

1 Introduction
Co3O4 is an important antiferromagnetic p-type semiconductor with excellent properties such as gas-sensing, catalytic and electrochemical ones, and has been studied widely for applications in solid-state sensors, electrochromic devices and heterogeneous catalysts as well as lithium batteries [1–5]. When reduced down to the nanometer scale, Co3O4 were found to have interesting magnetic, optical, field emission and electrochemical properties that are attractive in device applications [6–8]. Thus tremendous efforts have been directed in recent years to the synthesis and investigation of properties of Co3O4 nanostructures.
Up to now, although many methods have been applied in the synthesis of Co3O4 nanoparticles [9–11], there are still several challenges in the synthetic route. One is the tendency of Co3O4 to grow into larger or irregular particles. To prevent these undesired processes, organic surfactants were used to control growth [12]. Using organic surfactants increases the cost and is not environment friendly. The other challenge is the phase purity control of the final product in the low-temperature synthesis of Co3O4 nanomaterials. According to the above note, the decomposition of compounds can be a useful method for the synthesis of nanomaterials [13]. These methods are complicated and/or require long-time thermal treatment. New methodologies are in great demand for the synthesis and fabrication of Co3O4 nanostructures.
Among the many methods applied for preparing metal oxide powders, the organometallic molecular precursor way has been regarded as one of the most convenient and practical techniques, because it not only enables us to avoid special instruments and complicated processes as well as severe preparation conditions, but it also provides good control over purity, composition, homogeneity, phase and microstructure of the resultant products [14–18].
In our group, we have been interested for a few years in the synthesis of metal, metal oxide and magnetic nanoparticles, using new inorganic precursors, taking profit of the tools of organometallic chemistry [19–23]. A major interest at the moment is in the development of organometallic or inorganic compounds for the preparation of nanoparticles. Using novel compounds can be useful and opens a new way for preparing nanomaterials with controlled shape, nanocrystal, and distribution size.
Herein, we have reported the simple synthesis of Co3O4 nanoparticles via the thermal treatment of an easily obtained solid organometallic molecular precursor, [bis(salicylaldehyde)ethylenediiminecobalt(II)]; Co(salen), as well as the characterization results of the products by X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FT-IR), transmission electron microscopy (TEM) and scanning electronic microscopy (SEM). Magnetic properties of the prepared sample were investigated by VSM.
2 Experimental
2.1 Materials
All the chemical reagents used in our experiments were of analytical grade and were used as received without further purification. Cobalt (II) acetate, salicylaldehyde and 1,2-ethylenediamine were obtained from Merck Co.
2.2 Characterization
Thermogravimetric analyses (TGA) were carried out using a thermal gravimetric analysis instrument (Shimadzu TGA-50H) with a flow rate of 20.0 mL min−1 and a heating rate of 10 °C min−1. XRD patterns were recorded using a Rigaku D-max C III, X-ray diffractometer using Ni-filtered Cu Kα radiation. Scanning electron microscopy (SEM) images were obtained using a Philips XL-30ESEM device equipped with an energy-dispersive X-ray spectroscopy system. Transmission electron microscopy (TEM) images were obtained on a Philips EM208 transmission electron microscope with an accelerating voltage of 100 kV. Fourier transform infrared (FT-IR) spectra were recorded on a Varian 4300 spectrophotometer on KBr pellets. The magnetic measurement was carried out in a vibrating sample magnetometer (VSM) (BHV-55, Riken, Japan) at room temperature. VIS-NIR was carried out using a PerkinElmer Lambda 950 UV/Vis/NIR spectrophotometer.
2.3 Preparation of bis(salicylaldehyde)ethylenediimine; H2salen [24]
A stoichiometric amount of salicylaldehyde (0.02 mol, 2.44 g) dissolved in methanol (25 ml) is added dropwise to a 1,2-ethylenediamine solution (0.01 mol, 0.61 g) in 25 ml of methanol. The mixture was refluxed for 3 h and a bright yellow precipitate of the symmetrical Schiff-base ligand (H2salen) was obtained. The yellow precipitate was separated by filtration, washed and dried under vacuum. It was then recrystallized from methanol to yield H2salen (92%). The elemental and spectroscopic analyses of the neat complex confirmed the molecular composition of the ligand.
2.4 Preparation of [Co(salen)]
[Bis(salicylaldehyde)ethylenediirninccobalt(II)], [Co(salen)] was prepared in a similar manner to that reported by Deiasi et al. [25]. The flask containing a stirred suspension of cobalt(II) acetate tetrahydrate (3.96 g, 0.016 mol) in propanol (100 cm3) was purged with nitrogen, and then warmed to 50 °C under nitrogen atmosphere. Bis(salicylaldehyde)ethylenediimine (4.29 g, 0.016 mol) was added in one portion, and the resulting black suspension was then stirred and heated under reflux under nitrogen atmosphere for 8 h. Then the mixture was cooled and filtered out under reduced pressure. The collected solid was washed with diethylether and dried in the air, to give a dark red-brown crystalline [Co(salen)] which was purified by recrystallization from chloroform.
2.5 Synthesis of Co3O4 nanoparticles
Black Co3O4 nanocrocrystals were produced by subjecting 0.01 mol of the as-prepared [bis(salicylaldehyde)ethylenediiminecobalt(II)] [Co(salen)] powders to heat treatment at a relatively low temperature (500 °C) in the air. An average temperature increase of 30 °C is recorded every minute, before the temperature reached 500 °C, and after keeping the thermal treatment at 500 °C for 5 h, it was allowed to cool unaffectedly at room temperature.
3 Results and discussion
Fig. 1a displays a typical XRD pattern of the precursor prepared at reflux for 8 h. All the reflection peaks in this pattern could be readily indexed to crystalline [Co(salen)] (JCPDS Card file No. 38-547). No obvious peaks of impurities were seen in this pattern. The XRD pattern shown in Fig. 1b corresponds to the sample obtained by thermal decomposition and oxidization of this compound at 500 °C for 5 h. All reflection peaks could be readily indexed compared to crystalline cubic phase Co3O4 with lattice constant a = 8.065 Å, which is consistent with the standard value a = 8.065 Å (JCPDS Card file No. 74-1656). The crystallite size of the as-synthesized product, Dc, was calculated from the major (111) diffraction peaks using the Scherrer formula (Eq. (1)) [26]:
| (1) |
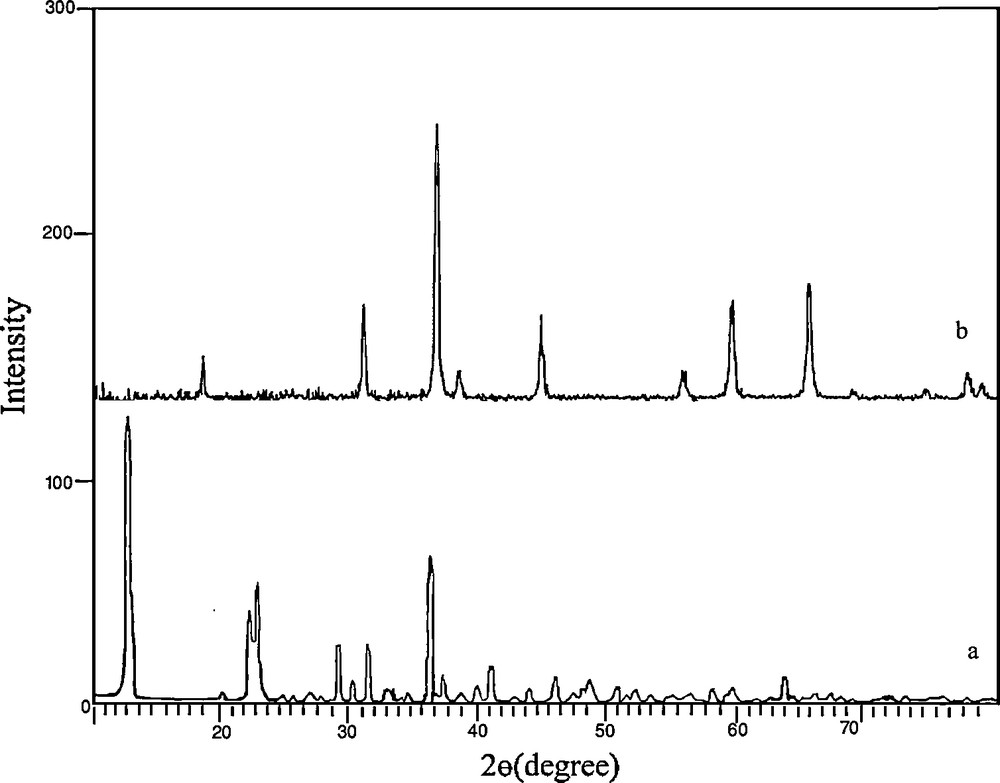
XRD patterns of (a) the precursor [Co(salen)] and (b) Co3O4 nanoparticles obtained by thermal treatment of the precursor at 500 °C for 5 h.
Fig. 2 shows that there is a sharp mass loss around 380 °C, indicating that the decomposition temperature of the precursor is situated at about 380 °C. The weight loss is about 74.23%, which is close to the theoretical value (77.0%). Upon the calcination of the obtained [Co(salen)] at 500 °C in the air, all the cobalt salen was converted into Co3O4 nanoparticles. The XRD pattern of the calcined sample is shown in Fig. 1b.
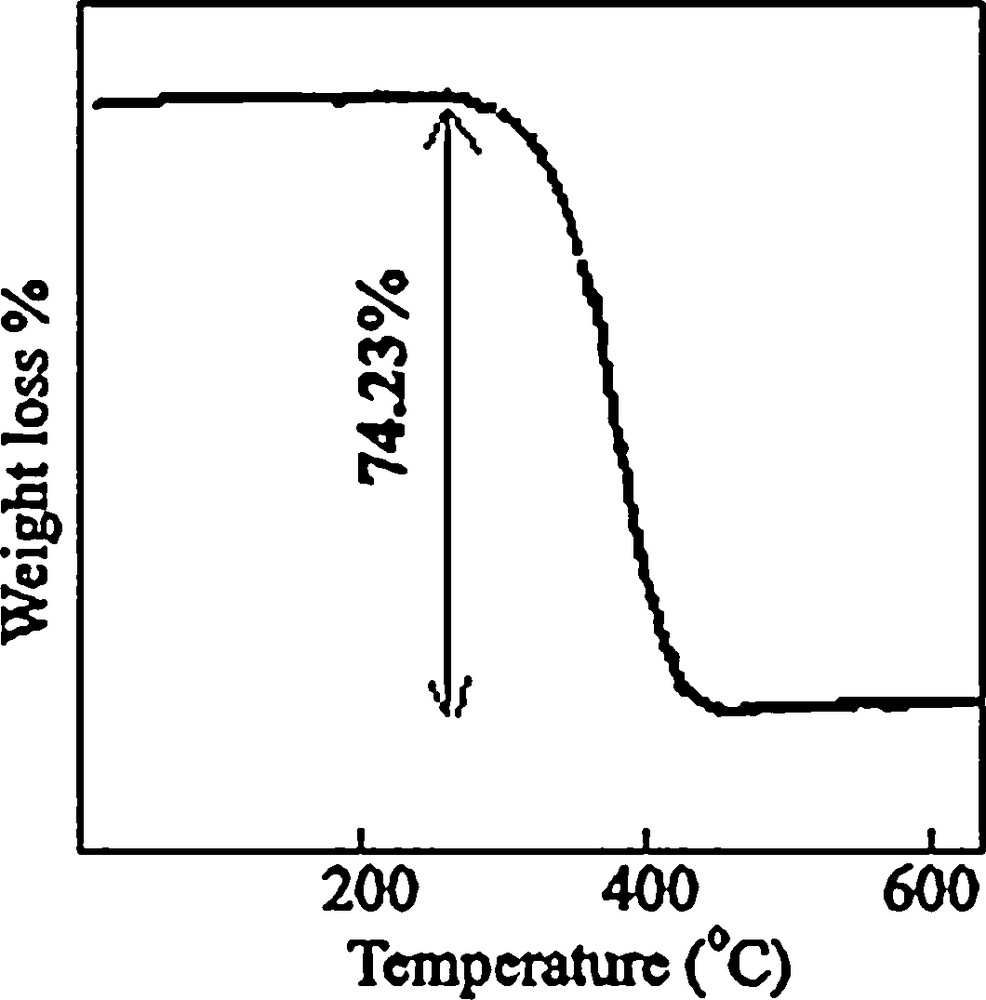
TGA curve of the cobalt salen complex.
The addition of H2salen to the propanol solution of cobalt acetate provokes the precipitation of a dark red–brown solid of cobalt Schiff-base as shown by FT-IR spectrum in Fig. 3a. The spectra of the [Co(salen)] showed evidences of the synthesis of the complex, since two bands at 1550 cm−1 (attributed to νC=C) and at 1600 cm−1 (attributed to νC=N), which are characteristic of the [Co(salen)] complex, appeared in the IR spectra of the precursor. The IR bands observed below 500 cm−1 are assigned to γCo–N and the band at 550 cm−1 to γCo–O(phenolic). The IR spectrum of Co3O4 (Fig. 3b) contains two strong absorption bands at 668.6 and 578.7 cm−1, which confirm the spinel structure of Co3O4. The former peak at 668.6 cm−1 is attributed to the stretching vibration mode of M–O, in which M is Co+2 and is tetrahedrally coordinated. The band at 581 cm−1 can be assigned to the M–O bound, in which M is Co+3, and so coordinates octahedrally [27,28]. The broad band at 3440.1 cm−1 is assigned to both υs(H–O–H) and υas(H–O–H) of hydration water. This result confirmed the formation of Co3O4 under the above-mentioned thermal treatment.
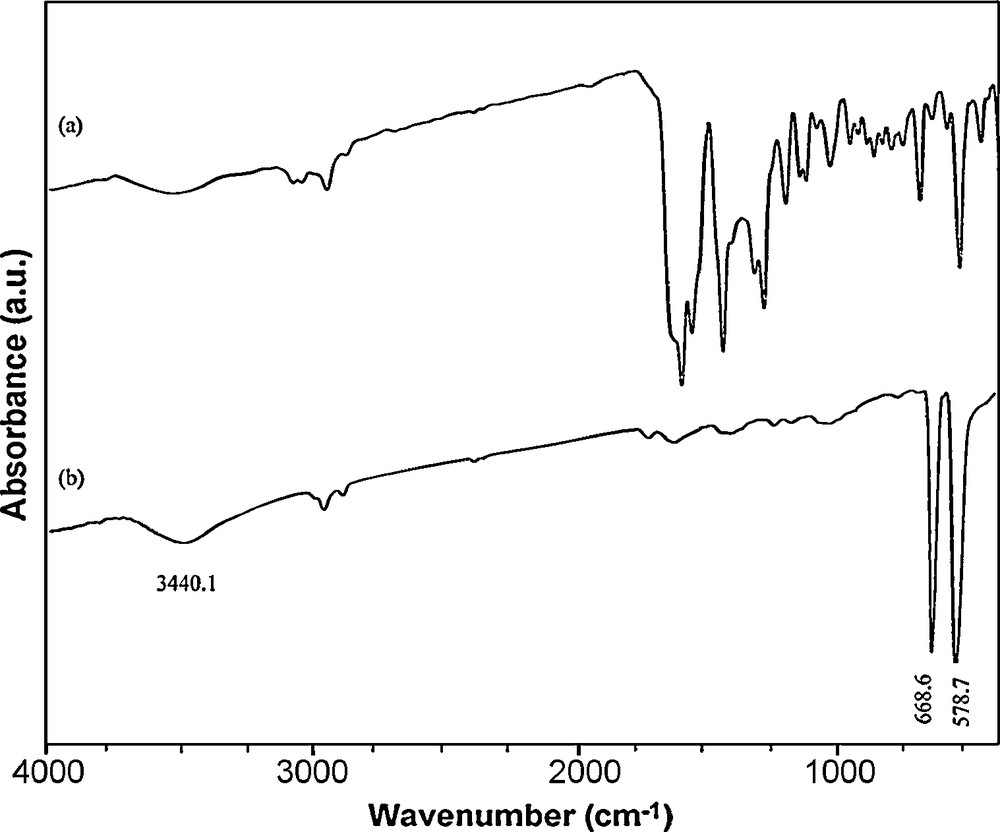
IR spectra of (a) Co(salen) and (b) the product obtained upon thermal treatment of the precursor at 500 °C for 5 h.
The SEM images of the precursor are shown in Fig. 4a and b. It can be seen that Co(salen) is formed of large particles. An overall view of the particles shows that their sizes are about 2–4 μ, with an irregular shape. Fig. 4c and d show SEM images of the product prepared with the solid state method, where we can see that Co3O4 nanoparticles exhibit a quasi-spherical morphology. By comparison, SEM images of nanoparticles and of the precursor are different. TEM images of the as-prepared sample are given in Fig. 4e and f. The particles have a regular shape, and their sizes are about 30–50 nm.

(a, b) SEM images of precursor, (c, d) SEM and (e, f) TEM images of Co3O4 nanoparticles.
The obtained Vis–NIR spectra for samples synthesized at 500 °C are reported in Fig. 5. The I band (λ = 1512 nm) was attributed to crystal field 4A2(F) → 4 T1(F) transitions in the Co3O4 structure [29]. The II signal (λ = 1264 nm) was assigned to an “intervalence” Co(II) ↔ Co(III) charge transfer, representing an internal oxidation–reduction process [30]. There are two absorption peaks (III, λ = 718 nm and IV, λ < 500 nm) obviously found in Fig. 5, which indicate ligand–metal O(II) → Co(III) and O(II) → Co(II) charge transfer events, respectively [9]. Co3O4 is a P-type semiconductor and its optical band gap can be obtained using the following equation (Eq. (2)):
| (2) |
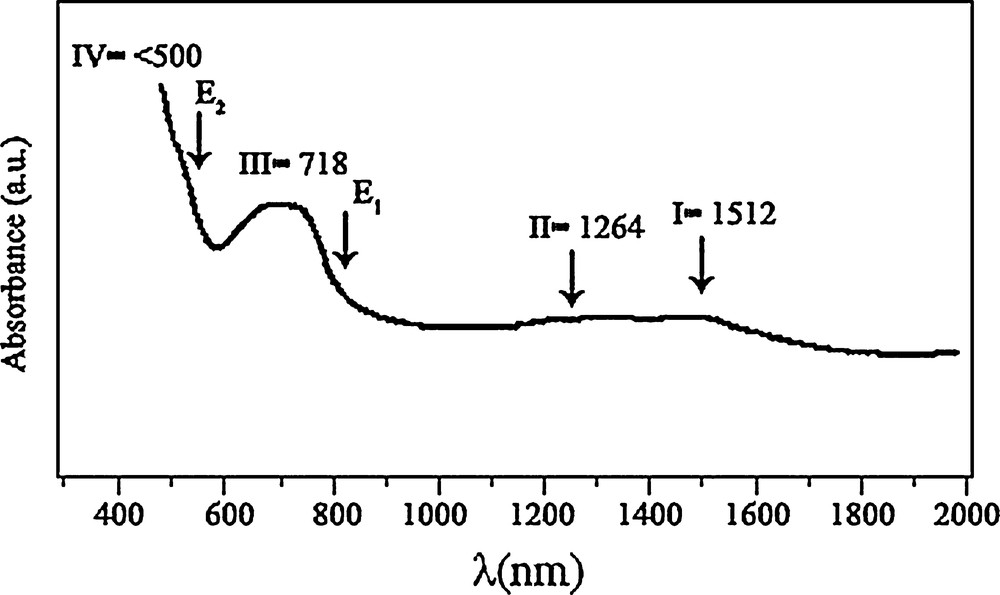
Optical absorption spectrum of the Co3O4 nanoparticles.
The hysteresis loops (Fig. 6) measured at room temperature show a ferromagnetic behavior of the Co3O4 nanoparticles. At 25 °C, the remanent magnetization (Mr) is 9.05 emu/g, the coercive field (Hc) is 700.7 Oe and the magnetization at saturation (Ms) is estimated to be only 27.02 emu/g (the saturation magnetization Ms was determined from the extrapolation of the curve giving H/M versus H) [32,33]. Cobalt oxide nanoparticles are made of the small crystalline domains. Each crystalline domain is characterized by its own magnetic moment oriented randomly. The total magnetic moment of the nanoparticles is the sum of these magnetic domains coupled by dipolar interactions. As a result, a low value of Ms is obtained. The magnetic properties of the nanomaterials are believed to be highly dependent on the sample's shape, crystallinity, magnetization direction, and so on.
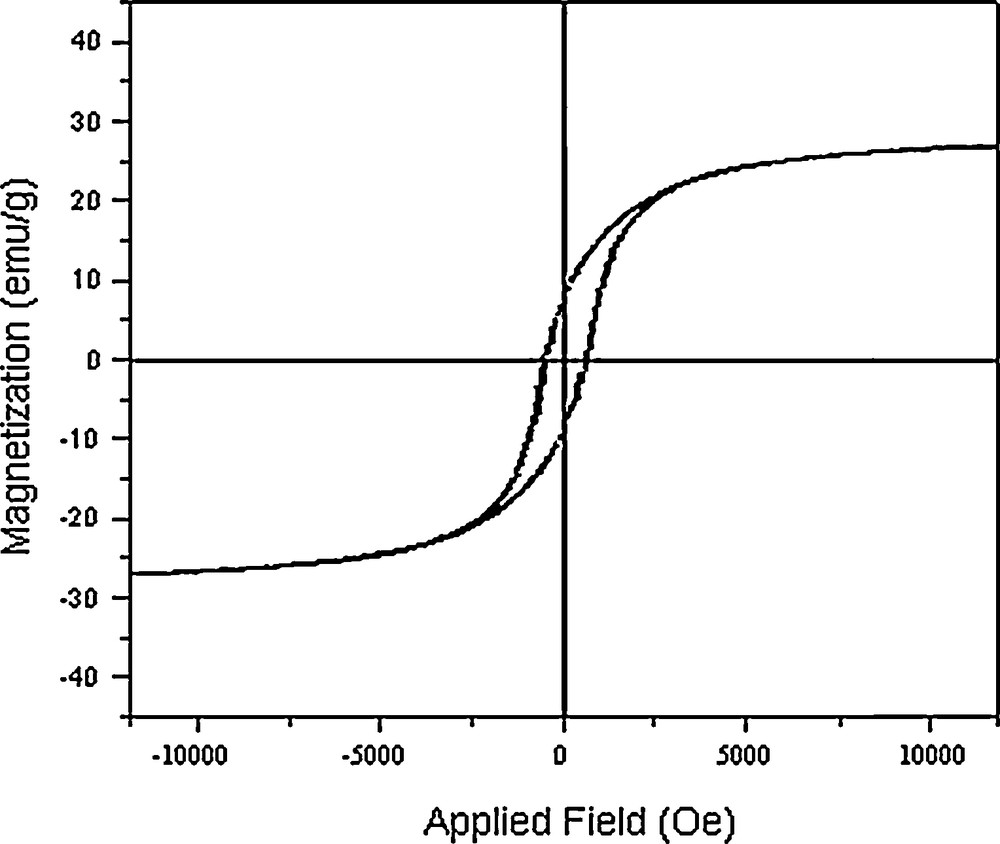
Magnetization versus applied field at 300 K for Co3O4 nanoparticles.
In addition, the cobalt oxide nanoparticles have been prepared under different condition (Figs. 7 and 8, Table 1). XRD results evidence that the products prepared through calcination of [Co(salen)] in air at 300–350 °C for 5 h consist of the precursor, and that the pure product has not been formed (Fig. 7a). By increasing the temperature, pure Co3O4 has been synthesized. The diffraction peaks in the XRD pattern can be indexed relative to cubic Co3O4 (Fig. 7b–d). Scanning electron micrographs of the products obtained at different annealing temperatures show that morphology and size are directly linked to the temperature. The aggregation of the nanoparticles is visible in these images. The micrograph of Co(salen) calcined at 350 °C in Fig. 8a shows that the oxide has an irregular shape. According to the XRD results, the product obtained at this temperature is a mixture of Co(salen) and Co3O4. The micrograph of the precursor calcined at 400 °C (Fig. 8b) shows that the shape of nanoparticles is semi-spherical; they are about 80 nm in size. Fig. 8c shows the micrograph of the precursor calcined at 450 °C; the oxide has a spherical particle shape with a 50-nm-size distribution, while SEM imaging gives an overall view of the product, revealing the surface morphology of the Co3O4 spheres. It can be seen that the products are tiny, aggregated nanoparticles with a spherical shape. Increasing the calcination temperature from 500 °C on, the kinetic energy increases, then nanoparticles agglomerate and the size of the nanoparticles becomes larger (Fig. 8d).
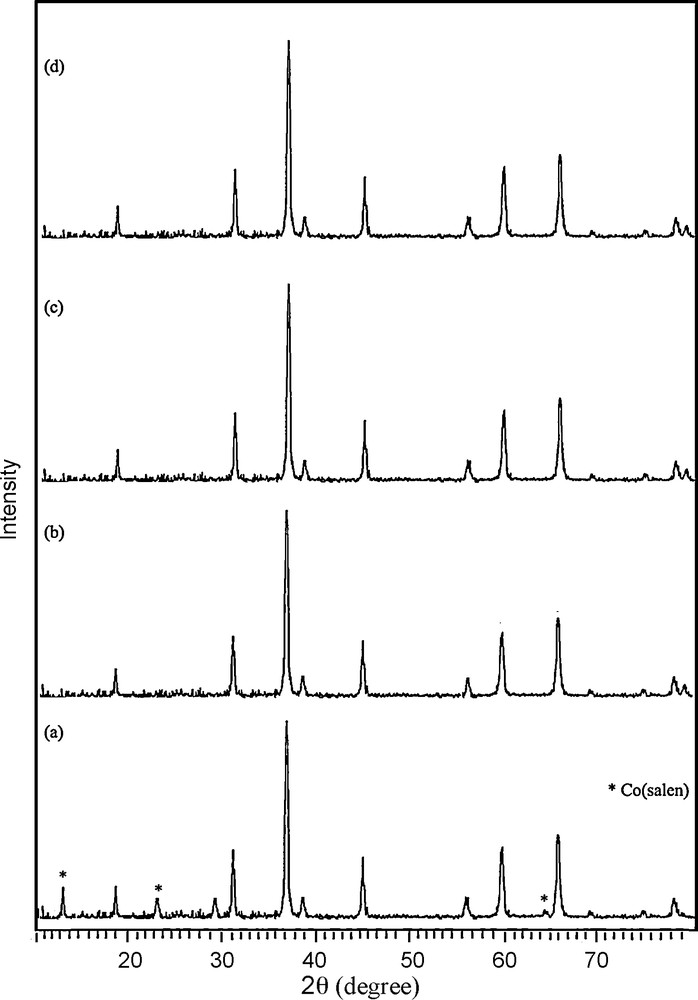
XRD patterns of the products synthesis in (a) 350 °C (b) 400 °C (c) 450 °C (d) 550 °C for 5 h in the air.

SEM images of the products synthesized at (a) 350 °C (b) 400 °C (c) 450 °C (d) 550 °C for 5 h in the air.
Reaction conditions and corresponding products.
| Reaction temperature (°C) | Composition of the products (XRD result) | Size of the nanoparticles (XRD result) |
| 300 | Co3O4 + Co(salen) (main) | |
| 350 | Co3O4 + Co(salen) (minor) | |
| 400 | Co3O4 | 80 nm |
| 450 | Co3O4 | 50 nm |
| 500 | Co3O4 | 30 nm |
| 550 | Co3O4 | 60 nm |
In our thermal method for the synthesis of Co3O4 powders, the used source material was only Co(salen). Like Schiff-base ligand, salen may also take the role of electron transfer agent, displaying moderate intensity reductive properties under high-temperature conditions. Especially, when salen was firstly chelated with Co(II) to form a relatively stable coordination compound, Co(salen), the short distances between cobalt and oxygen or nitrogen atoms in this solid molecular precursor were favorable for electrons to be transferred. Furthermore, the thermal system usually afforded a great driving force for the self-oxidation-reduction reaction processes to initiate. Thus, self-oxidation-reduction reactions of [Co(salen)] should take place to form Co3O4 under the current thermal conditions (500 °C during 5 h).
4 Conclusion
Adopting the self-prepared [Co(salen)] as the precursor, Co3O4 nanoparticles have been synthesized by the thermal treatment method. The proposed methods for the synthesis of Co3O4 nanoparticles are simple, mild and cheap, which makes them very suitable for scale-up productions. Furthermore, it is well expected that such techniques would be extended to prepare many other important semiconducting metal-oxide ultrafine powders, because many other metal ions, such as Zn(II), Cd(II), Ni(II), Cu(II), Mn(II), etc., can also easily form solid metal salen compounds, which are inclined to decompose into metal oxides upon thermal treatment. And the optical absorption properties of the Co3O4 nanoparticles were investigated. The results indicate that the nanoparticles are semiconducting, with direct transitions at 1.53 and 2.02 eV. The optical property test indicates that the absorption peak of the nanoparticles shifts towards short wavelengths. Moreover, the blue shift phenomenon might be ascribed to the quantum effect.
Acknowledgment
Authors are grateful to the council of Iran National Science Foundation and University of Kashan for supporting this work by Grant No. 159271/52.