

1 Introduction
Morpholines are widely used in organic synthesis, mostly as basic and nucleophilic reagents. Morpholines substituted at the ring carbon atoms, however, are fragments incorporated in the structure of both natural and synthetic compounds with antitumor [1]; endoprotease inhibiting [2]; HIV-protease inhibiting activity [3], etc. The polysubstituted morpholine 1 has shown anti-inflammatory activity [4,5], while aprepitant (2) is an antagonist of the neurotransmitter substance P (SP) and exerts antihistaminic and antiemetic activity [5,6]. It is known that peptides incorporating lactam rings have shown diverse biological activities [7]. Morpholin-3-ones, as δ-lactams, are considered as isosteric peptide analogs and are used as peptidomimetics [8].
In the frames of a broader program for the preparation of monocyclic lactams by means of the reactions between cyclic anhydrides and imines [9,10], we started an investigation of the reaction of diglycolic anhydride (1,4-dioxane-2,6-dione) (3) with various imines 4. Unlike the reactions of glutaric and succinic anhydride with imines, which attracted more attention [9–23], there are only two examples of reaction of anhydride 3 with imines so far [19]. A reaction of 3 with N-benzylidene-N-benzylamine (4a) and N-benzylidene-N-phenethylamine (4b) affording the corresponding 5-oxo-3-aryl-N-substituted morpholine-2-carboxylic acids 5a,b has been described in a patent claiming that morpholinones 5a,b possess analgesic, anti-inflammatory, and antihistaminic activity [19]. The work of Shetty, however, does not consider the steric course of the reaction and the relative configuration of the products [19] (Fig. 1).
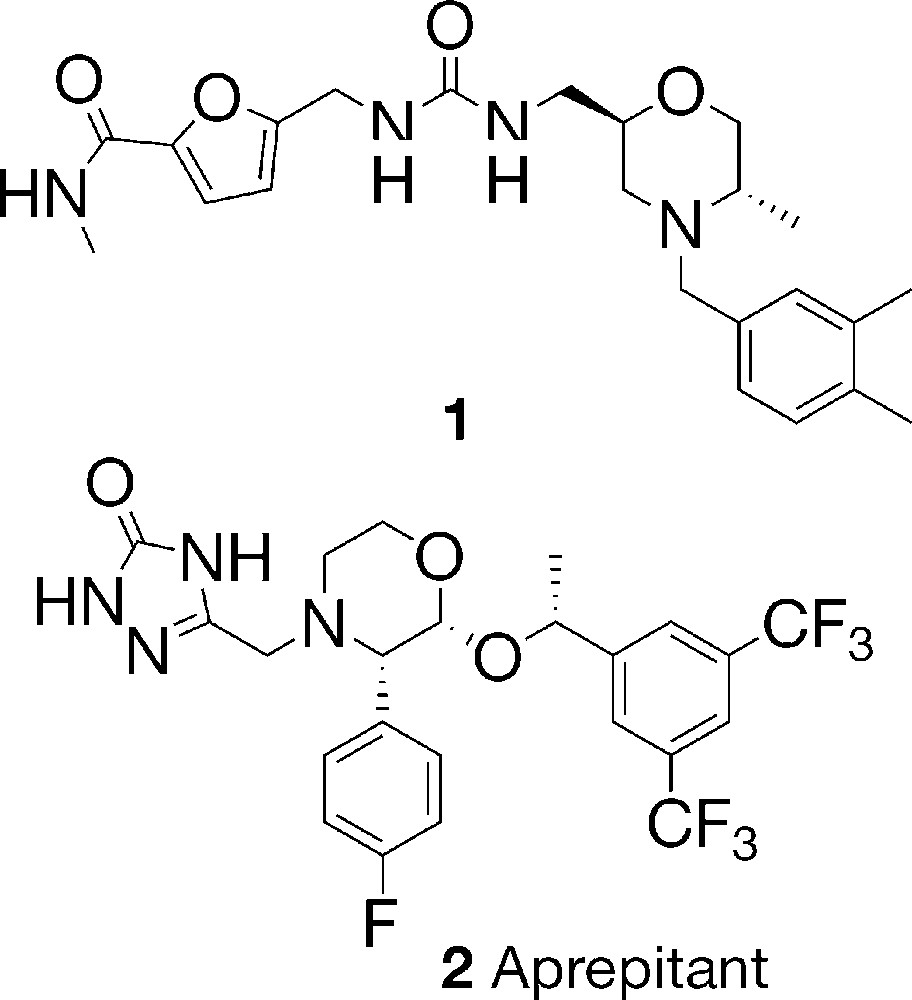
Chemical structures of compounds 1 and 2.
The aim of the present paper is to determine the steric course of the reaction of the anhydride 3 with a series of arylideneamines 4 and to establish the relative configuration of the newly prepared 3,4-disubstituted 5-oxomorpholin-2-carboxylic acids 5. Acids 5 are the starting compounds for the introduction of amino acid residues via a peptide bond to the morpholinone heterocycle in order to obtain model compounds for biological evaluation. The carboxylic acids 5 themselves are β-amino acid derivatives and as such they can be regarded as tools for medicinal chemistry [24,25]. Recently, we have shown that piperidinones containing peptide bond to the heterocycle exhibit antihistaminic activity [23], which gave us ground to continue our research with the bioisosteric morpholinones. We prepared two types of peptide derivatives of the morpholinones: type I (acylated aminomethylmorpholinones) and type II (2-carboxamides) (Fig. 2). The amino acids used were derivatives of proline, phenylalanine and tryptophan, in view to introduce pharmacophore substituents. l-Proline and l-phenylalanine moieties are incorporated in drugs with ACE inhibiting activity [26]. Protected l-tryptophan was selected, because esters of N-acetyl-l-tryptophan have been shown to possess high antihistaminic activity [23,27].
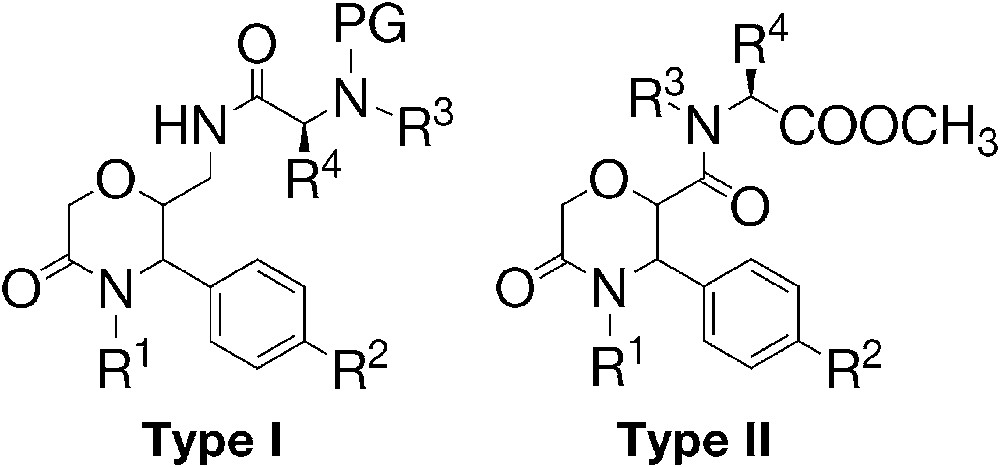
Type I and type II (PG: protective group).
2 Results and discussion
The reaction of diglycolic anhydride 3 with N-benzylidene-N-benzylamine 4a was repeated and acid 5a was obtained in 40% yield as a single diastereomer, which was established by 1H NMR spectroscopy (Scheme 1). We ascribed a trans relative configuration to this product, having in mind that the reaction conditions, i.e. long reflux at 140 °C, would favour the formation of the thermodynamically more stable diastereomer [9,11,12]. Further on, we carried out the reactions of anhydride 3 with N-(arylmethylene)-N-alkylamines 4c-g in p-xylene under reflux for 6 h (Scheme 1).
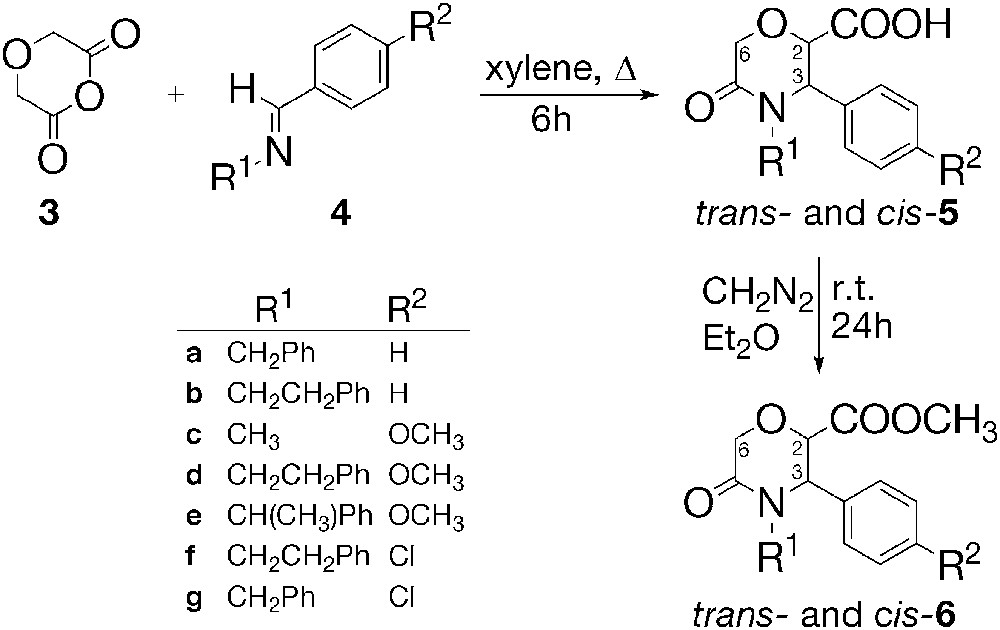
Yields are given in Table 1.
The acids 5c–g were isolated from the reaction mixtures either by filtration (5c,d,f) or extraction with aq. Na2CO3 (5a,e,g), followed by recrystallization. The 1H NMR spectroscopy of the crude products 5c–g showed signals for two diastereomeric products. The ratio of the diastereomeric acids 5c,d,f,g varied from 3:1 to 6:1. In analogy with trans-5a, we concluded that the predominant isomers of 5c–g are also trans isomers. The diastereomeric mixture 5c (trans:cis = 3:1) was separated by column chromatography and the individual diastereomers trans- and cis-5c were characterized. However, chromatographic separation of trans- and cis- acids 5d–g was difficult, so the diastereomeric mixtures were analyzed by means of 1H NMR spectra. This was possible, because the signals for H-2, H-3, H-6, as well as for CH3O in acids 5d,e, could be seen separately, which allowed the characterization of the two diastereomers in a mixture. The vicinal coupling constants 3J2,3 of the trans isomers appear in the range of 1.7–3.6 Hz, and for the cis-3J2,3 at about 3.4 Hz (Table 1). Acid 5e, which can be a mixture of eight enantiomers, gave a single product after recrystallization, to which a trans configuration was ascribed. Very small difference of 3J2,3 for the both diastereomers within a couple is observed, which makes difficult the application of the vicinal coupling constants 3J2,3 for the determination of the relative configuration. The recrystallization of the crude reaction products afforded only trans-5 in enough pure form for further transformations. Acids trans- and cis-5 are obtained as racemic mixtures of enantiomers and only one of them will be shown throughout the text for clarity.
Yields of acids 5 and their methyl esters 6.
| No. | 3Jtrans (Hz) | 3Jcis (Hz) | Trans/Cis | Yielda (%) |
| 5a | 2.6 | — | — | 40/0 |
| 5c | 3.6 | 3.4 | 3:1c | 35/12 |
| 5d | 3.2 | 3.4 | 6:1c | 51/0 |
| 5e | 1.7 | — | — | 22/0 |
| 5f | 2.8 | 3.4 | 4:1c | 48/0 |
| 5g | 2.6 | —b | 3:1c | 35/0 |
| 6a | 2.6 | — | — | 92/0 |
| 6c | 4.2 | 3.5 | 3:1d | 55/19 |
| 6d | 3.7 | 3.4 | 6:1d | 69/12 |
| 6e | 1.5 | — | — | 56/0 |
| 6f | 3.1 | 3.4 | 4.5:1d | 59/13 |
| 6g | 2.6 | 3.5 | 2:1d | 55/32 |
a The values are for the isolated trans/cis product.
b The value of 3Jcis-5g could not be measured because of overlap of signals.
c The ratio is determined by integral intensities of the protons in the 1H NMR spectra of the crude products.
d The ratio is determined using the yields of the esters.
In order to collect more data about the configuration of the studied compounds, we converted the crude acidic products 5 into the methyl esters 6 by means of reaction with diazomethane (Scheme 1). The mixtures of the methyl esters trans- and cis-6 were separated by column chromatography. Trans-6a,c,g were obtained as oily products, while trans-6d,e,f were crystalline. Cis esters 6c,d,f were oily compounds, and cis-6g a crystalline product. Cis-6e could not be isolated by means of chromatography, because of the complex mixture of products. 1H NMR spectroscopy analysis of the individual esters trans- and cis-6 showed vicinal coupling constants 3J2,3 for the trans esters in the range of 1.5–4.2 Hz, and for the cis-3J2,3 in the range of 3.4–3.5 Hz (Table 1). Trans isomers 6c,d,f,g exhibited COOCH3 singlet in a lower field than the corresponding cis isomers. Similar deshielding of the COOCH3 singlet signal was observed in the 1H NMR spectra of methyl trans-1,2-disubstituted 6-oxopiperidine-3-carboxylates [13,14]. The difference in the chemical shifts of COOCH3 singlet signal of the trans- and cis- methyl esters 6 can be used for the determination of the relative configuration, only when the two diastereomers are available. This is important because of the close values of 3J2,3 of the trans and cis isomers of a given diastereomeric couple.
It can be accepted that the morpholinone ring is in a half-chair conformation, due to the conjugation in the NCO fragment [28,29] (Fig. 3). The value of 2J for H-6 (ca. 16.5 Hz) corresponds to a conformation where the CO plane bisects the angle H–C(6)–H [28,29]. The 3J2,3 values of 5 and 6 might indicate either a trans isomer in which the substituents at C-2 and C-3 chiral centers occupy axial, resp. pseudoaxial positions in solution, i. e. the corresponding protons are equatorial (partial conformation II, Fig. 3), or a cis isomer, which should exist predominantly in a conformation with axial 3-Ar, i.e. with equatorial H-3 in solution. The conformation with axial 3-Ar allows less steric A1,2 strain between the aryl and the N-substituent, in analogy with other polysubstituted lactams [9,13] (partial conformation III, Fig. 3).
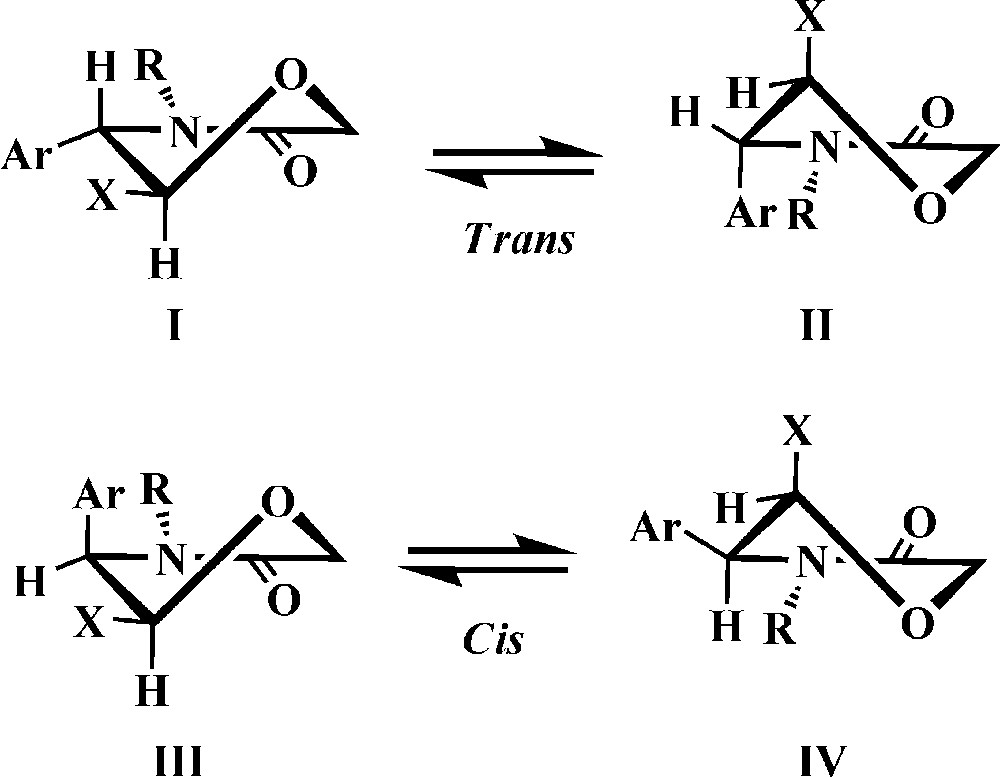
Partial conformations of trans and cis morpholinones.
X-ray diffraction analysis of a single crystal of the ester trans-6d confirmed the trans configuration of the 2- and 3-substituents (Fig. 4). The conformation of the morpholinone ring was determined as an envelope (puckering parameters [30]: θ = 48.6(2), φ = 64.7(3)°). In spite of the envelope shape of the ring, the spatial orientations of the COOCH3 group at C-2 and the 4-methoxyphenyl substituent at C-3 are respectively axial and pseudoaxial, which resembles conformation II (Fig. 3). The corresponding protons at C-2 and C-3 remain to be equatorial and pseudoequatorial, respectively. The dihedral angle between the above-mentioned protons is evaluated at 60°. Thus, the 1H NMR data on the conformation of compounds trans-5, 6 in solution coincide with the conclusion made through X-ray single-crystal analysis (Fig. 4).
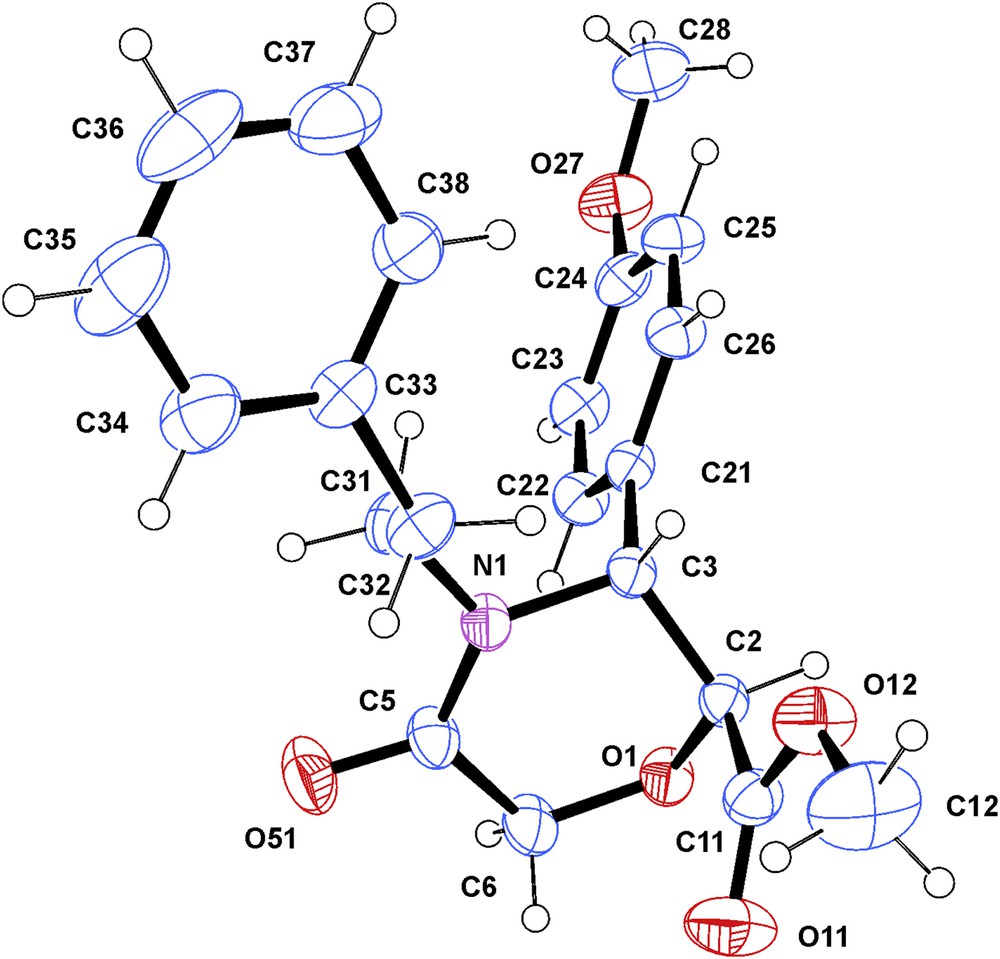
ORTEP view of compound 6d. Displacement ellipsoids are drawn at the 50% probability level (the hydrogen atoms are represented by circles of arbitrary radii).
The methyl esters trans-6d,f were obtained from the crude reaction products 5d,f, respectively, by esterification (reflux with MeOH/H2SO4) [9]. Trans-6d obtained by this method was subjected to further transformations. The planned transformations of the carboxylic group of trans-6d do not affect the stereogenic carbon atoms at the morpholinone ring, so, they are expected to give trans isomers.
The preparation of 5-aminomethylmorpholin-3-one from the acid trans-5d via alcohol 7 is depicted in Scheme 2. Ester trans-6d was converted selectively into 6-hydroxymethyl-3-morpholinone 7 by the reduction with LiBH4 in THF [9]. Crystalline alcohol 7 thus obtained was characterized by means of 1H NMR, which exhibited 3J5,6 = 9.4 Hz. Alcohol 7 was treated with phthalimide and glutarimide according to the Mitsunobu protocol (Ph3P; DEAD) [31], which gave rise to trans-2-((3-(4-methoxyphenyl)-5-oxo-4-phenethylmorpholin-2-yl)methyl)-isoindol-1,3-dione 8 and the respective 1-((trans-(3-(4-methoxyphenyl)-5-oxo-4-phenethylmorpholin-2-yl)methyl)-piperidin-2,6-dione 9. (Scheme 2) The reaction products 8 and 9 were isolated after chromatography in 60%, resp. 29% yields as crystalline materials.
Mitsunobu substitution of OH group (only one enantiomer is shown).
The higher yield of phthalimide 8 makes it a more suitable starting material for the Gabriel synthesis of the primary amine 10 after deprotection (Scheme 3). Best yields (85%) of sufficiently pure amine 10 were obtained by treatment of 8 with 40% aqueous methylamine at room temperature overnight [32]. The 1H NMR spectra of alcohol 7 and amine 10 exhibited 3J = 9.4, resp. 9.1 Hz for the protons at the stereogenic centers. This value is consistent with the trans configuration of alcohol 7 and amine 10 as well as with the preferred conformation in solution with diaxial H at C-5 and C-6 (partial conformation I, Fig. 3). This conclusion about the conformation in solution of compounds 7 and 10 is in agreement with the conformation of other 5,6-disubstituted morpholin-3-ones previously described by Stefanovsky et al. [28,29]. The preference for the conformation with dipseudoequatorial substituents (I, 7 and 10) can be attributed to the larger effective volume of the substituent at the C-6 stereocenter. The signals of H-5 and H-6 in the 1H NMR spectra of phthalimido derivative 8 and piperidinedione 9 overlapped with other multiplet signals, which did not allow us to determine the 3J value of the two compounds.
Peptide derivatives of type I (only one enantiomer of 8 and 10 is shown).
X-ray analysis of alcohol 7 (Fig. 5) and phthalimide 8 (Fig. 6) confirmed the trans configuration of the substituents at the morpholinone ring. The conformation of the morpholinone ring was determined as half-chair (puckering parameters 30 θ = 48.11(19) and 49.0(7), φ = 17.0(3) and 28.1(10)° for compounds 7 and 8 respectively), with pseudoequatorial, resp. equatorial substituents at the chiral C-5 and C-6. The respective H-5 and H-6 protons occupy the axial positions with a dihedral angle estimated at 170°, leading to a higher value of 3J. Thus, the conformation of compound 7 in solution as determined by 1H NMR spectroscopy coincides with that pointed by means of X-ray analysis.
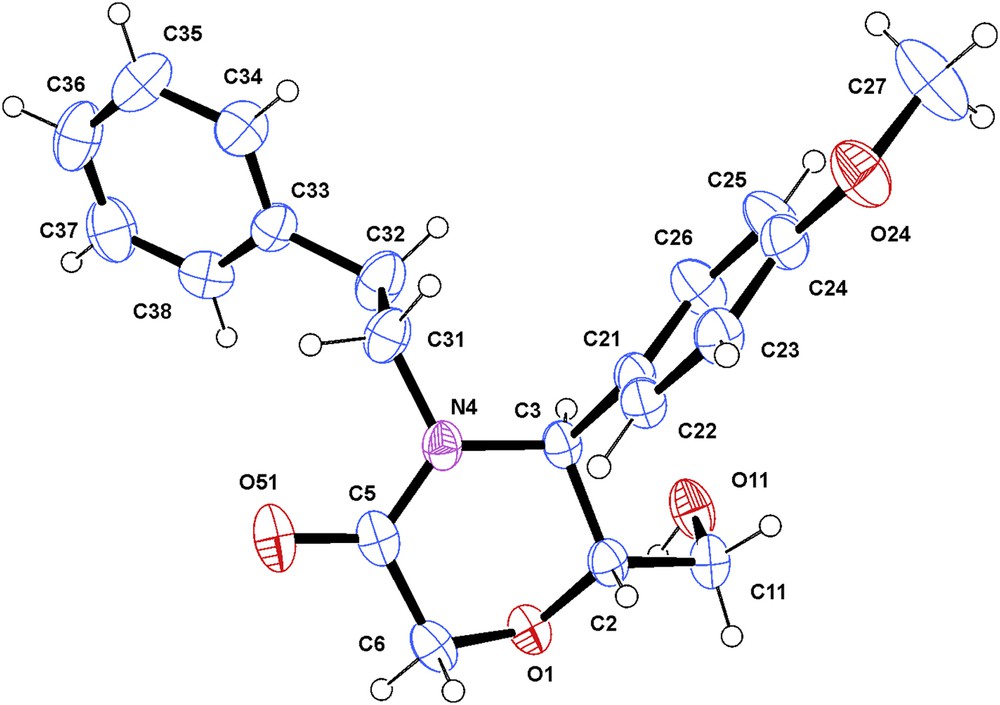
ORTEP view of compound 7. Displacement ellipsoids are drawn at the 50% probability level (the hydrogen atoms are represented by circles of arbitrary radii).
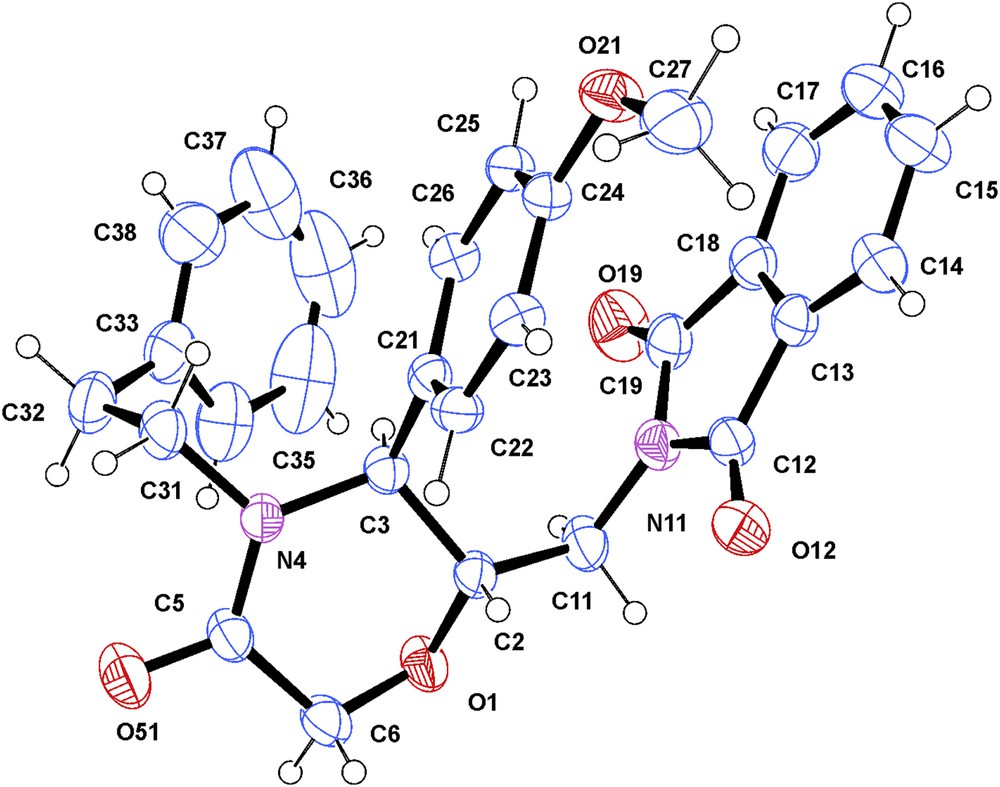
ORTEP view of compound 8. Displacement ellipsoids are drawn at the 50% probability level (the hydrogen atoms are represented by circles of arbitrary radii).
The introduction of a peptide bond into the side chain to the morpholinone ring was carried out from amine 10 to type-I peptides, and from acid trans-5d to type-II peptides in the presence of DCC [33], in order to obtain model compounds for the study of ACE inhibitory activity. Amine 10 was acylated by means of BOC-l-proline and N-trifluoroacetyl-l-phenylalanine, affording derivatives 11, resp. 12, in good yields after column chromatography (Scheme 3). Compounds 11 and 12 are 1:1 mixtures of α-S, (±)-trans diastereomers, according to their 1H NMR spectra. In spite of our efforts, we could not separate the diastereomeric couples of the compounds 11 and 12 by means of column chromatography. This made the description of the 1H NMR spectral data in the experimental part difficult, because the signals for the same protons in the different diastereomers are not shifted in one and the same direction. For this reason, in the experimental part, the signals for the same protons with higher chemical shift are noted with the “*” symbol, by analogy with a previous publication [33].
The methyl esters of l-tryptophan and l-proline (used as hydrochlorides) were N-acylated by means of trans-5d to the derivatives 13 and 14, in good yields (Scheme 4). As in the case of compounds 11 and 12, compounds 13 and 14 are 1:1 mixtures of α-S, (±)-trans diastereomers according to their 1H NMR spectra. The acylated tryptophan 13 was separated by means of fractional recrystallization into the two diastereomers, which are denoted in the experimental part as 13a (Rf 0.29) and 13b (Rf 0.36), referring to the TLC behavior of the isomers. Proline derivative 14 could not be separated into the individual diastereomers.
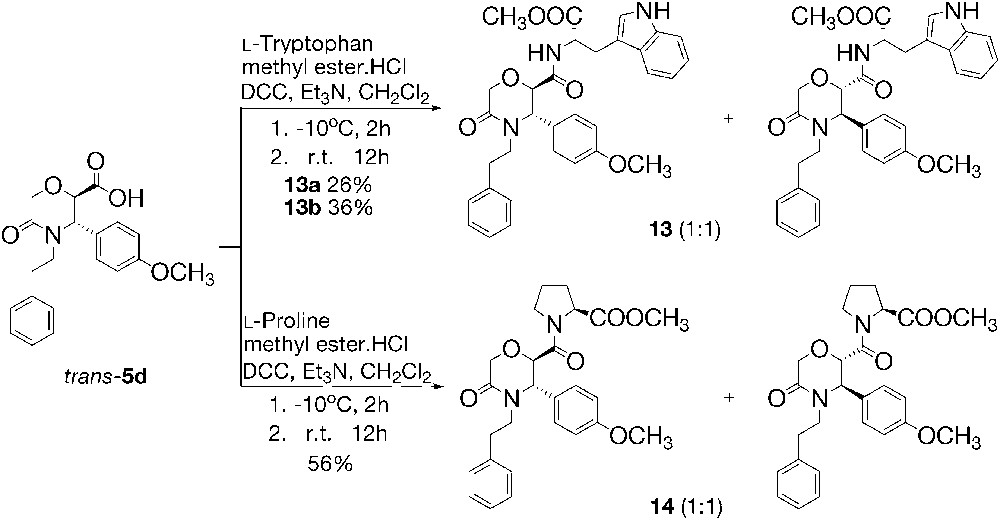
Peptide derivatives of type II.
The transformations done on α-amino acids to afford type-I and Type-II peptides took place with retention of their l-configuration. The trans configuration of the substituents at the morpholinone ring was ascribed to compounds 11–14, because the reactions employed for their preparation do not affect the chiral centers of the heterocycle. 1H NMR spectra of peptide derivatives 11–14 are in agreement with the stereochemistry. Thus, type-I peptide compounds 11 and 12 exhibit 3J within the 9.2–9.4 Hz range, similarly to the starting amine 10. Type-II derivatives 13 and 14 are characterized by a 3J value in the 3.8–4.3 Hz range, which is close to the value of 3J of the starting acid trans-5d (3.2 Hz). In the schemes, only one enantiomer of trans-5d and 10 is shown for clarity.
3 Experimental
3.1 General
Melting points were taken on a Boetius PHMK 0.5 microhot stage apparatus and are uncorrected. IR spectra were recorded on a Specord 75 instrument in Nujol. 1H NMR spectra (250.13 MHz) were obtained on a “Bruker Avance DRX-250” spectrometer; 1H NMR spectra (600 MHz) were taken on a Bruker AV 600 spectrometer in CDCl3 or DMSO-d6. The chemical shifts are given in parts per million (δ ppm) relative to tetramethylsilane as an internal standard. Multiplicity is indicated by s (singlet), br s (broad singlet), d (doublet), t (triplet), q (quartet) or m (multiplet). Coupling constants (J) are reported in Hz.
Microanalyses were carried out at the Faculty of Chemistry and Pharmacy, University of Sofia, Bulgaria, using a Vario EL III Elemental analyzer.
Mass spectra (MS) were recorded on a DFS high-resolution magnetic sector instrument Thermo Fisher Scientific GmbH (Bremen, Germany), as EI-mode (electron energy 70 eV, emission current 0.250 mA, source temperature 250 °C) or as CI-mode (electron energy 120 eV, emission current 0.450 mA and source temperature 150 °C).
Thin-layer chromatography (TLC) was performed on Merck 1.05554 silica gel 60 F254 aluminum plates. Column chromatography was carried out using MN Kieselgel 60 (0.063–0.2 mm) and Merck silica gel 60 (0.063–0.2 mm).
The preparation of the known starting imines 4 was carried out according to the procedure for N-benzylidene-N-benzylamine 4a [19].
3.2 X-ray structure determination
Colorless single crystals of compounds 6d, 7 and 8 were obtained by slow evaporation from ethyl acetate. Diffraction data were collected at 290 K the by ω-scan technique, on an Agilent Diffraction SuperNova Dual four-circle diffractometer equipped with an Atlas CCD detector using a mirror-monochromatized Mo Kα radiation from micro-focus source (λ = 0.7107 Å). The determination of cell parameters, data integration, scaling and absorption correction were carried out using the CrysAlis Pro program package [34]. The structures were solved by direct methods (SHELXS-97) and refined by full-matrix least-square procedures on F2 (SHELXL-97) [35]. Non-hydrogen atoms were refined anisotropically and hydrogen atoms were placed at idealized positions and refined using the riding model. Crystallographic data (excluding structure factors) for the structural analysis were deposited with the Cambridge Crystallographic Data Centre, CCDC Nos. 905074, 905075 and 905076. Copy of this information may be obtained free of charge from: The Director, CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK. Fax: +44 1223 336–033, e-mail: deposit@ccdc.cam.ac.uk, or www.ccdc.cam.ac.uk.
3.3 General procedure for preparation of (±)-trans- and cis-3,4-disubstituted 5-oxomorpholine-2-carboxylic acids 5
To a solution of N-arylidene-N-alkylamine (4a, 4c-g, 5 mmol) in dried p-xylene (10 mL), diglycolic anhydride (3, 0.58 g, 5 mmol) was added. The reaction mixture was refluxed for 6 h. Acids 5c,d,f crystallized from the reaction mixtures and were obtained after filtration. Acids 5a,e,g deposited as oily products which were extracted by means of 10% aq. Na2CO3 (3 × 5 mL), then once with water (5 mL) and the aqueous solutions were combined, acidified (10% HCl) and extracted with ethyl acetate (3 × 10 mL). The combined organic solutions were dried (Na2SO4) and the solvent was evaporated. The residue was triturated with ethyl acetate and filtered to give acids 5a,e,g. Recrystallization of crude acids 5d–g afforded trans-5d–g as single diastereomers. 1H NMR data for cis-5d,f were extracted from the spectra of the crude acids.
3.3.1 Trans-(±)-4-benzyl-3-phenyl-5-oxomorpholine-2-carboxylic acid 5a
The reaction of anhydride 3 and imine 4a yielded trans-5a (0.622 g, 40%) as white crystals. Rf 0.58 (light petroleum/ethyl acetate/HCOOH 2:3:0.05, two-fold development). Mp 177–179 °C. According to [19], mp of 5a is 177–181 °C. IR (Nujol): 2400–3200 (OH); 1720 (COOH); 1620 (CON) cm−1. 1H NMR (250 MHz, CDCl3 + DMSO-d6): δ 3.50 (d, 1H, NCHPh, J = 14.8 Hz); 4.33 (d, 1H, H-3, J = 2.6 Hz); 4.43 (d, 1H, H-6, J = 17.0 Hz); 4.79 (d, 1H, H-2, J = 2.6 Hz); 4.88 (d, 1H, H-6, J = 17.0 Hz); 5.47 (d, 1H, NCHPh, J = 14.8 Hz); 7.10–7.40 (m, 10H, arom. H); the COOH signal is not seen because of exchange. MS (EI) m/z: 311 (M, 25); 252 (14); 194 (12); 164 (30); 148 (100); 147 (48); 131 (16); 104 (12). Anal. calcd for C18H17NO4 (311.31): C 69.44%; H 5.50%; N 4.50%; found: C 69.69%; H 5.19%; N 4.52%.
3.3.2 (±)-Trans- and cis-3-(4-methoxyphenyl)-4-methyl-5-oxomorpholine-2-carboxylic acid 5c
The reaction of anhydride 3 and imine 4c yielded 5c (0.833 g, 63%) as off-white crystals. Mp 173–175 °C. According to 1H NMR, the trans/cis ratio was 3:1. Chromatographic purification (light petroleum/ethyl acetate/HCOOH 2:3:0.05) and recrystallization from ethyl acetate-methanol yielded:
Trans-5c (0.464 g, 35%) as white crystals. Rf 0.20 (light petroleum/ethyl acetate/HCOOH 2:3:0.05, two-fold development). Mp 190–192 °C. IR (Nujol): 2300–3200 (OH); 1750 (COOH); 1590 (CON) cm−1. 1H NMR (250 MHz, DMSO-d6): δ 2.69 (s, 3H, NCH3); 3.76 (s, 3H, CH3O); 4.28 (d, 1H, H-6, J = 16.6 Hz); 4.39 (d, 1H, H-6, J = 16.6 Hz); 4.43 (d, 1H, H-3, J = 3.6 Hz); 4.82 (d, 1H, H-2, J = 3.6 Hz); 6.94–7.25 (m, 4H, arom. H); 13.31 (br s, 1H, COOH). MS (EI) m/z: 265 (M, 43); 232 (19); 221 (45); 190 (37); 177 (86); 161 (85); 146 (80); 136 (26); 121 (100); 105 (64). Anal. calcd for C13H15NO5 (265.26): C 58.86%; H 5.70%; N 5.28%; found: C 59.08%; H 5.51%; N 5.23%.
Cis-5c (0.159 g, 12%) as white crystals. Rf 0.04 (light petroleum/ethyl acetate/HCOOH 2:3:0.05, two-fold development). Mp 200–202 °C. IR (Nujol): 2300–3500 (OH); 1750 (COOH); 1590 (CON) cm−1. 1H NMR (250 MHz, DMSO-d6): δ 2.68 (s, 3H, NCH3); 3.74 (s, 3H, CH3O); 4.25 (d, 1H, H-6, J = 16.3 Hz); 4.36 (d, 1H, H-6, J = 16.3 Hz); 4.65 (d, 1H, H-3, J = 3.4 Hz); 4.79 (d, 1H, H-2, J = 3.4 Hz); 6.88–7.21 (m, 4H, arom. H); 13.11 (br s, 1H, COOH). Anal. calcd for C13H15NO5 (265.26): C 58.86%; H 5.70%; N 5.28%; found: C 59.10%; H 5.40%; N 5.35%.
3.3.3 (±)-Trans- and cis-3-(4-methoxyphenyl)-5-oxo-4-phenethylmorpholine-2-carboxylic acid 5d
The reaction of anhydride 3 and imine 4d yielded 5d (1.226 g, 69%) as white crystals. Mp 168–170 °C. According to 1H NMR, the trans/cis ratio was 6:1. The crude product was recrystallized from ethyl acetate to give trans-5d (0.908 g, 51%) as colorless crystals. Rf 0.42 (light petroleum/ethyl acetate/HCOOH 2:3:0.05, two-fold development). Mp 175–177 °C. IR (Nujol): 2400–3200 (OH); 1720 (COOH); 1600 (CON) cm−1. 1H NMR (600 MHz, DMSO-d6): δ 2.55–2.82 (m, 3H, NCHCH2); 3.76 (s, 3H, CH3O); 3.84–3.92 (m, 1H, NCH); 4.36 (d, 1H, H-6, J = 16.5 Hz); 4.45 (d, 1H, H-6, J = 16.5 Hz); 4.54 (d, 1H, H-3, J = 3.2 Hz); 5.00 (d, 1H, H-2, J = 3.2 Hz); 6.78–7.37 (m, 9H, arom. H); 13.50 (br s, 1H, COOH). MS (EI) m/z: 355 (20, M+); 264 (20); 251 (100); 207 (15); 177 (66); 146 (22); 121 (71); 105 (10). Anal. calcd for C20H21NO5 (355.38): C 67.59%; H 5.96%; N 3.94%; found: C 67.10%; H 5.85%; N 3.69%.
Cis-5d: Rf 0.08 (light petroleum/ethyl acetate/HCOOH 2:3:0.05, two-fold development). 1H NMR (600 MHz, DMSO-d6): δ 2.50–2.60 (3H, m, NCHCH2); 3.73 (3H, s, CH3O); 3.84–3.92 (1H, m, NCH); 4.31 (1H, d, H-6, J = 16.6 Hz); 4.42 (1H, d, H-6, J = 16.6 Hz); 4.55 (1H, d, H-3, J = 3.4 Hz); 4.68 (1H, d, H-2, J = 3.4 Hz); 6.78–7.37 (9H, m, arom. H); 13.50 (br s, 1H, COOH).
3.3.4 (±)-Trans-3-(4-methoxyphenyl)-5-oxo-4-(1-phenylethyl)morpholine-2-carboxylic acid 5e
The reaction of anhydride 3 and imine 4e yielded 5e (1.065 g, 60%) as oily product. Recrystallization from ethyl acetate yielded trans-5e (0.391 g, 22%) as a white powder. Rf 0.58 (light petroleum/ethyl acetate/HCOOH 2:3:0.05, two-fold development). Mp 212–216 °C. IR (Nujol): 2400–3200 (OH); 1730 (COOH); 1590 (CON) cm−1. 1H NMR (250 MHz, DMSO-d6): δ 1.10 (d, 3H, CH3, J = 7.2 Hz); 3.78 (s, 3H, CH3O); 4.21 (d, 1H, H-3, J = 1.7 Hz); 4.41 (d, 1H, H-6, J = 16.7 Hz); 4.58 (d, 1H, H-2, J = 1.7 Hz); 4.61 (d, 1H, H-6, J = 16.7 Hz); 5.70 (q, 1H, NCHCH3, J = 7.2 Hz); 6.94–7.36 (m, 9H, arom. H); 13.13 (br s, 1H, COOH). MS (CI) m/z: 384 (M+ + 29, 9); 356 (M+ + 1, 100); 312 (10); 280 (14.); 252 (74); 234 (14); 208 (12); 178 (18); 105 (29). Anal. calcd for C20H21NO5 (355.38): C 67.59%, H 5.96%; N 3.94%; found C 67.61%; H 5.60%; N 3.96%.
3.3.5 (±)-Trans- and cis-3-(4-chlorophenyl)-5-oxo-4-phenethylmorpholine-2-carboxylic acid 5f
The reaction of anhydride 3 and imine 4f yielded 5f (1.343 g, 78%) as white crystals. Mp 183–186 °C. According to 1H NMR, the trans/cis ratio was 4:1. The crude product was recrystallized from ethyl acetate to give trans-5f (0.863 g, 48%) as colorless crystals. Rf 0.27 (light petroleum/ethyl acetate/HCOOH 2:3:0.05). Mp 222–225 °C. IR (Nujol): 2300–3200 (OH); 1720 (COOH); 1600 (CON) cm−1. 1H NMR (DMSO-d6): δ 2.67–2.92 (m, 3H, NCHCH2); 3.83–3.96 (m, 1H, NCH); 4.30 (d, 1H, H-6, J = 16.7 Hz); 4.44 (d, 1H, H-6, J = 16.7 Hz); 4.54 (d, 1H, H-3, J = 2.8 Hz); 5.10 (d, 1H, H-2, J = 2.8 Hz); 7.03–7.52 (m, 9H, arom. H); 13.10 (br s, 1H, COOH). Anal. calcd for C19H18ClNO4 (359.81): C 63.42%; H 5.04%; N 3.89%; found C 63.34%; H 5.09%; N 3.93%.
Cis-5f: Rf 0.05 (light petroleum/ethyl acetate/HCOOH 2:3:0.05). 1H NMR δ (250 MHz, DMSO-d6): 2.51–2.62 (3H, m, NCHCH2); 3.68–3.77 (m, 1H, NCH); 4.30 (d, 1H, H-6, J = 16.2 Hz); 4.41 (d, 1H, H-6, J = 16.2 Hz); 4.64 (d, 1H, H-3, J = 3.4 Hz); 4.72 (d, 1H, H-2, J = 3.4 Hz); 7.03–7.52 (m, 9H, arom. H); 13.13 (br s, 1H, COOH).
3.3.6 (±)-Trans- and cis-4-benzyl-3-(4-chlorophenyl)-5-oxomorpholine-2-carboxylic acid 5g
The reaction of anhydride 3 and imine 4g yielded 5g (0.945 g, 57%) as off-white crystals. Mp 176–180 °C. According to 1H NMR trans/cis ratio was 3:1. The crude product was recrystallized from 2-propanol to give trans-5g (0.604 g, 35%) as colorless crystals. Rf 0.30 (light petroleum/ethyl acetate/HCOOH 2:3:0.05). Mp 205–207 °C. IR (Nujol): 2300–3200 (OH); 1740 (COOH); 1620 (CON) cm−1. 1H NMR (250 MHz, DMSO-d6): δ 3.59 (d, 1H, NCH, J = 15.1 Hz); 4.40 (d, 1H, H-6, J = 16.8 Hz); 4.48 (d, 1H, H-3, J = 2.6 Hz); 4.58 (d, 1H, H-6, J = 16.8 Hz); 4.81 (d, 1H, H-3, J = 2.6 Hz); 5.10 (d, 1H, NCH, J = 15.1 Hz); 7.01–7.59 (m, 9H, arom. H); 13.45 (br s, 1H, COOH). HRMS: 345.0762; calcd for C18H16O4N35Cl (345.0768).
Cis-5g: Rf 0.07 (light petroleum/ethyl acetate/HCOOH 2:3:0.05).
3.4 General procedure for the preparation of methyl esters of (±)-trans- and cis-3,4-disubstituted 5-oxomorpholine-2-carboxylic acids 6
3.4.1 Method A
Acid 5a,c–g (1 mmol) was dissolved in methanol and dichloromethane 1:1 (4 mL), diazomethane (ethereal solution) was added and the mixture was left at room temperature overnight. The solution was concentrated and the residue was purified by column chromatography or recrystallization (for 6e).
3.4.2 Method B
Acid 5d,f (3 mmol) was dissolved in dry methanol (6 mL) and concentrated H2SO4 (0.16 mL, 3 mmol) was added. After 3 h under reflux, the reaction mixture was poured into a saturated aq. NaCl solution (10 mL). The solution was neutralized with 10% aq. Na2CO3 till pH = 8 and extracted with ethyl acetate (3 × 10 mL). The combined organic solutions were dried (Na2SO4) and the solvent was evaporated. The residue was purified by means of column chromatography and/or recrystallization.
3.4.3 Methyl (±)-trans-4-benzyl-3-phenyl-5-oxomorpholine-2-carboxylate 6а (method A)
Chromatographic purification (cyclohexane/ethyl acetate 3:2) gave trans-6a (0.299 g, 92%) as colorless oil. Rf 0.67 (light petroleum/ethyl acetate 2:3). IR (CHCl3): 1730 (COOCH3); 1640 (CON) cm−1. 1H NMR (250 MHz, CDCl3): δ 3.38 (d, 1H, NCH, J = 14.6 Hz); 3.52 (s, 3H, COOCH3); 4.37 (d, 1H, H-3, J = 2.6 Hz); 4.45 (d, 1H, H-6, J = 17.0 Hz); 4.70 (d, 1H, H-2, J = 2.6 Hz); 4.86 (d, 1H, H-6, J = 17.0 Hz); 5.60 (d, 1H, NCH, J = 14.6 Hz); 6.66–7.07 (m, 10H, arom. H). MS (CI) m/z (%): 354 (M+ + 29, 10); 326 (M+ + 1, 100); 294 (2); 266 (2); 248 (3); 162 (3); 119 (2). Anal. calcd for C19H19NO4 (325.30): C 70.14%; H 5.89%; N 4.31%; found: C 70.38%; H 6.10%; N 4.67%.
3.4.4 Methyl (±)-trans- and cis-4-methyl-3-(4-methoxyphenyl)-5-oxomorpholine-2-carboxylate 6c (method A)
Chromatographic purification (light petroleum/ethyl acetate 2:3) gave trans- and cis-6c.
Trans-6c (0.154 g, 55%) as colorless oil. Rf 0.28 (light petroleum/ethyl acetate 2:3). IR (CHCl3): 1740 (COOCH3); 1660 (CON) cm−1. 1H NMR (250 MHz, CDCl3): δ 2.83 (s, 3H, NCH3); 3.78 (s, 3H, OCH3); 3.82 (s, 3H, COOCH3); 4.34 (d, 1H, H-3, J = 4.2 Hz); 4.35 (d, 1H, H-6, J = 16.8 Hz); 4.59 (d, 1H, H-6, J = 16.8 Hz); 4.80 (d, 1H, H-2, J = 4.2 Hz); 6.91–6.94 (m, 2H, arom. H); 7.17–7.20 (m, 2H, arom. H). Anal. calcd for C14H17NO5 (279.29): C 60.21%; H 6.14%; N 5.02%; found C 60.58%; H 6.10%; N 4.97%.
Cis-6c (0.080 g, 19%) as colorless oil. Rf 0.10 (light petroleum/ethyl acetate 2:3). IR (CHCl3): 1760 (COOCH3); 1660 (CON) cm−1. 1H NMR (250 MHz, CDCl3): δ 2.86 (s, 3H, NCH3); 3.57 (s, 3H, COOCH3); 3.80 (s, 3H, OCH3); 4.57 (d, 1H, H-3, J = 3.5 Hz); 4.64 (d, 1H, H-6, J = 16.7 Hz); 4.60 (d, 1H, H-6, J = 16.7 Hz); 4.72 (d, 1H, H-2, J = 3.5 Hz); 6.85–6.89 (m, 2H, arom. H); 7.15–7.18 (m, 2H, arom. H). Anal. calcd for: C14H17NO5 (279.29): C 60.21%; H 6.14%; N 5.02%; found (%): C 60.82%; H 6.28%; N 4.69%.
3.4.5 Methyl (±)-trans- and cis-3-(4-methoxyphenyl)-5-oxo-4-phenethylmorpholine-2-carboxylate 6d
3.4.5.1 Method A
Chromatographic purification (light petroleum/ethyl acetate 3:2) afforded trans- and cis-6d.
Trans-6d (0.255 g, 69%) as colorless crystals. Rf 0.48 (light petroleum/ethyl acetate 2:3). Mp 124–126 °C (ethyl acetate). IR (Nujol): 1735 (COOCH3); 1630 (CON) cm−1. 1H NMR (250 MHz, CDCl3): δ 2.73–2.89 (m, 3H, NCHCH2); 3.75 (s, 3H, OCH3); 3.82 (s, 3H, COOCH3); 4.10–4.19 (m, 1H, NCH); 4.37 (d, 1H, H-3, J = 3.7 Hz); 4.36 (d, 1H, H-6, J = 16.9 Hz); 4.64 (d, 1H, H-6, J = 16.9 Hz); 4.82 (d, 1H, H-2, J = 3.7 Hz); 6.90–7.30 (m, 9H, arom. H). MS (EI) m/z: (%): 369 (M+, 15), 310 (7), 278 (20), 265 (41), 221 (48), 177 (100). Anal. calcd for C21H23NO5 (369.16): C 68.28%; H 6.28%; N 3.79%; found: C 68.49%; H 6.65%; N 3.58%.
Cis-6d (0.042 g, 12%) as colorless oil. Rf 0.19 (light petroleum/ethyl acetate 2:3). IR (CHCl3): 1760 (COOCH3); 1640 (CON) cm−1. 1H NMR (250 MHz, CDCl3): δ 2.64–3.01 (m, 3H, NCHCH2); 3.55 (s, 3H, COOCH3); 3.81 (s, 3H, OCH3); 3.97–4.42 (m, 1H, NCH); 4.30 (d, 1H, H-3, J = 3.4 Hz); 4.37 (d, 1H, H-6, J = 16.7 Hz); 4.52 (d, 1H, H-2, J = 3.4 Hz); 4.61 (d, 1H, H-6, J = 16.7 Hz); 6.83–6.90 (m, 2H, arom. H); 7.08–7.15 (m, 2H, arom. H); 7.17–7.38 (m, 5H, arom. H). MS (CI) m/z: 398 (M+ + 29); 370 (M+, 100); 338 (20); 310 (1); 262 (16); 177 (3). Anal. calcd for C21H23NO5 (369.16): C 68.28%; H 6.28%; N 3.79%; found: C 68.59%; H 6.55%; N 3.50%.
3.4.5.2 Method B
Recrystallization from ethyl acetate yielded trans-6d (0.609 g, 67%) of colorless crystals. Mp 124–126 °C. TLC (light petroleum/ethyl acetate 3:2) with a sample of trans-6d obtained by method A showed that the products are identical; mixed mp with the sample obtained by method A was not depressed.
3.4.6 Methyl (±)-trans-3-(4-methoxyphenyl)-5-oxo-4-(1-phenylethyl)morpholine-2-carboxylate 6e (method A)
Chromatographic purification (cyclohexane/ethyl acetate 5:2) and recrystallization from ethyl acetate gave trans-6e (0.208 g, 56%) as colorless crystals. Rf 0.55 (light petroleum/ethyl acetate 2:3). Mp 137–139 °C. IR (Nujol): 1740 (COOCH3); 1640 (CON) cm−1. 1H NMR (250 MHz, CDCl3): δ 1.14 (d, 3H, CH3CH, J = 7.3 Hz); 3.39 (s, 3H, COOCH3); 3.83 (s, 3H, OCH3); 4.14 (d, 1H, H-3, J = 1.5 Hz); 4.47 (d, 1H, H-6, J = 17.1 Hz); 4.51 (d, 1H, H-2, J = 1.5 Hz); 4.91 (d, 1H, H-6, J = 17.1 Hz); 6.14 (q, 1H, NCH, J = 7.4 Hz); 6.82–7.43 (m, 9H, arom. H). MS (EI) m/z 369 (4, M+); 238 (4); 193 (14); 192 (100); 162 (8); 105 (9). Anal. calcd for C21H23NO5 (369.16): C 68.28%; H 6.28%; N 3.79%; found: C 68.00%; H 6.60%; N 3.90%.
3.4.7 Methyl (±)-trans- and cis-3-(4-chlorophenyl)-5-oxo-4-phenethylmorpholine-2-carboxylate 6f
3.4.7.1 Method A
Chromatographic purification (cyclohexane/2-propanol 8:1) gave trans-and cis-6f.
Trans-6f (0.199 g, 59%) as colorless crystals. Rf 0.65 (light petroleum/ethyl acetate 2:3). Mp 130–133 °C (ethyl acetate). IR (Nujol): 1720 (COOCH3); 1620 (CON) cm−1. 1H NMR (250 MHz, CDCl3): δ 2.72–2.87 (m, 3H, NCHCH2); 3.77 (s, 3H, COOCH3); 4.05–4.23 (m, 1H, NCH); 4.35 (d, 1H, H-3, J = 3.1 Hz); 4.36 (d, 1H, H-6, J = 17.0 Hz); 4.64 (d, 1H, H-6, J = 17.0 Hz); 4.85 (d, 1H, H-2, J = 3.1 Hz); 7.10–7.38 (m, 9H, arom. H). MS (CI) m/z (%): 402 (M+ + 29, 17); 374 (M+ + 1, 100); 342 (8); 314 (1); 282 (2); 262 (2). Anal. calcd for C20H20NO4Cl (373.84): C 64.26%; H 5.36%; N 3.75%; found: C 64.13%; H 5.27%; N 3.77%.
Cis-6f (0.046 g, 13%) as an oily product. Rf 0.38 (light petroleum/ethyl acetate 2:3). IR (CHCl3): 1760 (COOCH3); 1630 (CON) cm−1. 1H NMR (250 MHz, CDCl3): δ 2.68–3.03 (m, 3H, NCHCH2); 3.56 (s, 3H, OCH3); 4.03–4.13 (m, 1H, NCH); 4.29 (d, 1H, H-3, J = 3.4 Hz); 4.38 (d, 1H, H-6, J = 16.9 Hz); 4.52 (d, 1H, H-2, J = 3.4 Hz); 4.61 (d, 1H, H-6, J = 16.8 Hz); 7.10–7.39 (m, 9H, arom. H). MS (CI) m/z (%) 402 (M+ + 29); 374 (M+ +1, 100); 342 (5); 282 (2); 262(1); 181 (1). Anal. calcd for C20H20NO4Cl (373.84): C 64.26%; H 5.36%; N 3.75%; found: C 64.43%; H 5.05%; N 3.90%.
3.4.7.2 Method B
Recrystallization from ethyl acetate yielded trans-6f (0.649 g, 58%) as colorless crystals. Mp 130–132 °C. TLC (light petroleum/ethyl acetate 3:2) with a sample of trans-6f obtained by method A showed that the products are identical; mixed mp with a sample obtained by method A was not depressed.
3.4.8 Methyl (±)-trans- and cis-4-benzyl-3-(4-chlorophenyl)-5-oxomorpholine-2-carboxylate 6g (method A)
Chromatographic purification (cyclohexane/ethyl acetate 5:1) gave trans-and cis-6g.
Trans-6g (0.197 g, 55%) as colorless oil. Rf 0.63 (light petroleum/ethyl acetate 2:3). IR (film): 1750 (COOCH3); 1650 (CON) cm−1. 1H NMR (250 MHz, CDCl3): δ 3.39 (d, 1H, NCH, J = 14.6 Hz); 3.80 (s, 3H, COOCH3); 4.35 (d, 1H, H-3, J = 2.6 Hz); 4.70 (d, 1H, H-2, J = 2.6 Hz); 4.46 (d, 1H, H-6, J = 17.1 Hz); 4.88 (d, 1H, H-6, J = 17.1 Hz); 5.61 (d, 1H, NCH, J = 14.6 Hz); 6.82–7.48 (m, 9H, arom. H). MS (EI) m/z (%): 359 (20, M+); 300 (15); 228 (6); 198 (31); 197 (16); 196 (100), 167 (10); 162 (29); 125 (11); 104 (5); 91 (40). Anal. calcd for C19H18NO4Cl (359.84): C 63.42%; H 5.04%; N 3.89%; found: C 63.13%; H 5.29%; N 3.63%.
Cis-6g (0.115 g, 32%) as colorless crystals. Rf 0.41 (light petroleum/ethyl acetate 2:3). Mp 116–118 °C (ethyl acetate). IR (Nujol): 1760 (COOCH3); 1650 (CON) cm−1. 1H NMR (250 MHz, CDCl3): δ 3.36 (d, 1H, NCH, J = 14.7 Hz); 3.52 (s, 3H, COOCH3); 4.45 (d, 1H, H-6, J = 16.9 Hz); 4.52 (d, 1H, H-3, J = 3.5 Hz); 4.59 (d, 1H, H-2, J = 3.5 Hz); 4.69 (d, 1H, H-6, J = 16.9 Hz); 5.49 (d, 1H, NCH, J = 14.7 Hz); 7.04–7.43 (m, 9H, arom. H). Anal. calcd for C19H18NO4Cl (359.84): C 63.42%; H 5.04%; N 3.89%; found: C 63.23%; H 5.32%; N 3.60%.
3.5 (±)-Trans-6-(hydroxymethyl)-5-(4-methoxyphenyl)-4-phenethylmorpholin-3-one (7)
To a stirred suspension of LiCl (0.509 g, 12 mmol) and KBH4 (0.647 g, 12 mmol) in dry THF (5 mL), a solution of 6d (1.478 g, 4 mmol) in dry THF (20 mL) was added dropwise for 20 min. The reaction mixture was stirred at room temperature for 35 h. The solvent was removed under reduced pressure and the residue was poured in water (50 mL). The suspension was extracted with ethyl acetate (3 × 10 mL) and the organic phase was dried (Na2SO4). After removal of the solvent, the residue was purified by recrystallization from ethyl acetate–cyclohexane to give 7 (1.008 g, 74%) as white crystals. Rf 0.35 (light petroleum/ethyl acetate 2:3). Mp 100–102 °C. IR (Nujol): 3400 (OH); 1620 (CON) cm−1. 1H NMR (600 MHz, CDCl3): δ 2.17 (s, 1H, OH); 2.65 (ddd, 1H, CHAr, J = 5.2, 8.8, 12.9 Hz); 2.74 (ddd, 1H, NCH, J = 6.7, 8.8, 13.5 Hz); 2.87 (ddd, 1H, CHAr, J = 6.7, 9.2, 12.9 Hz); 3.37 (ddd, 1H, CH2OH, J = 5.5, 6.2, 12.4 Hz); 3.52 (ddd, 1H, CH2OH, J = 2.7, 6.2, 12.0 Hz); 3.63 (ddd, 1H, H-6, J = 2.8, 5.3, 9.4 Hz); 3.83 (s, 1H, OCH3); 4.01 (ddd, 1H, NCH, J = 5.1, 9.2, 13.5 Hz); 4.26 (d, 1H, H-5, J = 9.4 Hz); 4.31 (d, 1H, H-2, J = 16.3 Hz); 4.43 (d, 1H, H-2, J = 16.3 Hz); 6.89–6.92 (m, 2H, arom. H); 7.05–7.07 (m, 2H, arom. H); 7.09–7.12 (m, 2H, arom. H); 7.19–7.22 (m, 1H, arom. H); 7.25–7.28 (m, 2H, arom. H). MS (EI) m/z (%): 341 (13, M+); 250 (38); 237 (39); 220 (11); 188 (9); 162 (23); 135 (13); 121 (100); 105 (18). Anal. calcd for C20H23NO4 (341.16): C 70.36%; H 6.79%; N 4.10%; found: C 70.78%; H 6.62%; N 4.00%.
3.6 (±)-Trans-2-((3-(4-methoxyphenyl)-5-oxo-4-phenethylmorpholin-2-yl)methyl)isoindoline-1,3-dione (8)
To a stirred solution of alcohol 7 (1.023 g, 3 mmol), Ph3P (0.717 g, 3 mmol), and phthalimide (0.441 g, 3 mmol) in dry THF (11 mL) under argon, a solution of diethyl azodicarboxylate (1.8 mL, 40% in toluene, 4 mmol) was added dropwise for 10 min at room temperature. The reaction mixture was sonicated for 90 min. The solvent was evaporated and the residue was recrystallized from ethyl acetate to give 8 (0.846 g, 60%) as white crystals. Rf 0.48 (light petroleum/ethyl acetate 2:3). Mp 161–162 °C. IR (Nujol): 1770 (CON); 1700 (CON); 1650 (CON) cm−1. 1H NMR (250 MHz, CDCl3): δ 2.37–2.99 (m, 3H, NCHCH2); 3.46 (dd, 1H, CH2Phth, J = 3.7, 14.4 Hz); 3.76 (s, 3H, OCH3); 3.82 (dd, 1H, CH2Phth, J = 7.6, 14.4 Hz); 3.89–4.03 (m, 1H, NCH); 4.07–4.19 (m, 2H, H-2, H-3); 4.21 (d, 1H, H-6, J = 16.5 Hz); 4.41 (d, 1H, H-6, J = 16.5 Hz); 6.82–6.91 (m, 2H, arom. H); 7.04–7.29 (m, 7H, arom. H); 7.64–7.83 (m, 4H, arom. H). MS (EI) m/z (%): 470 (13, M+); 379 (33); 335 (32); 311 (26); 310 (100); 280 (45); 271 (12); 232 (58); 219 (11); 176 (17); 174 (42); 160 (97); 146 (24); 121 (45); 103 (31). Anal. calcd for C28H26N2O5 (470.52): C 71.47%; H 5.57%; N 5.95%; found: C 71.61%; H 5.50%; N 5.87%.
3.7 Trans-1-((3-(4-methoxyphenyl)-5-oxo-4-phenethylmorpholin-2-yl)methyl)piperidine-2,6-dione (9)
To a stirred solution of alcohol 7 (0.342 g, 1 mmol), Ph3P (0.239 g, 1 mmol) and glutarimide (0.119 g, 1.05 mmol) in dry THF (4 mL) under argon, a solution of diethyl azodicarboxylate (0.59 mL, 40% in toluene, 1.3 mmol) was added dropwise for 10 min at room temperature. The reaction mixture was sonicated for 190 min. The solvent was evaporated and the residue was purified by column chromatography (cyclohexane/2-propanol 8:1) and recrystallized from ethyl acetate to give 9 (0.127 g, 29%) as white crystals. Rf 0.13 (light petroleum/ethyl acetate 2:3). Mp 134–138 °C. IR (Nujol): 1730(CON); 1680(CON); 1650(CON) cm−1. 1H NMR (250 MHz, CDCl3): δ 1.78–1.83 (m, 2H, CH2); 2.54 (t, 4H, CH2, J = 6.5 Hz); 2.61–2.95 (m, 3H, NCHCH2); 3.49 (dd, 1H, NCH, J = 3.4, 12.8 Hz); 3.82 (s, 3H, OCH3); 3.86–4.20 (m, 5H, NCH, H-2, H-3, H-6); 4.40 (d, 1H, H-6, J = 16.5 Hz); 6.85–7.31 (m, 9H, arom. H). MS (CI) m/z (%): 465 (M++ 29, 18); 437 (M++ 1, 100); 345 (1); 324 (4); 258 (2); 216 (1). Anal. calcd for C25H28N2O5 (436.51): C 68.79%; H 6.47%; N 6.42%; found: C 68.93%; H 6.35%; N, 6.44.
3.8 (±)-Trans-6-(aminomethyl)-5-(4-methoxyphenyl)-4-phenethylmorpholin-3-one (10)
To a solution of methylamine in water (15.5 mL, 40%, 180 mmol), phthalimide derivative 8 (0.564 g, 1.2 mmol) was added and the mixture was magnetically stirred at room temperature for 3 h until homogeneous. The solution was extracted with dichloromethane (3 × 10 mL) and the combined organic solutions were dried (Na2SO4). The solvent was evaporated to give amine 10 (0.347 g, 85%) as an oily product, which was sufficiently pure for further experiments. Rf 0.10 (ethyl acetate/2-propanol 2:1). IR (film): 3390 (NH2); 3310 (NH2); 1640 (CON) cm−1. 1H NMR (250 MHz, CDCl3): δ 1.51 (br s, 2H, NH2); 2.54–2.97 (m, 5H, NCHCH2, CH2N); 3.50–3.60 (ddd, 1H, H-6, J = 4.8, 5.0, 9.1 Hz); 3.85 (s, 3H, CH3O); 3.97–4.06 (m, 1H, NCH); 4.10 (d, 1H, H-5, J = 9.1 Hz); 4.30 (d, 1H, H-2, J = 16.3 Hz); 4.44 (d, 1H, H-2, J = 16.3 Hz); 6.88–6.97 (m, 2H, arom. H); 7.01–7.12 (m, 2H, arom. H); 7.16 -7.33 (m, 5H, arom. H). MS (CI) m/z (%): 341 (M+ + 1, 13); 312 (100); 279 (3); 204 (2); 162 (1). Anal. calcd for C20H24N2O3 (340.42): C 70.56%; H 7.11%; N 8.23%; found: C 70.28%; H 7.37%; N 8.02%.
3.9 General procedure for the synthesis of acylated derivatives of (±)-trans-6-(aminomethyl)-5-(4-methoxyphenyl)-4-phenethylmorpholin-3-one 11 and 12
To a stirred solution of amine 10 (0.170 g, 0.5 mmol) and of the corresponding N-protected l-amino acid (0.5 mmol) in dry dichloromethane (2 mL) cooled to −10 °C, a solution of dicyclohexylcarbodiimide (DCC, 0.134 g, 0.65 mmol) in dry dichloromethane (2 mL) was added dropwise. The mixture was stirred for 2 h at −10 °C and then for 12 h at room temperature. The precipitated urea derivative was filtered out and discarded. The filtrate was evaporated under reduced pressure and the resulting oil was dissolved in dichloromethane (10 mL). The solution was successively washed with 10% HCl, water, 10% Na2CO3 and brine. The organic layer was dried (Na2SO4) and the solvent was removed in vacuo. The residue was purified by means of column chromatography.
3.9.1 (2S)-tert-Butyl-2-(((±)-trans-3-(4-methoxyphenyl)-5-oxo-4-phenethylmorpholin-2-ylmethyl)carbamoyl)pyrrolidin-1-carboxylate (11)
From amine 10 and Boc-l-proline. Column chromatography purification (cyclohexane/ethyl acetate 1:2) gave 11 (0.197 g, 75%) as an oily product. Rf 0.18 (cyclohexane/ethyl acetate 1:2). IR (CHCl3): 3420 (NH); 1670 (COOC(CH3)3); 1650 (CON); 1610 (CON) cm−1. 1H NMR (250 MHz, CDCl3): δ 1.46 (s, 2 × 9H, C(CH3)3, C(CH3)3*); 1.86 (m, 4H, CH2, CH2*); 2.05 (m, 4H, CH2N, CH2N*); 2.49–2.94 (m, 6H, NCHCH2, NCHCH2*); 3.26 (m, 4H, NCH2, NCH2*); 3.42 (m, 4H, NCH2, NCH2*); 3.69 (ddd, 2H, H-6, H-6*, J = 4.3, 5.5, 8.5 Hz); 3.81 (s, 6H, OCH3, OCH3*); 3.85–4.02 (m, 2H, NCH, NCH*); 4.21 (m, 2H, COCHN, COCHN*); 4.25 (d, 2H, H-2, H-2*, J = 16.3 Hz); 4.37 (d, 2H, H-2, H-2*, J = 16.3 Hz); 4.20–4.35 (m, 2H, H-5, H-5*); 6.32 (m, 2H, NHCO, NHCO*); 6.98–7.33 (m, 18H, arom. H). Anal. calcd for C30H39N3O6 (537.28): C 67.02%; H 7.31%; N 7.82%; found: C 67.32%; H 7.21%; N 7.52%.
3.9.2 (S)-N-(((±)-trans-3-(4-methoxyphenyl)-5-oxo-4-phenethylmorpholin-2-yl)methyl)-3-phenyl-2-(2.2.2-trifluoroacetamido)-propanamide (12)
From amine 10 and N-trifluoroacetyl-l-phenylalanine. Column chromatography purification (cyclohexane/2-propanol 8:1) and subsequent recrystallization from ethyl acetate gave 12 (0.149 g, 51%) as white crystals. Rf 0.52 (cyclohexane/ethyl acetate 1:2). Mp 83–86 °C. IR (Nujol): 1710 (CON); 1620 (CON) cm−1. 1H NMR (250 MHz, CDCl3): δ 2.53–3.06 (m, 10H, NCHCH2, NCHCH2*, 2 × CH2, 2 × CH2*); 3.08–3.24 (m, 4H, NCH2, NCH2*); 3.30 (ddd, 1H, H-6, J = 3.4, 7.2, 9.4 Hz); 3.54 (ddd, 1H, H-6*, J = 3.4, 7.4, 9.2 Hz); 3.82 (s, 3H, CH3O); 3.83 (s, 3H, CH3O*); 3.87–4.01 (m, 4H, H-5, H*-5, NCH, NCH*); 4.05 (d, 1H, H-2, J = 16.3 Hz); 4.17 (d, 1H, H-2*, J = 16.3 Hz); 4.24 (d, 1H, H-2, J = 16.3 Hz); 4.29 (d, 1H, H-2*, J = 16.3 Hz); 4.45–4.58 (m, 2H, COCHN, COCHN*); 6.87–6.95 (m, 4H, NHCO, NHCO*, NHCH, NHCH*); 6.96–7.38 (m, 18H, arom. H). MS (CI), m/z (%): 612 (M+ + 29, 14); 584 (M+ + 1, 100); 498 (6); 405 (6); 334 (8); 324 (19); 305 (60); 280 (10); 252 (48); 225 (58); 192 (13); 126 (36). Anal. calcd for C31H32F3N3O5 (583.23): C 63.80%; H 5.53%; N 7.20%; found: C 63.55%; H 5.80%; N 7.29%.
3.10 General procedure for the preparation of 2-((±)-trans-3-(4-methoxyphenyl)-5-oxo-4-phenethylmorpholin-2-yl)carbonyl)amino derivatives 13 and 14
To a magnetically stirred solution of acid trans-5d (0.365 g, 1 mmol), l-α-amino acid methyl ester hydrochloride (1.07 mmol) and triethylamine (0.15 mL, 1.07 mmol) in dry dichloromethane (5 mL), DCC (0.275 g, 1.3 mmol) was added portionwise at –15 °C. The mixture was stirred for 2 h at –10 °C and then 12 h at room temperature. The resulting precipitate of dicyclohexylurea was filtered and discarded. The filtrate was evaporated under reduced pressure and the residue was dissolved in ethyl acetate (30 mL). The solution was successively washed with 10% HCl, water, 10% Na2CO3 and brine. The organic layer was dried (Na2SO4) and the solvent was removed in vacuo. The residue was purified by means of column chromatography and/or recrystallization.
3.10.1 Methyl (2S)-3-(1H-indol-3-yl)-2-((((±)-trans-3-(4-methoxyphenyl)-5-oxo-4-phenethylmorpholin-2-yl)carbonyl)amino)propanoate (13)
From methyl ester of l-tryptophan hydrochloride. Recrystallization from ethyl acetate gave isomer 13a. Concentration of the mother liquor gave white crystals of 13b. Total yield of 13 is 62%.
13a (0.143 g, 26%) as white crystals. Rf 0.29 (light petroleum/ethyl acetate 2:3). Mp 166–168 °C. IR (Nujol): 3365 (NH); 1700 (COOCH3); 1660 (CON); 1640 (CON) cm−1. 1H NMR (250 MHz, CDCl3): δ 2.62–2.92 (m, 3H, NCHCH2); 3.30 (dd, 1H, CH2Ind, J = 5.3, 15.1 Hz); 3.36 (dd, 1H, CH2Ind, J = 5.5, 15.1 Hz); 3.71 (s, 3H, COOCH3); 3.78 (s, 3H, OCH3); 3.82–3.14 (m, 1H, NCH); 4.19 (d, 1H, H-6, J = 16.7 Hz); 4.20 (d, 1H, H-3, J = 4.3 Hz); 4.30 (d, 1H, H-6, J = 16.7 Hz); 4.91 (ddd, 1H, CH, J = 5.3, 5.5, 7.9 Hz); 4.96 (d, 1H, H-2, J = 4.3 Hz); 6.80–6.94 (m, 4H, arom. H); 7.04–7.25 (m, 9H, arom. H); 7.35 (d, 1H, arom. H, J = 8.0 Hz); 7.48 (d, 1H, NHCO, J = 7.9 Hz); 8.19 (br s, 1H, NH of indole ring). Anal. calcd for C32H33N3O6 (555.24): C 69.17%; H 5.99%; found: C 69.31%; H 6.30%.
13b (0.198 g, 36%) as white crystals. Rf 0.36 (light petroleum/ethyl acetate 2:3). Mp 108–110 °C. IR (Nujol): 3340 (NH); 1710 (COOCH3); 1660 (CON); 1630 (CON) cm−1. 1H NMR (250 MHz, CDCl3): δ 2.60–2.89 (m, 3H, NCHCH2); 3.31 (dd, 1H, CH2-Ind, J = 6.4, 14.8 Hz); 3.40 (dd, 1H, CH2-Ind, J = 5.5, 14.8 Hz); 3.72 (s, 3H, COOCH3); 3.78 (s, 3H, OCH3); 4.01 (d, 1H, H-6, J = 16.7 Hz); 4.02–4.13 (m, 1H, NCH); 4.14 (d, 1H, H-6, J = 16.7 Hz); 4.25 (d, 1H, H-3, J = 4.1 Hz); 4.88 (ddd, 1H, CH, J = 5.5, 6.4, 7.6 Hz); 4.93 (d, 1H, H-2, J = 4.1 Hz); 6.76–6.94 (m, 4H, arom. H); 7.01–7.29 (m, 9H, arom. H); 7.36 (d, 1H, arom. H, J = 7.4 Hz); 7.53 (d, 1H, NHCO, J = 7.6 Hz); 8.27 (br s, 1H, NH of indole ring). Calcd for C32H33N3O6 (555.24): C 69.17%; H 5.99%; N 7.56%; found: C 69.00%; H 6.17%; N 7.91%.
3.10.2 Methyl (2S)-1-((±)-trans-3-(4-methoxyphenyl)-5-oxo-4-phenethylmorpholine-2-carbonyl)pyrrolidine-2-carboxylate 14
From methyl ester of l-proline hydrochloride. Column chromatography purification (light petroleum/2-propanol 9:1) afforded 14 (0.310 g, 56%) as an uncrystallizable oil. Rf 0.33 and 0.42 (light petroleum/ethyl acetate 2:3, two-fold development). IR (Nujol): 1740 (COOCH3); 1640 (CON) cm−1. 1H NMR (250 MHz, CDCl3): δ 1.75–2.25 (m, 8H, 2 × CH2, 2 × CH2*, proline-H); 2.71–2.97 (m, 6H, NCHCH2; NCHCH2*); 3.24–3.44 (m, 2H, NCH2, proline-H); 3.54–3.67 (m, 2H, NCH2*, proline-H); 3.71 (s, 3H, COOCH3); 3.74 (s, 3H, COOCH3*); 3.80 (s, 3H, OCH3) 3.81 (s, 3H, OCH3*); 3.92–4.58 (m, 10H, 2 × H-6, 2 × H-6*, NCH, NCH*, H-3, H-3*, proline-NCH, prolin-NCH*); 5.00 (d, 1H, H-2, J = 3.8 Hz); 5.06 (d, 1H, H-2*, J = 4.1 Hz); 6.85–6.92 (m, 4H, arom. H, arom. H*); 7.10–7.24 (m, 14H, arom. H, arom. H*). Anal. calcd for C26H30N2O6 (466.21): C 66.94%; H 6.48%; N 6.00%; found: C 66.82%; H 6.57%; N 5.90%.
Acknowledgment
The authors are grateful to the FP7 project Beyond Everest. The financial support of the National Science Fund of Bulgaria at the Ministry of Education and Science (project TK-X-1706/07 and project UNION, DO-02–82/2008) is acknowledged.