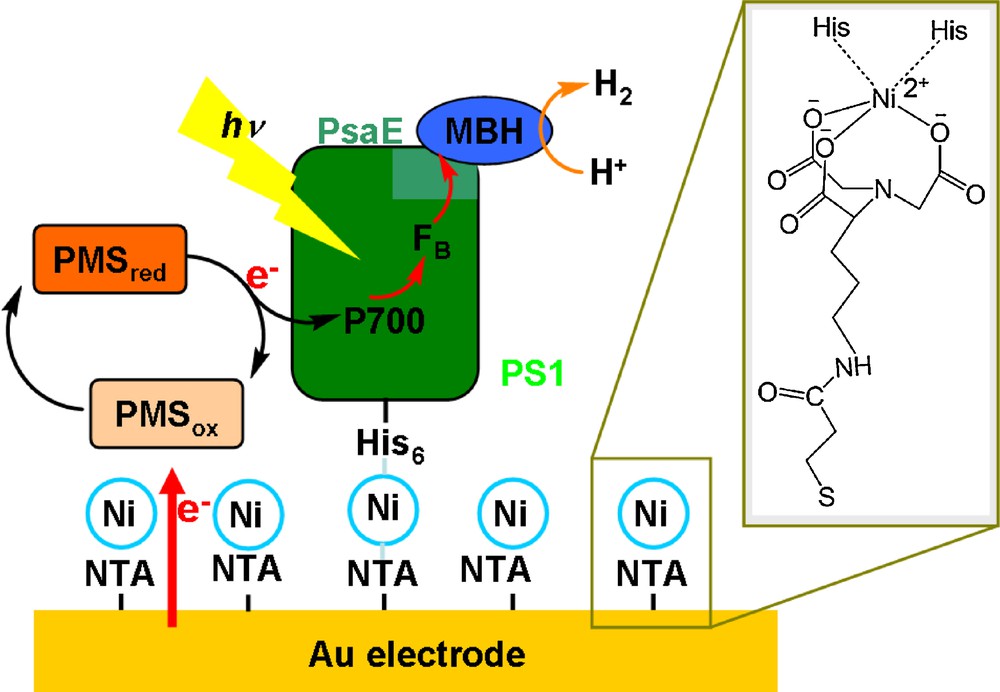
Structure of the biochemical H2-evolving photoelectrode designed by Heberle et al. [48].
In a slightly different approach F. A. Armstrong et al. adsorbed the [NiFeSe]-hydrogenase from Desulfovibrio baculatum, a very efficient H2 producer and rather O2-resistant enzyme, onto a thin film of nanocrystalline TiO2, sensitized by a dye (a light-absorbing Ru-tris-diimine complex in which a bipyridine ligand contains phosphonic acid groups providing a stable chemical linkage to Ti atoms) and deposited on a conducting ITO-coated glass support (Fig. 5) [49,50]. Irradiation with visible-light in the presence of a sacrificial electron donor provided an efficient generation of H2, with initial turnover frequency of 50 s−1 and turnover numbers of 10,000 and 100 based on hydrogenase and photosensitizer, respectively, after 8 h irradiation. However, some photoinstability of the system is observed with time. Similarly, the [NiFe]-hydrogenase from Thiocapsa roseopersicina, adsorbed on CdS particles that function as the photosensitizer, catalyzes formate decomposition into H2, CO and CO2 [51].
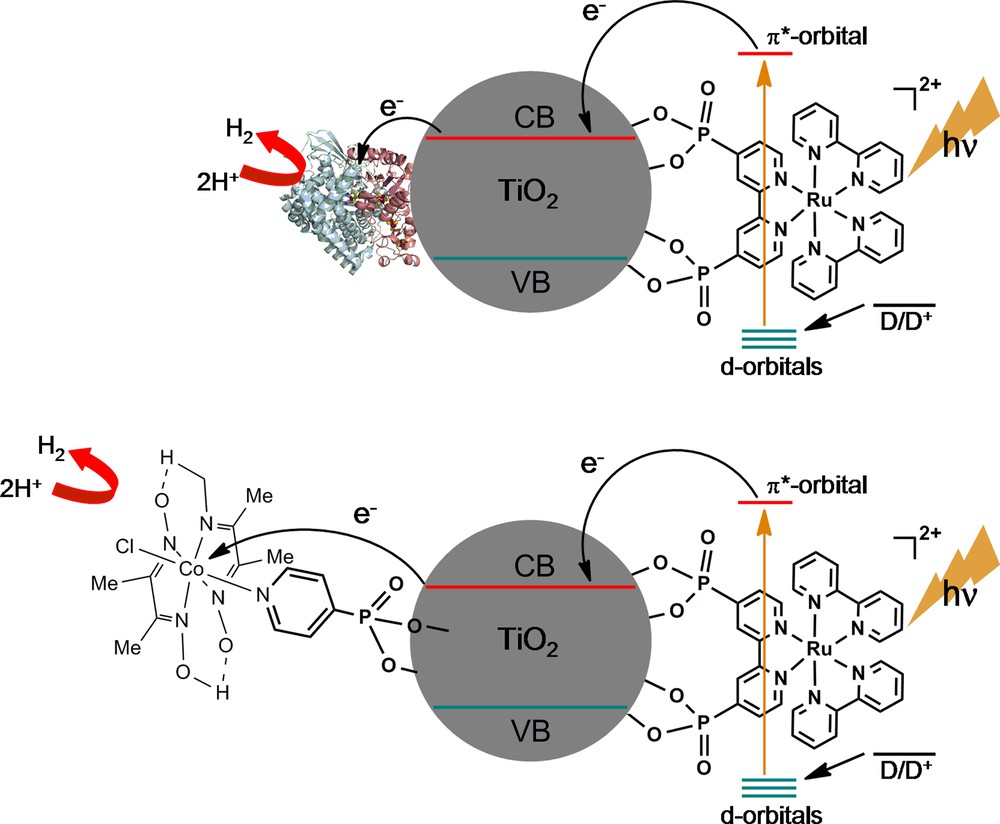
Structure of the TiO2-based material for H2 photoproduction using surface-immobilized hydrogenase [49,50] or cobaloxime as catalyst [61].
4.2 Synthetic H2-evolving photocatalytic systems
The last 5 years have seen the growing development of synthetic photocatalytic assemblies for hydrogen evolution [40,52] combining a photosensitizing unit (a metal-diimine complex) and an H2-evolving catalyst. Here we will focus on supramolecular systems based on first-row transition metal catalysts since they constitute a first step towards the construction of photocathode materials. The first supramolecular system using non-noble transition metal catalysts, in that case cobaloximes [2,53], has been reported by our group in 2008 only (Fig. 6) [40–42,54]. Similar systems were later on reported by the groups of M. Wang and L. Sun in Dalian [54] and K. Mulfort and D. Tiede in Argonne [55]. Such assemblies display activities far superior to those of the previously known photocatalytic systems, based quite exclusively on noble metals such as platinum as H2-evolving catalysts. Another strategy recently developed by Sakai et al. relies on the spontaneous self-assembly of bipy-appended cyclometallated Ir PS in the presence of CoII ions, thus in situ generating [Co(bipy-l-Ir)n]2+–type species (Fig. 6) [56]. In the presence of triethanolamine (TEOA), as an electron donor, these systems mediate light-driven H2 production in CH3CN/H2O mixtures with up to 20 turnovers. Multi-component systems tested under the same experimental conditions proved to be twice less efficient, again pointing to the importance of a supramolecular design.
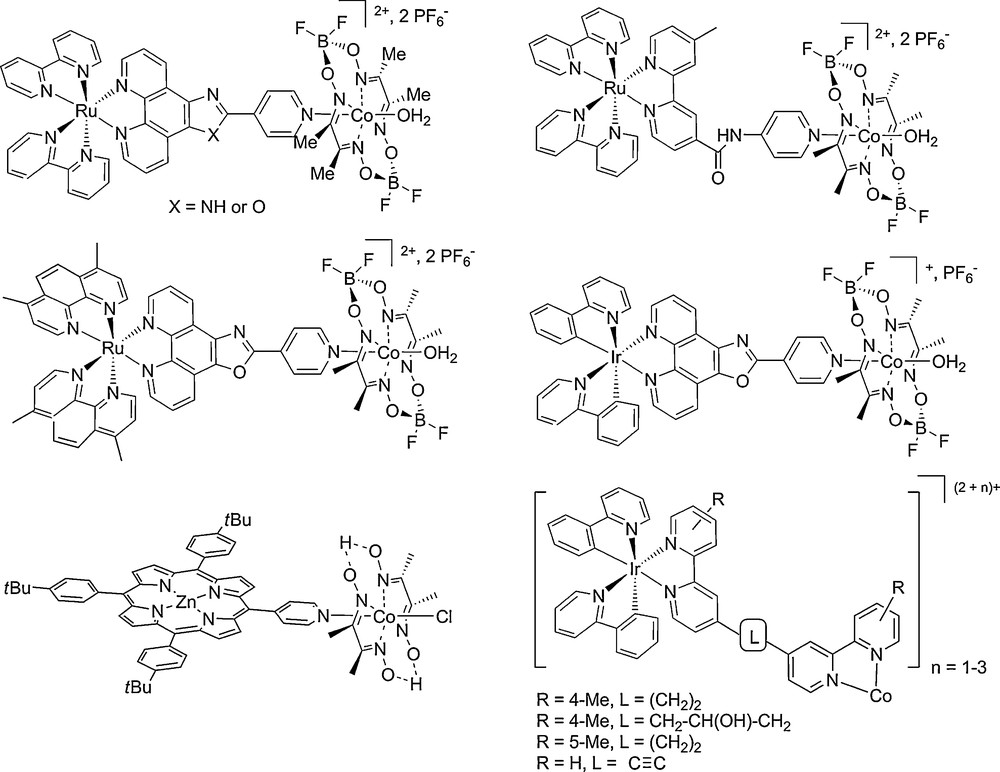
Structure of selected supramolecular photocatalysts for light-driven hydrogen production [41,42,54,56,57].
Finally, noble-metal free supramolecular devices have been reported by the group of Zhang et al. [57]. They have assembled pyridyl-functionalized zinc porphyrin photosensitive units to a cobaloxime complex via axial Co coordination (Fig. 6). TONs up to 22 are achieved in a 5-h experiment. Under the same experimental conditions, a multi-component catalytic system does not evolve any detectable amount of H2. Other supramolecular noble metal-free systems based on a porphyrin sensitizer and diiron mimics of the active site of FeFe hydrogenase have been reported but with very low H2-evolving activity [58,59].
So far, none of these systems has been immobilized on surfaces. Kölle and Grätzel have reported on a {Cp*Rh(bipy)} catalyst grafted on TiO2 particles via carboxylate anchors, catalyzing H2 evolution under UV-light in the presence of sacrificial electron donors [60]. More recently, a cobaloxime has been electronically connected to the photosensitizer [Ru(bipy)2(2,2′ bipyridine-4,4′-diylbis(phosphonic acid)]2+, with a ratio PS/catalyst = 3, via a TiO2 particle (Fig. 5), with phosphonate anchoring groups on both molecules allowing tight grafting onto the surface of TiO2 [61]. Hydrogen (53 TON) is evolved when this system is placed in pH 7 TEOA aqueous buffer and exposed to visible light. This design takes advantage of the ultrafast electron injection from the excited state of the photosensitizer into the conduction band of TiO2, from which they are further transferred to the catalyst.
Until very recently, the only systems for hydrogen photoproduction based on heterogeneized functional molecular H2-evolving catalysts use semi-conductors for light harvesting and charge separation: coating of a p-type silicon material with polystyrene functionalized with 1-methyl-[1.1]ferrocenophane (Fig. 7) provided a H2-evolving photocathode: under illumination, the voltage photogenerated within the silicon layer allows proton reduction to occur with an underpotential of 300 mV, i.e. the proton reduction occurs at a potential 300 mV more positive than the standard apparent potential of the H+/H2 couple [62].
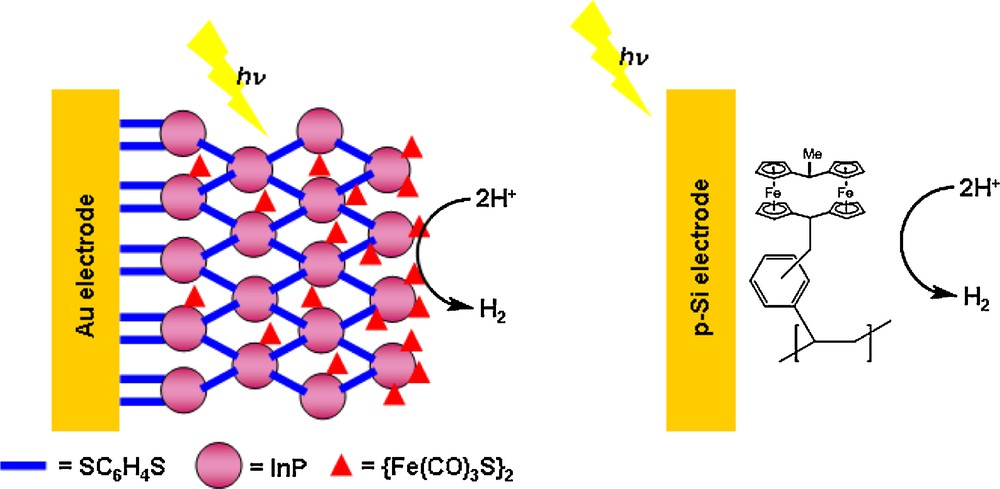
Structure of the H2-evolving photocathode based on InP nanocrystals and a diiron hexacarbonyl complex [63] and of that based on a p-type Si electrode covered with a [1.1] ferrocenophane derivative [62].
A similar approach has been recently reported by Nann et al. aiming at building a H2-evolving photoelectrode (Fig. 7). Nanocristals of InP, a semiconductive material absorbing a significant part of the visible spectrum, have been deposited and connected, through interactions with 1,4-benzenedithiolate, on a gold electrode using a layer-by-layer procedure. [Fe2(μ-S)2(CO)6] has then been adsorbed as a H2-evolving catalyst on this material and photocurrents (∼250 nA cm−2), associated with H2 evolution from neutral aqueous solution, could be measured under illumination with 395 nm LED, while the electrode was poised at −500 mV vs Ag/AgCl, thus 250 mV more positive than the standard potential of the H+/H2 couple [63].
5 Photosystem II and model complexes
In nature, the photo-driven oxidation of water to oxygen is catalyzed by Photosystem II, present in photosynthetic organisms, and occurs at a unique metal cluster (the Oxygen-Evolving Centre [OEC]), which has the potential to accumulate, one by one, four oxidizing equivalents that can be transferred to two metal-bound molecules of water. Excellent review articles, describing how our understanding of this catalyst has progressed, most often with controversies, are available [64–67]. In summary, even though the structure of the cluster is incompletely established, the most recent evidence strongly suggests that it is constituted of a mixture of manganese and calcium ions in a Mn4Ca stoichiometry, likely in the arrangement of a Mn3Ca cubane where ions are bridged by oxo groups, with a fourth manganese ion linked to this cluster [37].
Badura et al. succeeded in attaching a Photosystem II modified with an His tag and isolated from the cyanobacterium Thermosynechococcus elongatus onto gold electrodes modified with thiolates bearing terminal Ni(II)-nitrilotriacetic (NTA) acid groups. The surface coverage was found to correspond to one monolayer and, upon illumination in the presence of 2,6-dichloro-1,4-benzoquinone as the electron mediator, generation of a photocurrent up to 14 μA cm−2 was observed [68], corresponding to the highest activity measured for PSII in solution under similar conditions. Higher amounts of the enzyme could be immobilised on the electrode via osmium-containing redox polymers based on poly(vinyl)imidazole. In that case, the redox polymer works as both the immobilization matrix and the electron acceptor for the enzyme. Thus catalytic currents as high as 45 μA cm−2 can be measured under illumination (2.65 mW cm−2) without the use of any soluble redox mediator [69].
Manganese, the “biological solution”, thus appears as a very relevant target for water oxidation catalysis and thus extensive research is carried out to find efficient Mn-based synthetic systems and utilize them in technological applications. Unfortunately so far, functional analogy to the OEC has been only rarely achieved with Mn-based systems and in all cases small turnover numbers have been obtained.
An interesting model system, reported by Dismukes et al., is a synthetic Mn4O4L6 cubane-like complex, bearing diarylphosphinate l ligands (Fig. 8), which experiences conversion of its bridging oxides into O2 upon photooxidation in the gas phase [70]. Binding of the oxidized form of this complex, [Mn4O4L6]+, to a thin proton-conducting Nafion membrane deposited on a conducting (glassy carbon) electrode yields a robust catalyst that sustains the photoelectrooxidation of water to O2 [71–74]. When this electrode is polarized above the [Mn4]2+/[Mn4]+ potential at 1.00 V vs Ag/AgCl (electrical bias) and illuminated with filtered Xe light (λ > 275 nm), a large photocurrent is observed corresponding to the formation of O2, which was found by labelling experiments to derive exclusively from water. Catalytic turnover frequencies of 20–270 per Mn4 unit and per hour and turnover numbers > 1000 were reported. It was proposed that light activates the catalyst through photodissociation of a phosphinate ligand, which allows O2 formation, followed by reassembly of the cubane structure. Recently, as a variant of the tandem cell developed at EPFL [8], this photocatalyst was entrapped in Nafion polymer and deposited onto a nanostructured TiO2 electrode sensitized with a Ru-diimine dye. This photoanode, that uses two photons to sustain water oxidation, was coupled to a platinum electrode for the construction of a photo-electrocatalytic cell achieving overall water splitting (Fig. 8) [75,76]. This is the only active water oxidation photoelectrode material made from molecular mimics of the OEC so far. However, it has been recently found that the active species for water oxidation in this device is a disordered Mn(III/IV)-oxide phase [77]. Other manganese oxide materials, incorporating calcium ions, have also been shown to catalyze water oxidation efficiently [78].
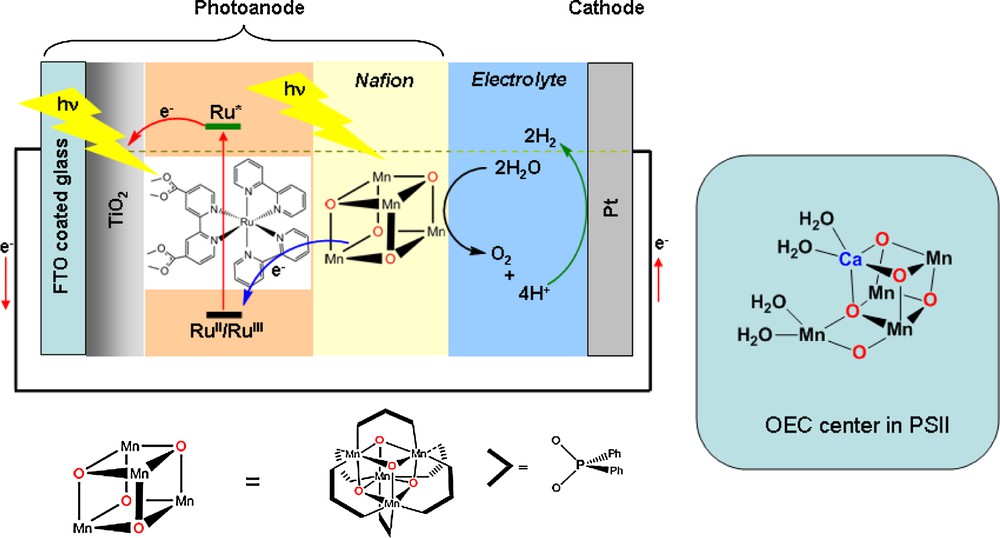
Structure of the structure of the oxygen-evolving center in PSII [37] and schematic representation of the photoelectrochemical device for overall water splitting reported by Spiccia et al. [76].
Catalysis of water oxidation using coordination compounds has been successful mostly with ruthenium complexes, following the pioneering work of T.J. Meyer on a family of dinuclear oxo-bridged Ru complexes containing di-imine and water ligands, the prototype being the so-called “blue dimer” containing bipyridine ligands (Fig. 9). This field has been recently discussed in few review articles [79–81]. Here, we discuss exclusively those, very few, which have been integrated within photochemical systems.
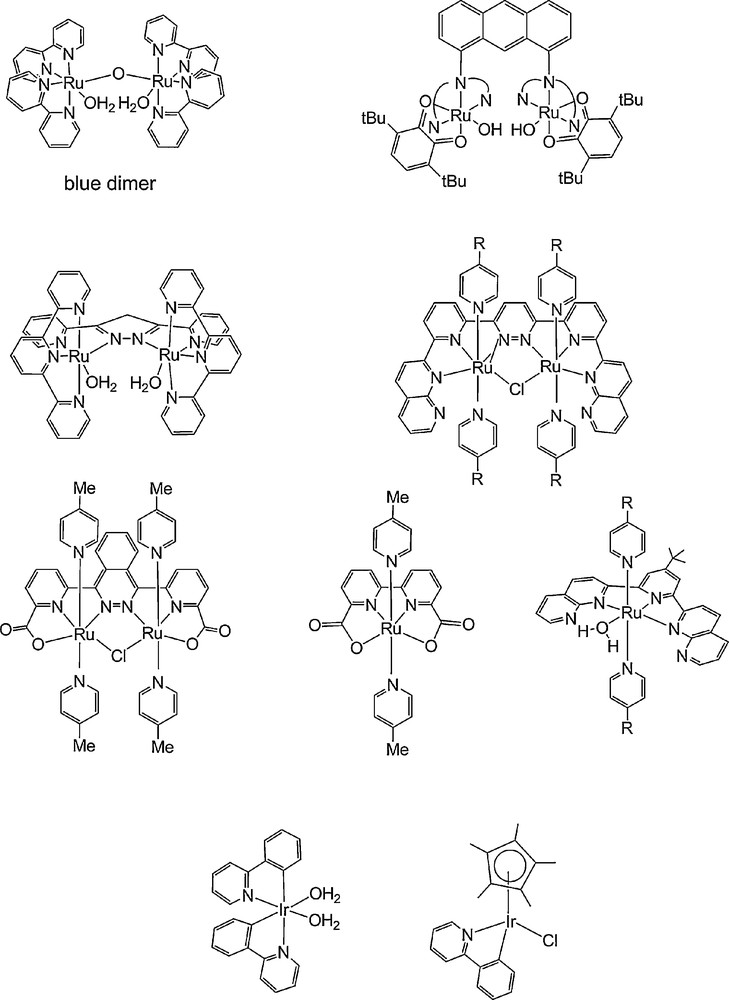
Selected Ru [84,86,108–111] and Ir-based [90,91] active catalysts for water oxidation.
The second generation of very active dinuclear Ru complexes incorporate an organic bridging ligand in place of the oxo bridges, thus providing more stability and more rigidity, favouring O–O bond formation. As prototypes for this class of catalysts, one can mention: (i) Tanaka's complex, in which two terpyridine (trpy) moieties, each one chelating a Ru atom, are attached to an anthracene unit and the coordination is completed by a bidentate chelating ligand such as a bipyridine or an orthoquinone; (ii) Llobet's complex, with a Hbpp (3,5-bis(2-pyridyl)pyrazole) bridging ligand and a trpy terminal ligand (Fig. 9) [82,83]. This type of system was further improved by Thummel et al. who integrated the different ligands into a polydentate dinucleating neutral ligand that contains two naphtyridyl groups coupled to a central bispyridylpyridazine unit (Fig. 9) [84] and by L. Sun et al. who introduced carboxylate ligands in order to lower oxidation potentials and stabilize higher oxidation states, resulting indeed in improved catalytic performances both during oxidation of water by Ce4+ or during light-driven water oxidation [85].
The third generation came with the discovery that there is no absolute requirement for polynuclear complexes for catalytic activity: a number of mononuclear Ru [86–89] and also Ir [90–92] complexes have been found to behave as excellent catalysts for conversion of water to O2 using chemical oxidants such as CeIV, or electrochemical oxidation. Turnover numbers ranging from 50 to 2500 have been reported. Some examples are shown in Fig. 9. Light-driven O2-evolving systems have been obtained based on such mononuclear Ru catalysts [88].
One should also mention the interesting use of tetranuclear ruthenium derivatives of polyoxometallate as catalysts for water oxidation [92–94]. For example, the complex [RuIV4(O)4(μ-OH)(H2O)4(γ-Si-W10O36)2]10− catalyzes the oxidation of water by CeIV with a turnover number of 385 and a yield of 90% during a 2-h reaction [94]. This is a remarkably stable activity probably related to the fact that there are no fragile organic ligands whose oxidation usually leads to catalyst deactivation. This catalyst has recently been coupled to PS in a light-driven water oxidation system [95–97].
Recently, Sun et al. reported on a photoelectrochemical device based on the same design as reported by Spiccia (Fig. 10). A molecular mononuclear Ru catalyst bearing carboxylate ligands was embedded in Nafion and deposited on a dye-sensitized nanostructured TiO2 film. This assembly provided the photoanode whereas a Pt foil was used for the cathode. Splitting of water from a neutral aqueous electrolyte is observed when a small voltage bias is applied [98]. Here again, one cannot fully exclude formation of ruthenium oxide in the course of the photocatalytic reaction. RuO2 is indeed well known as a water oxidation catalyst. In line with this comment is the recent report from the Yale group that iridium oxide materials are deposited onto the electrode surfaces during electrochemical measurements on molecular Cp*-iridium complexes [23].

Schematic representation of the voltage-biased photoelectrochemical device for overall water splitting reported by Sun et al. [98].
Inexpensive water oxidation catalysts are needed to develop photocatalytic solar cells and produce fuel from water and sunlight. Recently, cobalt catalysts appeared promising for the splitting of water [2]. Light-driven water oxidation mediated by cobalt salts was initially reported by Shafirovich et al. in 1980 [99]. Irradiation of a phosphate-buffered (pH 5–7) aqueous solution of [Ru(bipy)3]2+ and [Co(NH3)5Cl] generates low yields of O2. In that case, the hydrolysis of [Co(NH3)5Cl] liberates CoII ions which then behave as precursors of the active species [100]. Recently, Styring et al. characterized the colloidal catalyst produced under these conditions. Addition of methylenediphosphonate to the photolyzed solution generated a more homogeneous dispersion (10–60 nm) of the particle sizes [101]. Harriman et al. investigated the spinels NiCo2O4 and Co3O4 with [Ru(bipy)3]2+ as the photosensitizer and peroxodisulfate as the electron acceptor for light-driven water oxidation in deaerated aqueous Na2SO4 solution (pH 5) [102]. Recently H. Frei's group showed that suspensions of silica particles containing spinel Co3O4 nanocrystals (4% loading) in pH 5.8 aqueous solution were able to oxidize water to O2 upon visible light illumination (18% quantum efficiency), in the presence of [Ru(bipy)3]2+ as a photosensitizer and peroxodisulfate as the electron acceptor [103]. Remarkable TOF values in the range of 1000s−1 per nanocluster were observed, about three orders of magnitude greater than those observed using micron-sized Co3O4 particles, as a consequence of both the larger surface area derived from the nanostructuration of the material and the higher activity of Co surface sites (TOF = 0.01 s−1 per surface Co site).
The most recent advance in the direction of molecular catalysts for water oxidation based on cobalt has been provided by C.L. Hill et al. The polyoxometalate [Co4(H2O)2(PW9O34)2]10−, with a tetracobalt core, initially prepared in Paris in 1973 [104] (Fig. 11) has been shown to behave as an excellent catalyst for the oxidation of water by [Ru(bpy)3]3+ [105] as well as for the oxygen evolution in a homogeneous photon-driven system using [Ru(bpy)3]2+ as a photosensitizer and S2O82− as a sacrificial electron acceptor [106]. Under the last conditions, at pH 8, a high (30%) photon-to-O2 yield and a turnover number of 220 has been reported. Further exploitation of this interesting compound will depend on whether it can be integrated into a photoanode material.
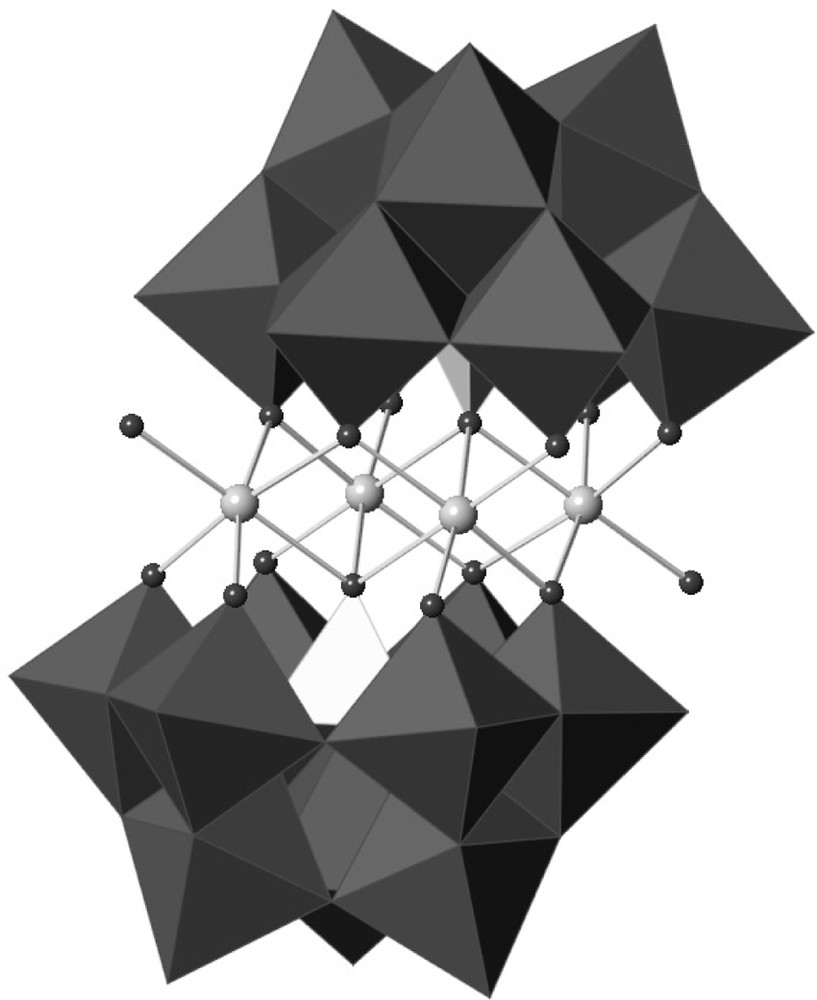
Structure of the tetracobalt polyoxometallate derivative [Co4(H2O)2{α-PW9O34}2]10−. The {α-PW9O34} subunits are represented as polyhedra with dark-grey {WO6} octahedral and light-grey {PO4} tetrahedra. Cobalt ions are depicted as grey spheres and oxygen atoms coordinated to Co are depicted as dark grey spheres.
To date, the only molecular-engineered O2-evolving photoelectrode using non-noble metals is based on cobalt. Abe et al. indeed demonstrated that a bilayer composed of a perylene derivative, acting as a photosensitizer, and cobalt phthalocyanine, the O2-evolving catalyst, can act as a water oxidation photoanode in basic aqueous solutions under visible-light irradiation when poised at 0.3 V vs Ag/AgCl (Fig. 12) [107]. Several thousands of turnovers are reported.
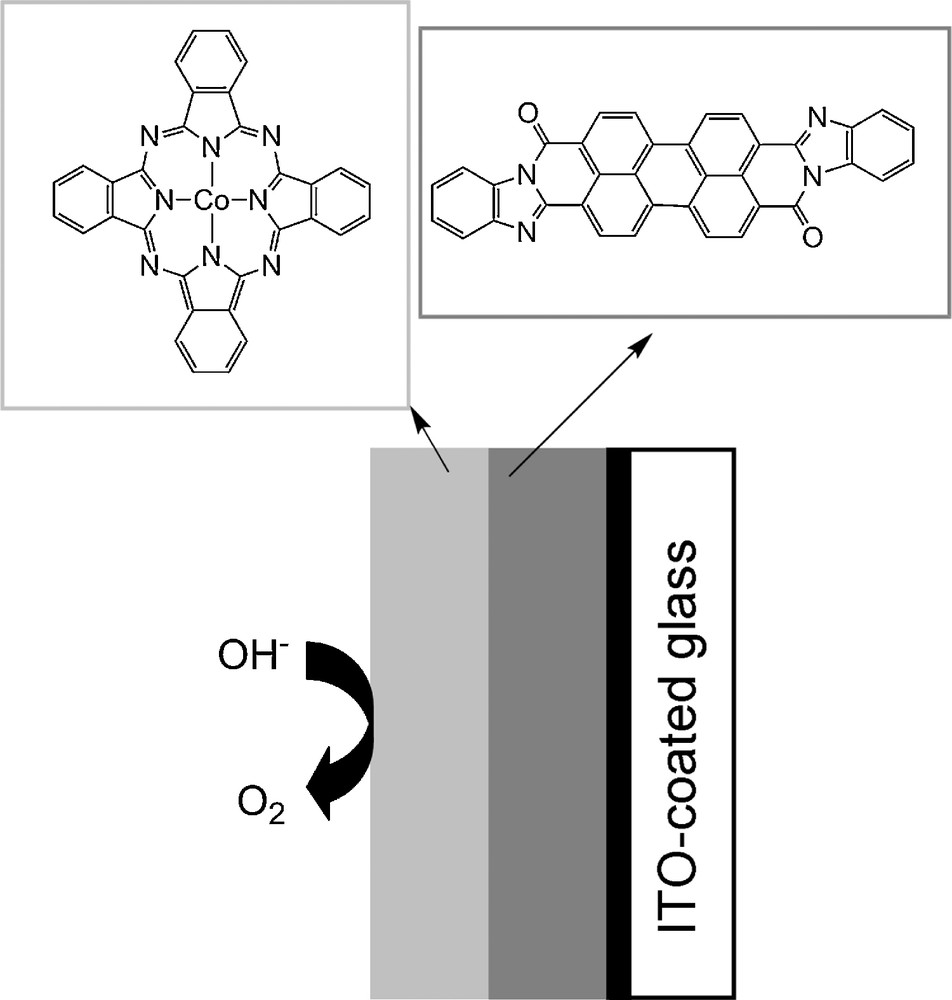
Structure of the perylene derivative/cobalt phthalocyanine bilayer photoanode for water photocatalytic oxidation described by Abe et al. [107].
6 Conclusion
From this short survey of the recent developments in the field of artificial photosynthesis and light-driven water splitting, at first sight one might feel quite disappointed. Indeed, so far only very few electrodes, which could practically be implemented within a photoelectrochemical cell such as that shown in Fig. 1, have been assembled. Furthermore, the rare ones described here have still limited performances. Finally, electrode materials with non noble metals are still scarce.
On the other hand, there are many good reasons for being optimistic regarding the assembly of an efficient photoelectrolyzer sooner than initially anticipated. First, it is only very recently, when the combined issues of the limitation of fossil fuel resources and global warming came into the scene, that this research field has received attention and support from research agencies and governmental programs. Yet, in a few years, remarkable progress have been obtained in terms of: (i) stability and turnover frequencies of molecular and solid (metal oxide) catalysts, as shown, for example, from the huge increase of the turnover number value obtained with molecular catalysts for water oxidation, including based on non noble metals such as cobalt, within a couple of years; (ii) efficiency of semiconducting materials and molecular PS; (iii) robustness of the methods for nanostructuring electrode materials and for grafting molecular compounds at their surface; (iv) diversity of synthetic supramolecular assemblies combining a catalyst unit and a photosensitizer; (v) efficient exploitation of enzymes (hydrogenases, photosystem I and II). Second, in a few years our understanding of how natural water splitting functions, in particular through a increasingly detailed structural, physicochemical and mechanistic characterization of the key enzymes of the process as well as our understanding of the basic, thermodynamic and kinetic, principles on which such a photo-electrochemical cell has to be built have dramatically increased. Third, whereas the pieces of the puzzle are elaborated in different laboratories all over the world, too much independently, awareness of the importance of international collaboration has increased. Recent initiatives provide a clear illustration of this move.
It is thus likely that, through this collaborative international effort, the first photo-electro-catalytic system, absolutely free of noble metals, for overall water splitting will appear in the very next future. This will open the way to unexpected developments towards practical technological devices.