

1 Introduction
Multinuclear transition metal complexes have attracted much interest because of their interesting molecular architecture, intriguing magnetic properties [1–4], fascinating biological activities [5–7], potential applications as functional materials in catalysis [8–10], and so on. For a multinuclear transition metal complex, it contains a variety of forces, including some strong interactions (i.e. metal–ligand coordination bonds) and other weak interactions such as hydrogen bonds [11–13] and π-π stacking interactions [10,14,15]. Although these weak interactions are relatively weak, they play critical roles in the construction of high-dimensional supramolecular networks [10]. Hence, research on weaker interactions is vital to the development of supramolecular chemistry [16].
Ligand H3dcp (H3dcp = 3,5-pyrazoledicarboxylic acid) is a versatile ligand, which contains a number of N or O coordination sites and rich coordination modes [17,18]. The deprotonated anion of dcp3− could provide six donor sites from two carboxylate groups and one pyrazole ring [19]. The carboxylate groups of dcp3− have a strong ability to bond various metal ions and influence the coordinated configurations, even change the dimensional number [20–24]. Till now, some H3dcp complexes with alkaline, transition, and lanthanide metals have been reported [25–28]. Moreover, ligand H3dcp contains abundance N or O atoms, thus the coordinated structural motifs of H3dcp complexes readily form hydrogen-bonded networks [29,30]. Herein, we reported a new trinuclear nickel(II) complex [Ni3(dcp)2(H2O)10] (1) with dcp3− as bridge ligands. The crystal structure and magnetic properties of complex 1 have also been investigated.
2 Experimental
2.1 Materials and instrumentations
All chemicals and solvents used for synthesis were purchased from Alfa, were of reagent grade and used without further purification. Elemental analyses (C, H, N) were performed on a Perkin-Elmer 240 C analyzer and the FT-IR spectra were recorded with a Bruker Tensor 27 Fourier transform infrared spectroscopy device in the 4000–400 cm−1 regions on KBr disks. Variable-temperature magnetic susceptibilities were measured on a Quantum Design MPMS-7 SQUID magnetometer from 2 to 300 K in a magnetic field of 2000 G. Diamagnetic contributions of all constituent atoms were made with Pascal's constants [31].
2.2 Preparation of [Ni3(dcp)2(H2O)10] 1
Complex 1 was synthesized by reaction of Ni(NO3)2·6H2O with H3dcp via hydrothermal reaction. A mixture of Ni(NO3)2·6H2O (0.0206 g, 0.1 mmol), H3dcp (0.0112 g, 0.1 mmol), NaOH (0.004 g, 0.1 mmol), 3 mL acetonitrile and 8 mL aqueous solution was sealed in a 25 mL stainless steel Teflon liner reactor. The reaction mixture was directly heated to 140 °C and kept for three days, and then cooled to room temperature at a rate of 6 °C per hour. The resulting green rectangular-shaped crystals were filtered off, washed with distilled water and dried at ambient temperature. (Yield: 78% based on Ni.) Anal. Calc. for C5H11N2Ni1.5O9 (331.22): C, 18.13%; H, 3.35%; N, 8.46%; Found: C, 18.20%; H, 3.12%; N, 8.37%. IR data (KBr, cm−1): 3369 (vs), 2363 (m), 1567 (m), 1368 (vs), 1236 (m), 1119 (w), 857 (w), 783 (s), 738 (w), 678 (w).
2.3 X-ray structure determination and refinement
A crystal of dimensions 0.22 mm × 0.32 mm × 0.12 mm for complex 1 was mounted on a Bruker Smart 1000 area detector using graphite monochromated Mo Kα radiation (λ = 0.71073 Å) at 293(2) K. The ω-φ scan technique was employed. Structure of the crystal was solved primarily by direct method followed by Fourier difference techniques (SHELXS-97) and refined by the full-matrix least-squares on F2 with anisotropic thermal parameters for all non-hydrogen atoms (SHELXL-97) [32]. Crystallographic data and experimental details for structural analyses are summarized in Table 1.
Crystal data and structure refinement for complex 1.
| Empirical formula | C5H11N2Ni1.5O9 |
| Formula weight | 331.22 |
| Temperature/K | 293(2) |
| Crystal system | Monoclinic |
| space group | C2/c |
| Unit cell dimensions | a = 12.183(3) Å |
| b = 8.8520(3) Å, β = 103.23(3)°C | |
| c = 19.674(4) Å | |
| V | 2065.4(7)Å3 |
| Z | 8 |
| Calculated density | 2.130 Mg/m3 |
| Absorption coefficient | 2.809 mm−1 |
| F(000) | 1352 |
| Crystal size (mm) | 0.32 × 0.22 ×0.12 |
| θ range for data collection | 3.33 to 24.99° |
| Limiting indices | -14 ≤ h ≤ 12, − 0 ≤ k ≤ 10, − 23 ≤ l ≤ 21 |
| Reflections collected/unique | 4843/1742 [Rint = 0.1565] |
| Completeness to θ = 27.89 | 95.8% |
| Absorption correction | Semi-empirical from equivalents |
| Max. and min. transmission | 0.7292 and 0.4668 |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 1742/64/172 |
| Goodness-of-fit on F2 | 1.116 |
| Final R indices [I >2σ(I)] | R1 = 0.0937, wR2 = 0.1369 |
| R indices (all data) | R1 = 0.1715, wR2 = 0.1600 |
| Largest diff. peak and hole | 0.672 and − 0.633 e Å−3 |
Crystallographic data (excluding structure factors) for the structural analysis have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication No. 826001. Copies of the data can be obtained free of charge from The Director, CCDC, 12 Union Road, Cambridge CB21EZ, UK (fax: +44 1223 336 033; e mail: deposit @ccdc cam ac uk.
3 Results and discussion
3.1 Description of crystal structure
Single-crystal X-ray diffraction analysis reveals that complex 1 crystallizes in the monoclinic space group C2/c. The crystal structure consists of centrosymmetric [Ni3(pdc)2(H2O)10] molecules. Selected bond lengths and angles are given in Table 2 and the perspective view of the compound with atomic numbering scheme is shown in Fig. 1. In complex 1, two pdc3− ligands behave as bis(bidentate) ligands binding three Ni ions (Ni(1), Ni(1A) and Ni(2)), forming a trinuclear unit. The intramolecular Ni(1)···Ni(2) separations are 4.472 Å. The trinuclear unit consists of two crystallographically independent Ni atoms, namely, Ni(1) and Ni(2). All Ni(II) ions have slightly distorted octahedral geometries. However, there are two sets of coordination environments around the Ni atoms. The Ni(1) or Ni(1A) atom is coordinated to one L3− and four H2O molecules, giving a slightly distorted octahedral geometry with an O5N donor set, while Ni(2), located at the center of the trinuclear unit, has an O4N2 donor set originating from two L3− ligands and two H2O molecules. The average Ni–O and Ni–N distances are 2.058(0) and 2.039(8) Å, respectively, which are all within the normal range.
Selected bond lengths (Å) and angles (°) for complex 1.
| Ni(1)–N(1) | 2.0514(6) | Ni(1)–O(2) | 2.0220(3) |
| Ni(1)–O(3) | 2.0692(3) | Ni(1)–O(4) | 2.0814(3) |
| Ni(1)–O(5) | 2.0766(6) | Ni(1)–O(6) | 2.0004(3) |
| Ni(2)–O(7) | 2.0786(3) | Ni(2)–O(8) | 2.0646(6) |
| Ni(2)–N(2) | 2.0280(3) | ||
| O(6)–Ni(1)–N(1) | 105.367(7) | O(2)–Ni(1)–N(1) | 80.352(6) |
| O(6)–Ni(1)–O(3) | 87.713(6) | O(2)–Ni(1)–O(3) | 89.787(6) |
| N(1)–Ni(1)–O(3) | 91.147(7) | O(6)–Ni(1)–O(5) | 86.488(6) |
| O(2)–Ni(1)–O(5) | 87.713(6) | N(1)–Ni(1)–O(5) | 167.947(13) |
| O(3)–Ni(1)–O(5) | 87.106(7) | O(6)–Ni(1)–O(2) | 173.773(6) |
| O(6)–Ni(1)–O(4) | 91.587(6) | O(2)–Ni(1)–O(4) | 90.751(5) |
| O(3)–Ni(1)–O(4) | 177.718(7) | O(5)–Ni(1)–O(4) | 90.700(6) |
| N(2)–Ni(2)–N(2)#1 | 180.000 | N(2)–Ni(2)–O(8) | 80.316(6) |
| N(2)–Ni(2)–O(8)#1 | 99.684(6) | Ni(1)–O(4)–H(4A) | 125.680(11) |
| Ni(1)–O(3)–H(3A) | 131.892(12) | N(2)–Ni(2)–O(7) | 87.316(5) |
| O(8)–Ni(2)–O(7) | 90.047(6) | O(7)–Ni(2)–O(7)#1 | 180.000 |
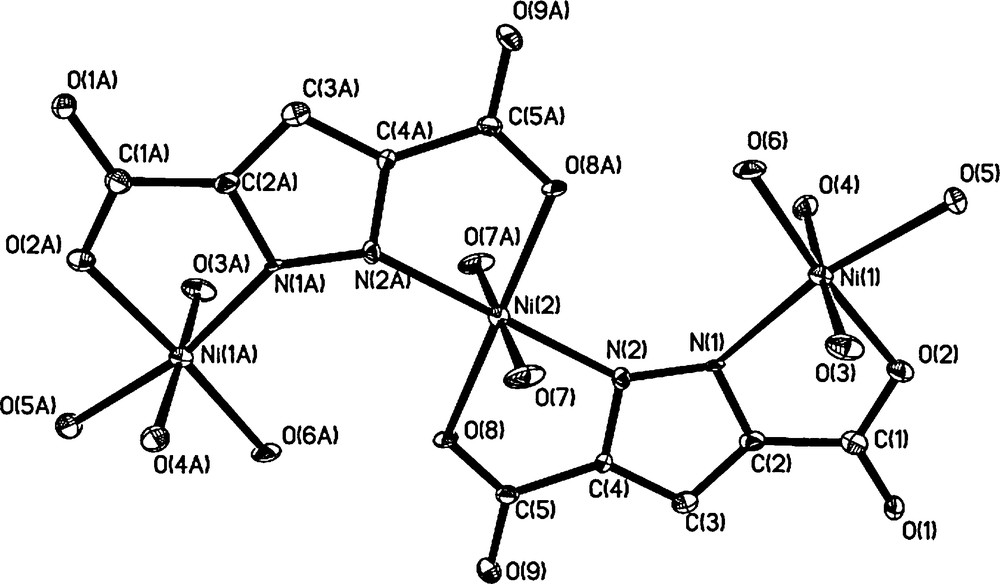
Molecular structure for [Ni3(pdc)2(H2O)10] with the atomic numbering scheme.
Fig. 2 shows the packing diagram of complex 1. There are two kinds of O-H···O hydrogen bonds in the title complex (Table 3). The first one is a hydrogen bond between the oxygen atoms from carboxylate groups and coordinated water molecules (O(3)···O(9), O(4)···O(9), O(5)···O(1), O(6)···O(8), O(4)···O(2) and O(7)···O(1)), in which the coordinated water molecules act as hydrogen donors to form hydrogen bonds with oxygen atoms of carboxylate moieties. The second one is the O–H···O hydrogen bond between coordinated water molecules (O(3)···O(5), O(4)···O(5)). As a result, these hydrogen bonds extend the trinuclear structure into a three-dimensional network. Moreover, Fig. 3 shows that the plane-to-plane distance of pyrazole rings are 4.205 Å and 3.902 Å, respectively, indicating the possible existence of π-π stacking interactions in the system. These O–H···O hydrogen bonds and π-π stacking interactions are likely to play an important role in the stabilization of the crystal system [24].
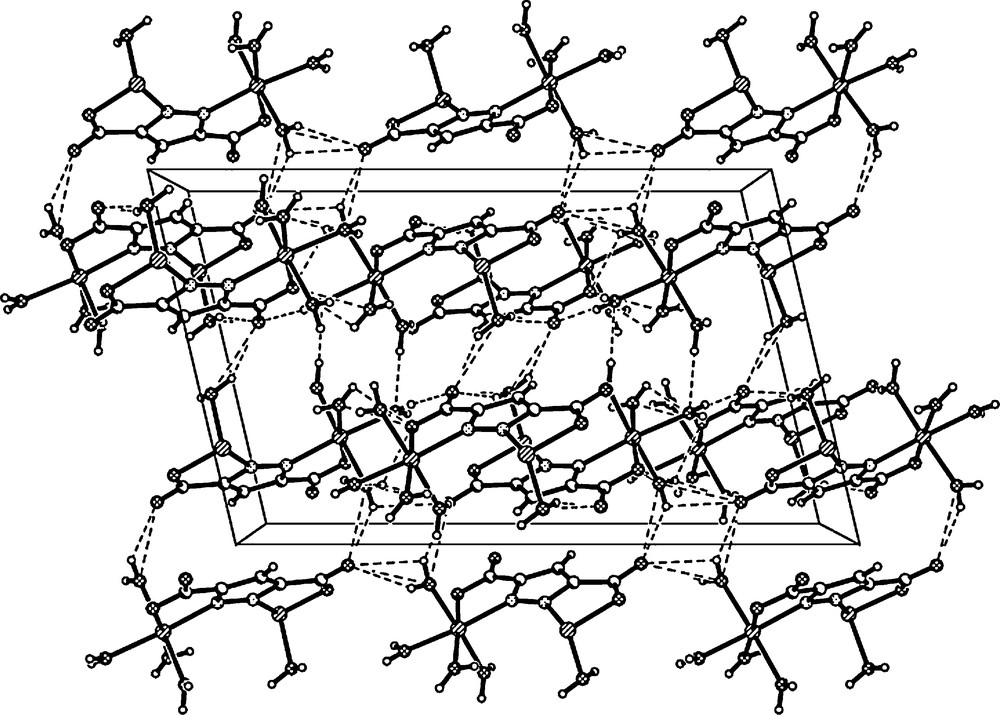
A view of intermolecular O–H···O hydrogen bonding interactions between adjacent molecules that lead to the 3D network (black dashed lines present the H–bond).
Hydrogen bonds (Å and°) in complex 1.
| D–H---A | D–H | H---A | D---A | < DHA | Symmetry code on A |
| O(3)–H(3A)---O(5) | 0.8658(2) | 2.0905(5) | 2.956 | 178.216(13) | –0.5+x,1.5–y,–0.5+z |
| O(3)–H(3B)---O(9) | 0.8692(2) | 1.9507(4) | 2.791(2) | 162.252(9) | 1.5–x,–0.5+y,1.5–z |
| O(4)–H(4A)---O(9) | 0.8635(1) | 2.4289(5) | 2.812(6) | 107.556(15) | –0.5+x,0.5+y,–1+z |
| O(4)–H(4A)---O(2) | 0.8635(1) | 2.4508(7) | 3.088(4) | 131.171(11) | –0.5+x,0.5+y,z |
| O(4)–H(4A)---O(5) | 0.8635(1) | 2.4731(5) | 3.004(4) | 120.447(8) | –0.5+x,0.5+y,z |
| O(4)–H(4B)---O(9) | 0.8706(1) | 2.1218(7) | 2.767(1) | 130.452(12) | 1.5–x,0.5+y,1.5–z |
| O(5)–H(5A)---O(1) | 0.8483(2) | 1.8470(3) | 2.689(1) | 171.558(13) | –0.5+x,0.5+y,z |
| O(5)–H(5B)---O(4) | 0.8467(1) | 2.3200(4) | 3.004(4) | 138.171(9) | 1–x,1–y,1–z |
| O(6)–H(6A)---O(2) | 0.8606(2) | 2.4230(6) | 2.731(9) | 101.789(13) | –0.5+x,0.5+y,z |
| O(6)–H(6B)---O(8) | 0.8555(1) | 1.7957(3) | 2.651(1) | 178.484(13) | 1–x,1+y,1.5–z |
| O(7)–H(7A)---O(1) | 0.8501(3) | 2.0787(7) | 2.863 | 153.158(15) | –0.5+x,1.5–y,–0.5+z |
| O(7)–H(7B)---O(1) | 0.8480(1) | 1.9538(3) | 2.751(7) | 156.347(11) | 1.5–x,–0.5+y,1.5–z |
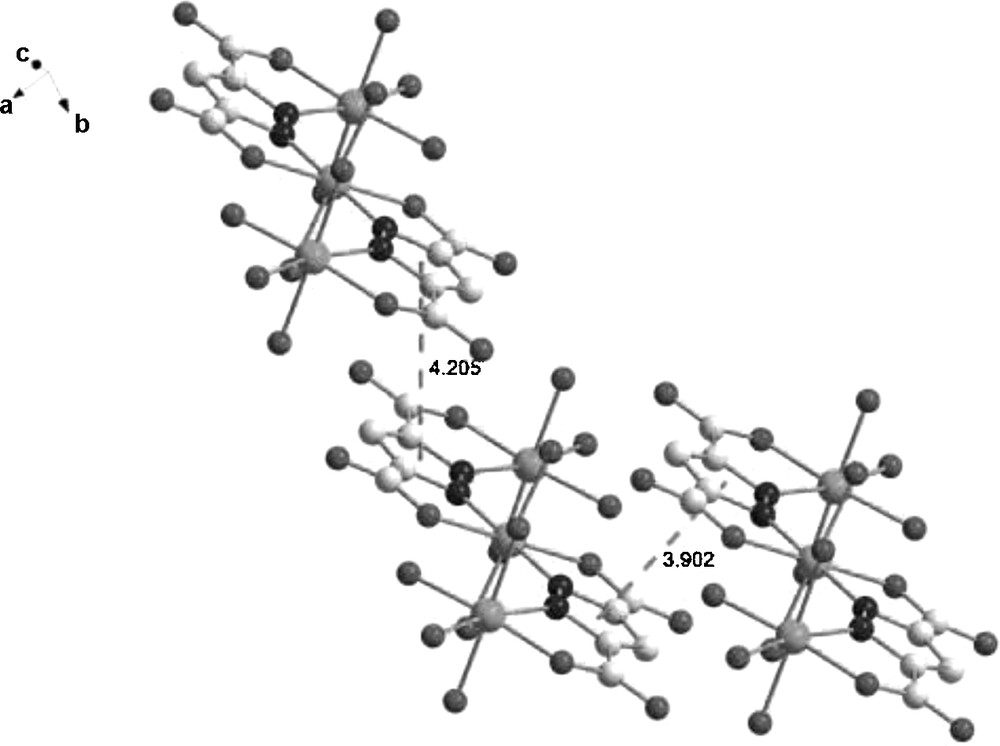
The plane-to-plane distance of pyrazole rings.
3.2 IR spectra
The FT-IR spectra for complex 1 is consistent with other pyrazoledicarboxylic compounds [19,33,34]. A band at 3369 cm−1 can be assigned to the O–H stretching frequency of the water molecule, which is approximately close to the value of 3359 cm−1 in the extended water tapes [35]. Bands at 1567 cm−1 can be assigned to the pyrazole ring CN bond stretching modes. The strong absorptions at 1368 cm−1 [υasym(CO2)] and 1236 cm−1 [υsym(CO2)] are attributable to the characteristic of the carboxylate group [24,36,37]. Additional characteristic bending vibrations can be identified in the region 857–678 cm−1.
3.3 Magnetic properties
The temperature dependence of χM and χMT for complex 1 in the range of 2–300 K is shown in Fig. 4 (χM is the molar magnetic susceptibility per Ni3 unit). As shown in Fig. 4, the value of χMT for complex 1 at room temperature is 3.07 cm3K mol−1, which is very close to the spin-only value (3.00 cm3K mol−1) for three isolated nickel(II) ions (S = 1, g = 2.0). When decreasing the temperature, the χMT plot decreases very slowly till 55 K and then begins to drop down sharply and reaches a minimum value of 0.57 cm3K mol−1 at 2 K, suggesting that the dominant antiferromagnetic interactions are propagated between nickel(II) ions. There is no peak in the χM vs T plot. Therefore, no long-range magnetic ordering is found.
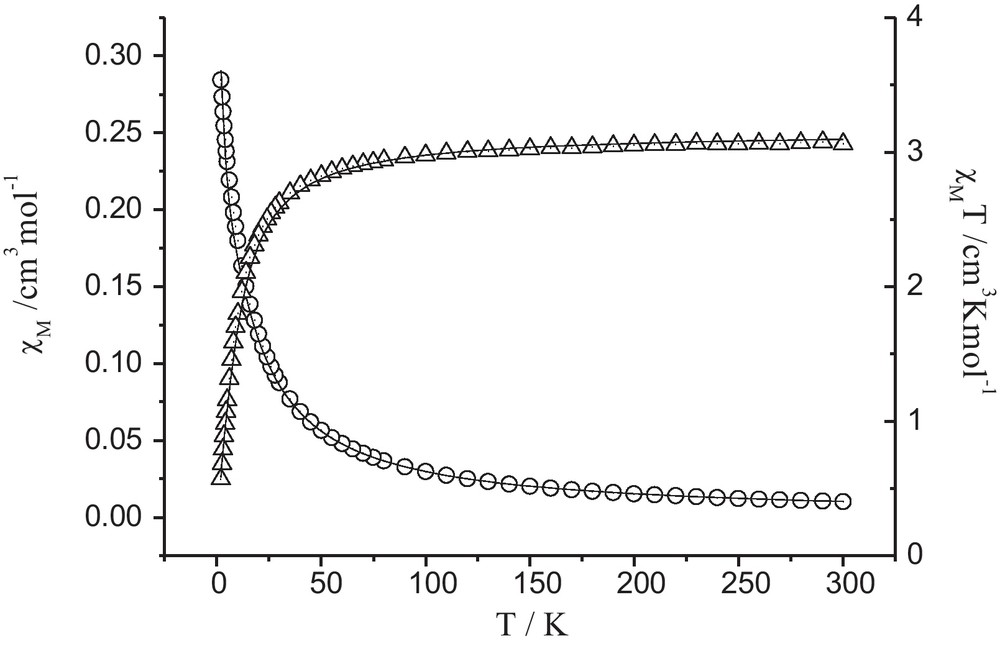
Experimental and calculated variations of χM(O) and χMT (Δ) versus T for complex 1.
In order to analyze the experimental data, we tried to simulate the data by taking into account zero-field splitting of Ni(II) ions by parameter D, and treating exchange interaction (J) between Ni(II) ions in the following Hamiltonian given in eq. (1):
| (1) |
By using the computational program of MAGPACK [38,39], we have obtained an excellent simulation of the experimental data from the following parameters D = 5.27 cm−1, J = −2.19 cm−1 and g = 2.05, where is the z component of the operator, J is the magnetic coupling constant between Ni(II) ions in the trimer, D is zero-field splitting parameter, g is the Landé factor of the Ni(II) ion. The small negative value of J also indicates that a weak antiferromagnetic interaction is operative between the nickel(II) ions. The obtained D parameter is close to previously reported values in similar nickel(II) complexes [40].
4 Conclusions
In this paper, we have presented the synthesis, spectroscopy, crystal structure and magnetic properties of a new nickel(II) compound [Ni3(dcp)2(H2O)10] 1 (H3dcp = pyrazoledicarboxylic acid). The crystal structure indicates that three Ni(II) ions exist in a line arrangement, with two dcp3− as bridge ligands and ten coordinated water molecules as terminal ligands. Complex 1 is further packed into a three-dimensional structure via intermolecular hydrogen bonds and π-π stacking interactions. Magnetic susceptibility measurements show that a weak antiferromagnetic interaction is operative in this nickel(II) complex.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 21071085, 21071084, 21101096 and 90922032), Tianjin Natural Science Foundation (No. 11JCYBJC03500) and Research Fund for the Doctoral Program of Higher Education 20100031120013.