

1 Introduction
The N-terminal domain of E. coli Ada protein contains a zinc ion tightly bound to four cysteinate residues [1]. The protein repairs methyl phosphotriesters in DNA by direct irreversible transfer of the methyl group to one of its cysteines, Cys69 (zinc-bonded cysteinate). The thioether thus formed, according to a multinuclear NMR study of the active site of the protein, remains coordinated to zinc [2]. Another example where the thioether resulting from alkyl group transfer apparently remains linked to the zinc centre is farnasyl transferase [3]. The bonding situation in the Zn2+(Cys)2 enzyme methionine sythease, which transfers a methyl group from methylcobalamine to homocysteinate [4], has not yet been revealed. Model studies concerning this question have been carried out, indicating that thioether complexation can be achieved in the case of chelating ligands [5–12], while the thioether is released in the case of methylation of Zn complexes containing monofunctional thiolates [13–25]. As part of our studies in mimicking the active site in thiolate alkylating enzymes [26–34], we have started a research program involving the design, synthesis, characterization, study of the structural properties and the reactivity of several pseudotetrahedral thiolate complexes as structural and functional models for enzymes with this feature, such as Ada repair protein.
The diamagnetic d10 zinc(II) center of the native enzyme offers little in terms of a spectroscopic probes. For this reason, the investigation of metal-substituted enzymes (e.g., Co, Ni, and Cu) has been actively pursued in order to provide information about both enzyme structure and activity [35–37]. Central to such research is the existence of a series of both structurally and spectroscopically well-characterized model complexes that thereby allow comparisons to be made with metal-substituted enzymes.
Toward this end, a monomeric cobalt(II)-bound thiolate complex (1) has been synthesized through changing the nature of the metal ion (Scheme 1), by the reaction of a methanolic solution of cobalt(II) nitrate hexahydrate with two equimolar amounts of the deprotonated form of tert-butyl N-(2-mercaptoethyl)carbamate (Boc-SH). The cobalt(II)-bound thiolate complex (1) was well-characterized in the solid state and in solution by using FT–IR, Raman, UV–visible, ESR, EI–mass spectroscopes, as well as X-ray structure determinations. The reaction of dithiolate cobalt(II) complex (1) with methyl iodide as a methylating agent was also studied; it typically leads to dithioether cobalt(II) complex [Co(Boc-SCH3)2]I2 (2) as a dication complex (Scheme 1). The result of this work may aid in our understanding of the mechanism of alkyl group transfer in metalloenzymes.
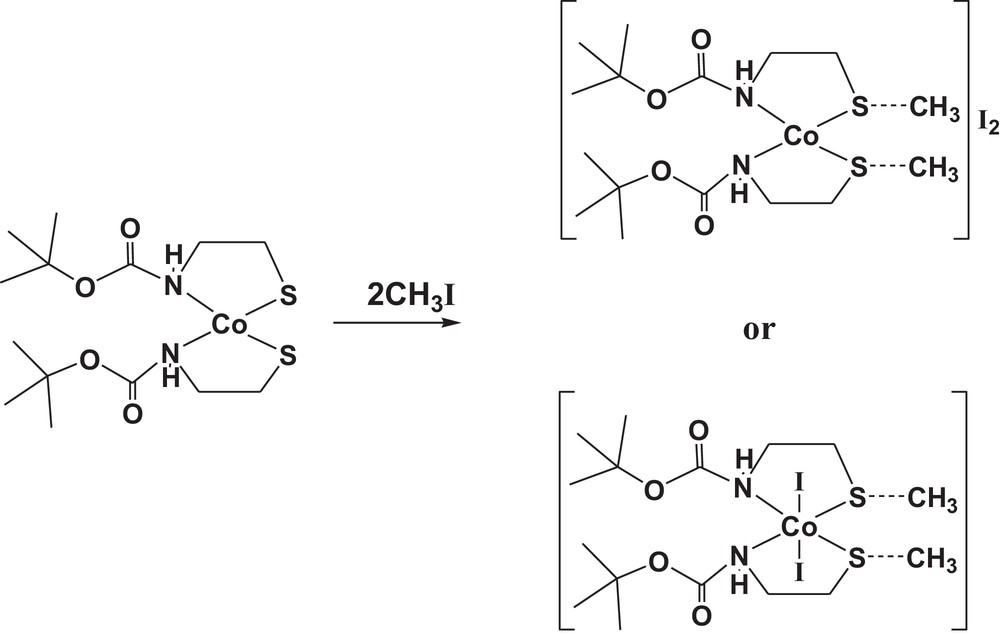
The methylation of cobalt(II) complex (1).
2 Experimental
2.1 Materials and general methods
The ligand tert-butyl N-(2-mercaptoethyl)carbamate (Boc-SH) was purchased from Aldrich and was used without further purification. The elemental analyses of the ligand and of its metal complexes were measured using Perkin-Elmer Series II CHNS/O Analyzer 2400. The IR spectra were recorded using Alpha-Atunated FT–IR Spectrophotometer, Bruker in the range of 400–4000 cm−1. Raman spectra were recorded with a Bruker FT-Raman equipped with a laser. All UV–visible measurements were made using a Perkin-Elmer Lambda 25 UV–Vis double-beam spectrophotometer. Thermal analyses of the complexes were recorded on a Netzsch STA 449F3 with a system interface device under nitrogen atmosphere. The temperature scale of the instrument was calibrated with high-purity calcium oxalate. The operational range of the instrument was from ambient temperature to 1200 °C. Accurately 5 mg of pure sample was subjected to dynamic TG scans at a heating rate of 10 °C·min−1. Molecular modeling calculations were done using computer program HyperChem version 8.0.
2.2 Syntheses
2.2.1 Synthesis of bis{tert-butyl N-(2-mercaptoethyl)carbamato}cobalt(II) (1)
The synthesis of the Co(II) complex was carried out under nitrogen atmosphere to exclude the possibility of oxidation of Co(II) to Co(III). It was prepared by stirring a solution of the ligand tert-butyl N-(2-mercaptoethyl)carbamate (Boc-SH) (5 mmol), containing NaOH (5 mmol) in MeOH (10 mL) with cobalt(II) nitrate hexahydrate (2.5 mmol) in MeOH (10 mL) for 1 h. The resulting solution was allowed to stand at room temperature and crystals of the obtained complexes were obtained after 2 days. They were filtered out, washed with diethylether, dried, and crystallized from MeOH:CH2Cl2 (1:3) to afford the dithiolate cobalt(II) complex [Co(Boc-S)2] (1), mp > 300 °C (dec.).
2.2.2 Synthesis of diiodo bis{S-methyl tert-butyl N-(2-mercaptoethyl)carbamato}-cobalt(II) (2)
Cobalt(II)-bound dithiolate complex (1) was reacted with an excess of methyl iodide in absolute methanol (10 mL). The reaction mixture was also stirred (for 2 h) under nitrogen atmosphere to exclude the possibility of oxidation of Co(II) to Co(III). The volatile compounds were removed in vacuo, and the residue was washed with diethylether, dried, and crystallized from MeOH:CH2Cl2 (1:3) to afford the dithiolate cobalt(II) complex [Co(Boc-SCH3)2]I2 (2), mp > 300 °C (dec.).
2.3 X-ray powder diffraction studies and zeta potential measurements
The X-ray diffraction study was carried out using a XRD-6000 (SHIMADZU) device. X-rays were detected using a fast counting detector based on silicon strip technology. The crystal structure and lattice parameter were analyzed by Bragg's law, 2 d sinθ = n λ, and the particle size was calculated using Debye–Scherrer's formula, t = 0.9 λ/B cosθ [38]. The particle charge measurements were conducted using a phase analysis light scattering with zeta potential analyzer (Zetapals–Brookhaven). The electrophoretic mobility of the dynamic light scattering samples in an applied electric field was measured in Malvern capillary plastic cells. Samples were prepared and measured at 25 °C in 10 mM in methanol pH = 4 (methanol was filtered through a 22-nm filter paper prior to use). Five zeta potential measurements were collected for each dispersion value, and the results were averaged. The zeta potential value was calculated directly from the Helmholtz–Smoluchowski equation using zeta potential analyzer software.
2.4 Electrochemical measurements
The electrochemical behaviors of the ligand Boc-SH and of its cobalt (II) complex were studied using cyclic voltammetry (CV) using an Auto Lab Potentiostat PGSTAT 302 (Eco Chemie, Utrecht, The Netherlands) driven by the General Purpose Electrochemical Systems data processing software (GPES, software version 4.9, Eco Chemie). The electrochemical cell used in this work comprises three electrodes: two platinum wires were used as the working and the counter electrode and Ag/AgCl as a reference electrode at room temperature. The potentials were scanned between –2.0 and +2.0 V at a scan rate of 100 mV·s−1. For recording cyclic voltammograms, 10–3 M of ligand or its complexes was dissolved in dimethyl sulfoxide (DMSO) in the presence of tetrabutylammonium perchlorate (TBAP) as a supporting electrolyte.
2.5 Kinetic measurements for the reaction of dithiolate cobalt(II) complex (1) with CH3I
All experiments were performed under pseudo-first-order conditions with a large excess of methyl iodide. In a typical experiment, 1 × 10−5 M of the cobalt(II)-bound thiolate complex (1) was dissolved in ClCl3, which was followed by the addition of 5–15 equivalents of MeI. All reactions were monitored by UV–visible spectroscopy at 300 K. The signal at 256 nm was used as a standard. The increase in the intensity (absorbance) of the synthesized [Co(Boc-SCH3)2]I2 (2) was recorded. The UV–visible measurements were done at intervals of times. The first-order rate constants were calculated from the slopes of the linear plots of ln(1 – At/A0) versus time. The second-order rate constants were calculated from the knowledge of the first-order rate constants at different concentrations.
3 Results and discussion
3.1 Characterization of [Co(Boc-S)2] (1) and [Co(Boc-SCH3)2]I2 (2)
The carbamate ligand Boc-SH behaves as a bidentate in the anionic form. The analytical results (Table 1) demonstrate that complex (1) has a 1:2 (metal/ligand) stoichiometry. The complex is a reddish brown solid, insoluble in H2O but soluble in common organic solvents, such as MeOH, EtOH, DMSO, and CHCl3. Its structure and properties were also characterized by FT–IR, Raman, UV–vis, and EI–mass spectroscopies, as well as by thermal analysis. Electrochemical measurements including cyclic voltammetry and electrical molar conductivity were also studied. Its molar conductivity is 4.3 Ω−1·cm2·mol−1, which is consistent with a non-electrolyte behavior [39]. Thermal analysis data have further confirmed the result of elemental analysis. The obtained thioether-containing iodod complex [Co(Boc-S)2]I2 (2) was also characterized by elemental analysis and IR spectroscopy. Complex (2) behaves as a 1:2 electrolyte with a molar conductance value of 84.0 Ω−1·cm2·mol−1, suggesting that iodide ions are not bound directly to the central cobalt(II) ion in solution.
Molecular formulae, elmental analyses, and conductivities of the obtained cobalt(II) complexes (1) and (2).
| Compound | Found (Calc.) % | ΛM (Ω–1·cm2·mol–1) | |||||
| % C | % H | % N | % S | % Co | |||
| [Co(Boc-S)2] C14H28N2O4S2Co | (1) | 40.23 (40.87) | 6.93 (6.86) | 7.01 (6.81) | 15.51 (15.58) | 14.43 (14.32) | 4.3 |
| [Co(Boc-S)2]I2 C16H34I2N2O4S2Co | (2) | 27.78 (27.64) | 4.85 (4.93) | 3.92 (4.03) | 9.43 (9.22) | 8.55 (8.48) | 84.0 |
3.2 IR and Raman spectra
The spectra of the ligand and of its coordination compound with Co(II) were carefully scrutinized to get information regarding the bonding sites of the ligand. The IR and Raman spectral data of the cobalt(II) complexes (1) and (2) (Table 2) display several significant spectral differences compared with the spectrum of the free carbamate. The absence of an infrared peak between 450 and 490 cm−1 (Fig. 1), which denotes the disulfide bond, indicates that Co(II) did not oxidize cysteine to form cystine. The disappearance of the thiol (SH) stretch at 2563 cm−1 in the complex and the obvious decrease of the thiol stretch in the complex indicate the deprotonation of the thiol group and subsequent binding of cobalt(II) to the ligand via the thiolate sulfur atom. The IR spectra of the complex also showed a characteristic band at 3166 cm−1, which is lower in comparison with free NH. Hence, it can be concluded that the nitrogen of the imino group is involved in metal coordination. No shift was observed in the asymmetric and symmetric stretching vibrations of carbonyl groups; this supports the non-involvement of carbonyl groups in metal coordination [40]. The Raman spectrum of complex (1) (Fig. 2) is well resolved and has a very high signal-to-noise ratio. The ν(Co–S) and ν(Co–N) symmetric stretching frequency for this complex is easily identified in the range of 360–320 and 410–465 cm−1. The peak due to ν(Co–O) at ∼ 500 cm−1 was not observed, indicating the non-coordination of oxygen in complexes. Thus from the IR spectra of the complexes, it is clear that the ligand binds to metal ions through two thiolate sulfur and two imino nitrogen atoms.
IR and Raman wave numbers (cm−1) and tentative assignement of the most important bands in the carbamate ligand and its complexes (1) and (3).
| Boc-SH | [Co(Boc-S)2] (1) | [Co(Boc-S)2]I2 (2) | Tentative assignments | ||
| IR | Raman | IR | Raman | IR | |
| 2563 | 2574 | − | − | − | νSH |
| 3341 | 3111 | 3166 | 3037 | 3165 | νNH |
| 2977 | 2933 | 2977 | 2928 | 2974 | νCH |
| 1687 | 1687 | 1691 | 1686 | 1693 | νCO |
| 1452 | 1451 | 1344 | 1451 | 1365 | δCH |
| 1509 | 1510 | 1511 | 1512 | 1509 | δNH |
| 1161 1249 | 1165 1250 | 1165 1297 | 1180 1239 | 1174 1288 | νC–O |
| − | − | 462 425 415 | 412 360 345 | 459 432 421 | νM–N/S |
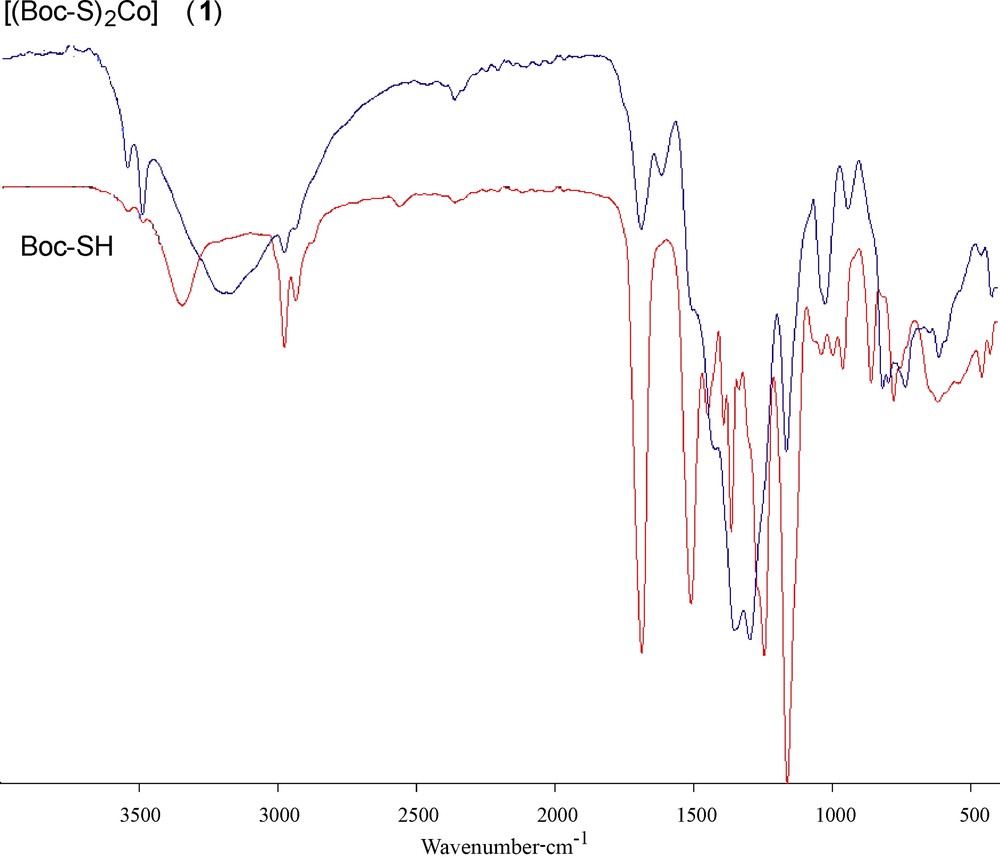
(Color online.) IR spectra of the ligand Boc-SH and of its dithiolate cobalt(II) complex (1).
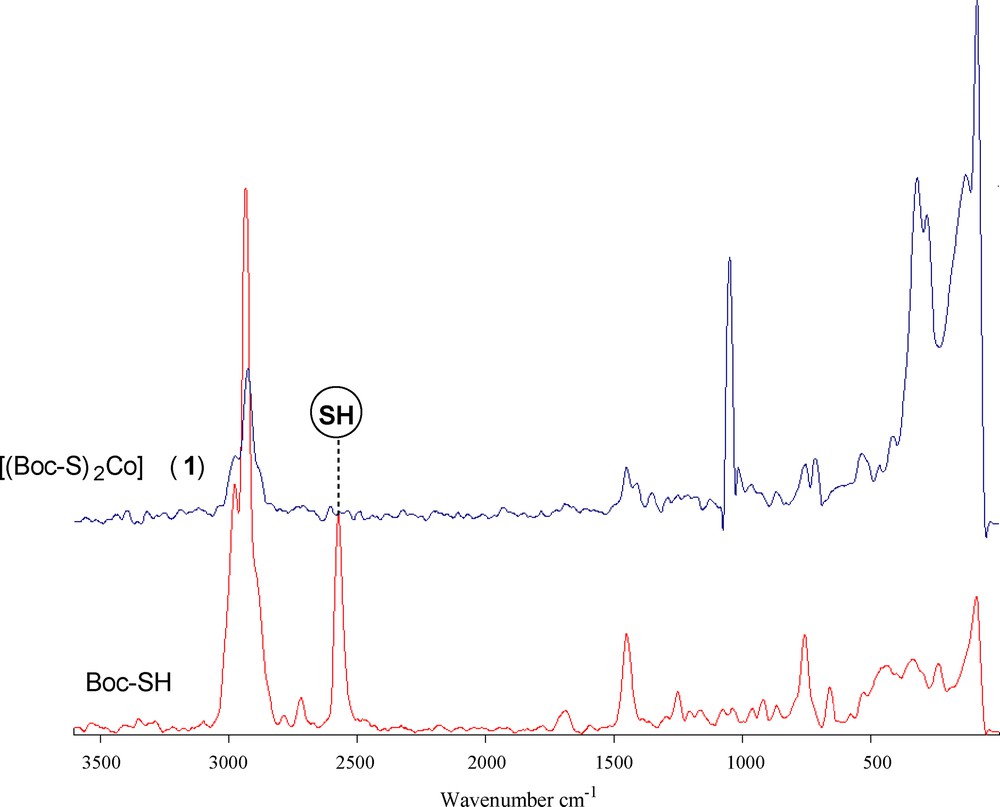
(Color online.) Raman spectra of the ligand Boc-SH and its dithiolate cobalt(II) complex (1).
3.3 Electronic absorption and ESR spectra
The electronic spectrum of the dithiolate cobalt(II) complex (1) in methanol and chloroform is shown in Figs. 3 and 4. The spectrum showed an intense peak in the UV region at 266 nm, due to the ligand field and charge transfer. An additional peak was observed at 422 nm (27,046 cm−1, ɛmax = 90 M−1·cm−1); it has been attributed to a charge-transfer transition. The peak at 560 nm (15,148 cm−1) (ɛmax = 41 M−1·cm−1), which can be assigned to (4A2(F) → 4T1(F)) and (4A2(F) → 4T1(P)) d–d transitions, suggests a tetrahedral structure around the cobalt (II) ion. The band positions, as well as the magnetic moment of 4.9 BM, are a further indication of the tetrahedral geometry. The ESR spectrum of the polycrystalline cobalt(II) complexes at room temperature does not show the characteristic ESR signal of the cobalt(II) complex species because its rapid spin lattice relaxation is short, which broadened the lines at the high temperature, i.e. room temperature. The suitable ESR spectrum of this complex must be recorded at liquid nitrogen's temperature, but this possibility is lacking in our laboratory. The presence of unpaired electrons from any source inside the molecule can be responsible for the appearance of these signals in the ESR spectra of these complexes.
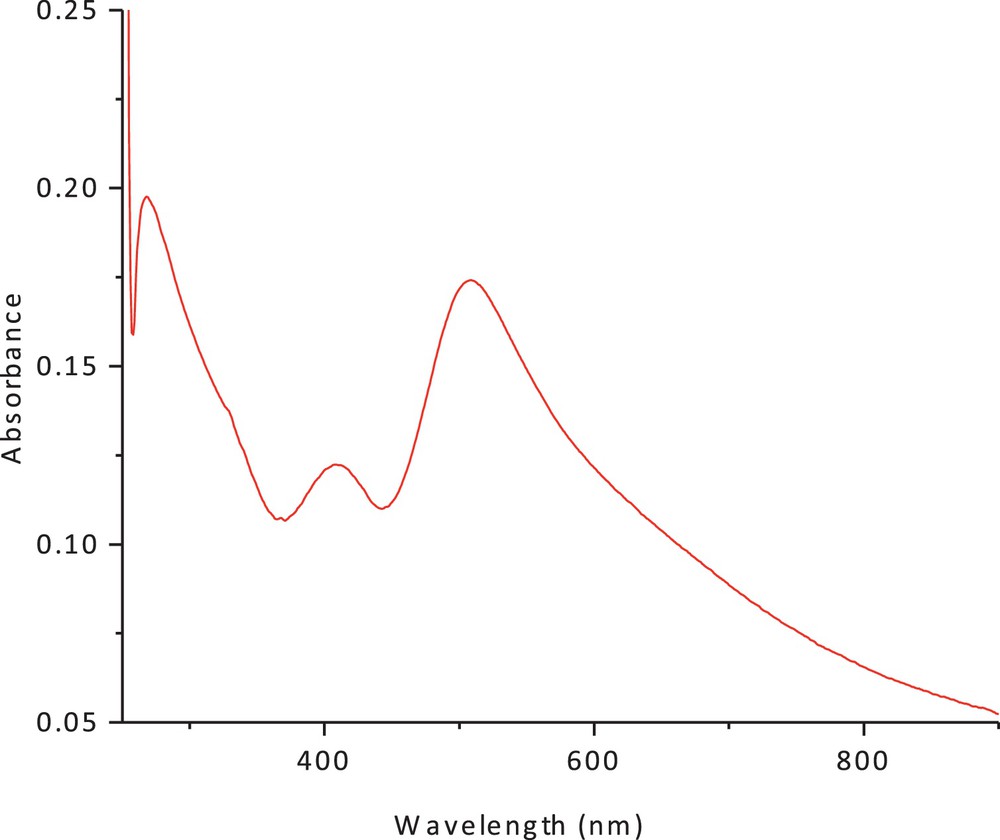
(Color online.) Electronic spectrum of cobalt(II) complex in methanol.
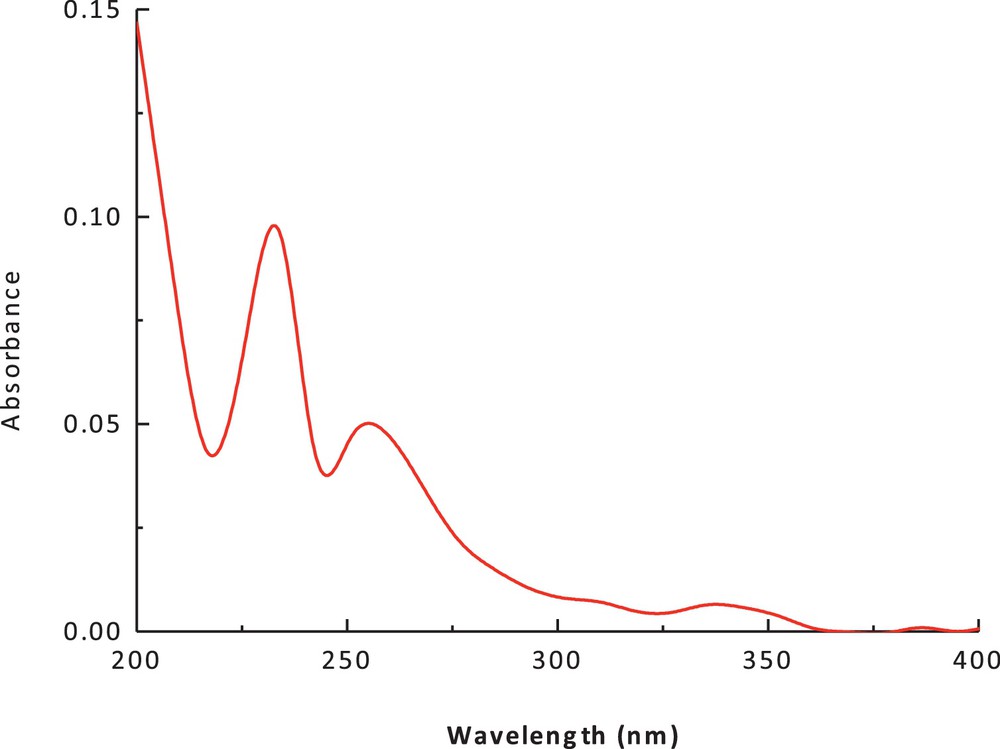
(Color online.) Electronic spectrum of cobalt(II) complex in chloroform.
3.4 Transmission electron microscopy
The TEM image (Fig. 5) of the final product after the incineration of complex (1) at its decomposition point shows that it is in a nano scale (50 nm). This is further substantiated by its XRD pattern using Scherrer's formula [41,42]: d = 0.9 λ/β cosθ, where λ is the wavelength, β is the full width at half maxima, and θ is the diffraction angle. The TEM image of the final residue shows its nanosized clusters, suggesting that cobalt(II) complex (1) may be used as a precursor for nano metal oxide [43].

TEM image of the decomposition product of cobalt(II) complex (1).
3.5 X-ray powder diffraction studies and zeta potential measurements
The XRD pattern of cobalt(II) complex (1) (Fig. 6) shows well-defined crystalline peaks indicating that the samples are crystalline in nature. The complex has specific d values that can be used for its characterization. XRD shows that Co(II) complexes have the average crystallite size of 47.86 nm, suggesting the complexes to be nanocrystalline.
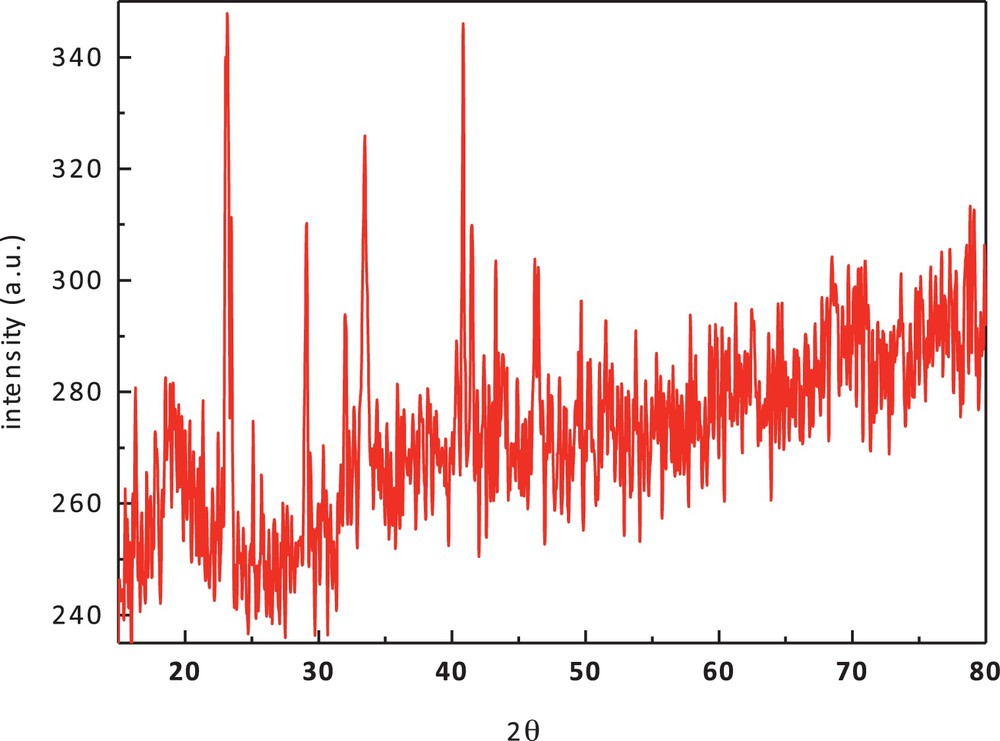
(Color online.) X-ray diffraction pattern of cobalt(II) complex (1).
The significance of zeta potential is that its value can be related to the stability of colloidal dispersions. The zeta potential indicates the degree of repulsion between adjacent and similarly charged particles in a dispersion. For molecules and particles that are small enough, a high zeta potential will confer stability, i.e., the solution or dispersion will resist aggregation [44,45]. From the result shown in Fig. 7, the zeta potential value of the cobalt(II) complex in methanol is 0.26 mV. This result is in accordance with XRD data.
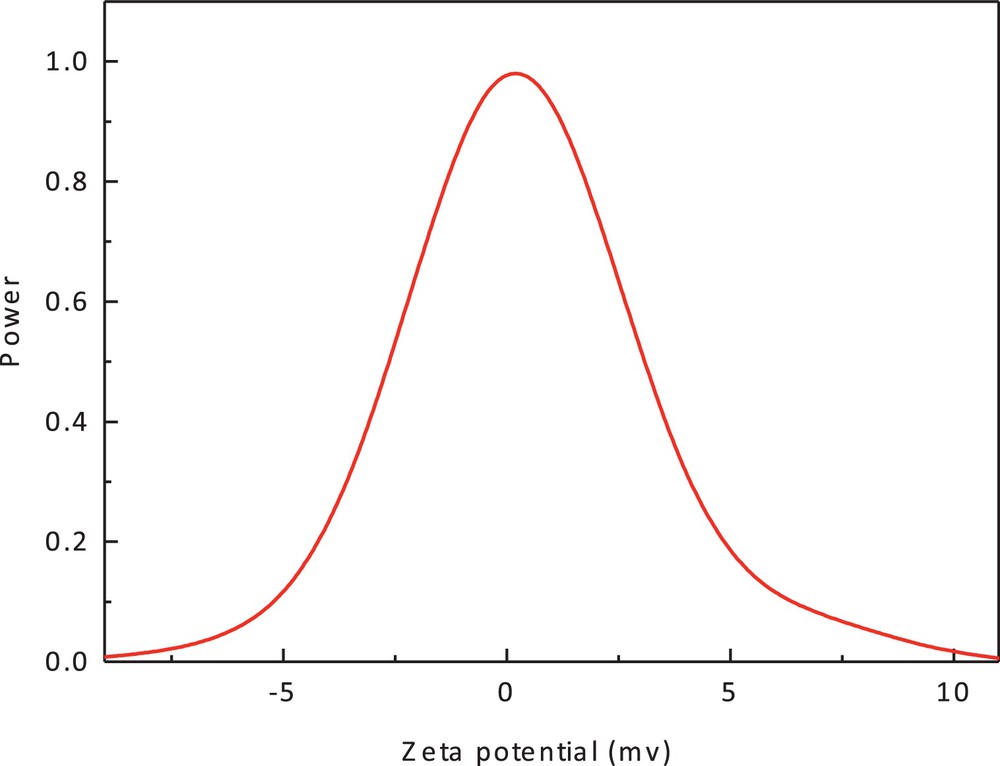
(Color online.) Zeta potential of cobalt(II) complex (1).
3.6 Thermal analysis
In order to get a deeper insight into the structure of the reported cobalt(II) complex 1, thermogravimetric (TGA) and differential thermal analysis (DTA) techniques have been used. The TGA and DTA curves for the decomposition of cobalt(II) complex 1 are shown in Fig. 8. The TGA curve shows several weight loss steps with endothermic peaks. These steps take place between 25 and 1200 °C and are accompanied by a total weight loss of 70.82% (Calc. 70.10), forming a mixture of CoO + 4C as final solid product (found 29.1%, Calc. 29.9%). From the DTA spectrum, the reaction is found to be exothermic in nature below 350 °C. Above this temperature, the reaction was found to be endothermic in nature. The kinetic parameters (ΔE#) and thermodynamic parameters ΔS#, ΔH#, and ΔG# (Table 3) also have been calculated for every step of the decomposition of the complex (1). The higher value of E for the second decomposition step as well as the third and fourth ones indicate that the ligands are strongly coordinated to cobalt(II) ion. The positive values of ΔS# in the second and the other following steps indicate that the activated complex has a less ordered structure than the reactants and that the reactants are slower than cobalt [46].
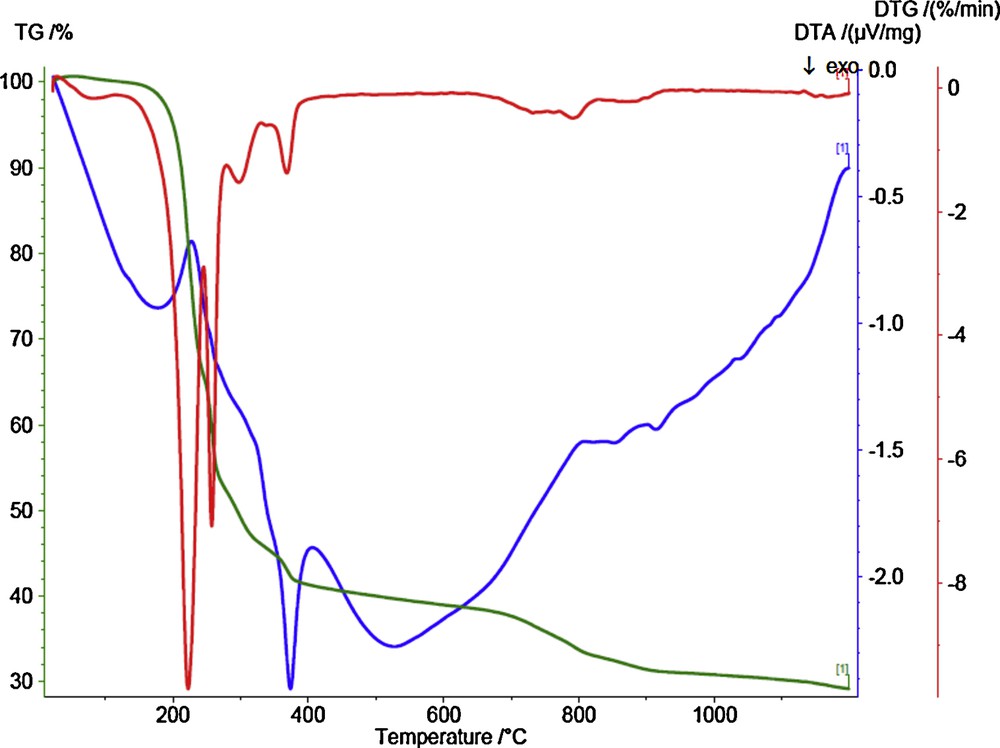
(Color online.) TGA–DTA curves of cobalt(II) complex (1).
Activation energies, frequency factor, coefficient of enthalpy, entropy, and free energy at a heating rate of 10 °C/min for cobalt(II) complex (1).
| 483 K | 509 K | 545 K | 606 K | 1027 K | |
| Activation energy (Ea)/kJ·mol–1 | 124.137 | 50.6923 | 106.756 | 8.68275 | 8.68275 |
| Frequency factor (A)/K | 4.4754 × 1011 | 790.7944 | 1.843618354.3 | 5630.002 | 5630.002 |
| Enthalpy/J·g–1 | 121.22 | 47.774 | 103.84 | 5.7645 | 70.977 |
| Entropy/J·g–1·K–1 | –0.0259 | –0.1939 | –0.0917 | –0.1790 | –0.2329 |
| Free energy (ΔG\)/kJ–1 | 133.73 | 146.45 | 153.82 | 144.32 | 310.09 |
3.7 Electrochemical studies of Boc-SH and its cobalt(II) complex (1)
The cyclic voltammetry of Boc-SH was examined in DMSO in the presence of 0.1 M of tetrabutylammonium perchlorate (TBAP) as the supporting electrolyte at a scan rate of 100 mV/s. The cyclic voltammogram of the ligand Boc-SH shows a well defined redox process (Fig. 9 and Table 4). One oxidation peak appeared at a +0.37 V (vs. Ag/AgCl) in the forward scan, due to the oxidation of Boc-SH, while in the reverse scan a reduction peak appeared at –0.27 V (vs. Ag/AgCl), due to the reduction of its oxidized form. This oxidation process occurs via the thiol group to disulfide according to the following mechanism [47,48]:
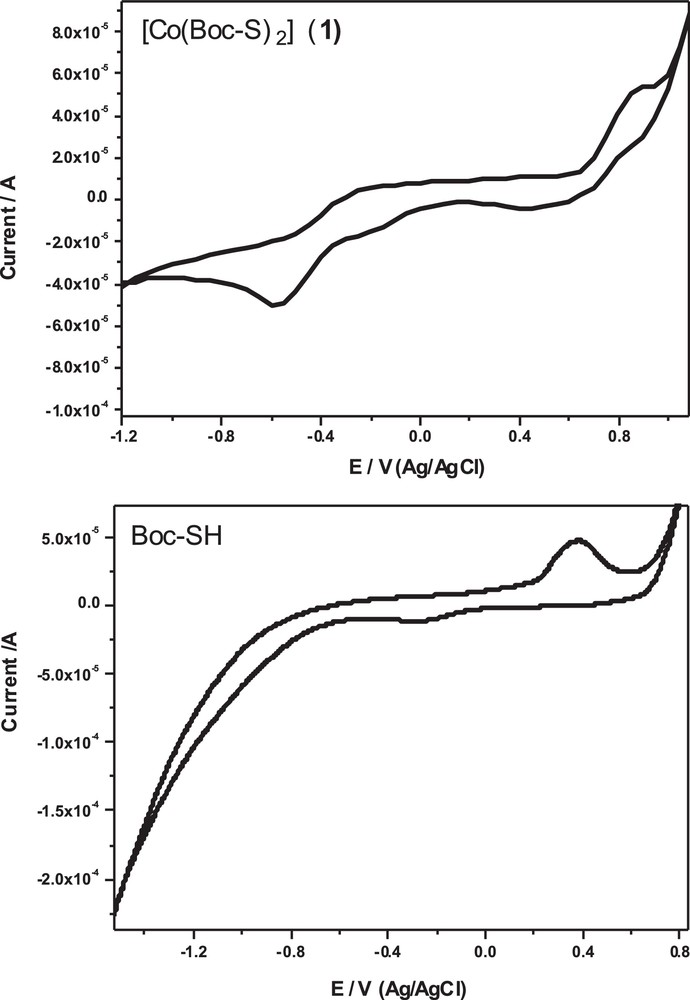
CV of the ligand Boc-SH and its dithiolate cobalt(II) complex (1) in DMSO with 0.1 M of tetrabutylammonium perchlorate (TBAP) as the supporting electrolyte at a scan rate of 100 mV/s.
Electrochemical data for the carbamate ligand Boc-SH and its cobalt(II) complex (1) in DMSO with 0.1 M of tetrabutylammonium perchlorate (TBAP) as the supporting electrolyte at a scan rate of 100 mV/s.
| Compound | Epa1 (mV) | Epc1 (mV) | ΔE1 (mV) | E1/2 (mV) | E1/2 (mV) | Epa2 (mV) | Epc2 (mV) | ΔE2 (mV) | ΔE2 (mV) | ipc2/ipa2 (A) |
| Boc-SH | +370 | –270 | +640 | +50 | 0.305 | – | – | – | – | – |
| [Co(Boc-S)2] (1) | –260 | –570 | +310 | –415 | 1.225 | +900 | +500 | +400 | +700 | 0.69 |
This equilibrium is forced to the right especially in the presence of transition metals because of the formation of insoluble metal sulfides. It is interesting to note that no free dithiocarbamic acid has been reported in the literature, whereas the free 4-methylpiperazine-1-carbodithioic acid has been found to be very stable. Transition metals are very reactive with proteins, particularly at the protein's sulfhydryl groups (–SH), which are free and unreduced in some of the bacterial enzymes, and are believed to bind protein molecules together by forming bridges between the groups.
The peak separation potential of ΔE = (Epa – Epc) between the anodic and cathodic peaks is 640 mV. This large peak separation potential suggests that the reaction is quasi-reversible, with a slow electron transfer. The cyclic voltammogram of the dithiolate cobalt(II) complex (1) (Fig. 9) showed two couple redox systems: two anodic peaks at –0.26 V and +0.9 V in the forward scan and another two cathodic peaks at –0.57 V and +0.5 V in the reverse scan. The peak separation potential ΔE = (Epa – Epc) between the first couple and second one is 310 and 400 mV, respectively. The higher peak separation potential between the two couples suggests that Co(III)/Co(II) and Co(II)/Co(I) couples involve a slow quasi-reversible one-electron transfer process.
3.8 Kinetic investigation for the methylation reactions
The reactions occurring at metal-bound sulfur atoms have been well demonstrated [49,50]. It seems that these reactions with methyl iodide take place without the rupture of the nickel–sulfur bond. The rates of reaction of some monomeric nickel(II)-bound dithiolate complexes with methyl iodide have been measured [50,51]. These reactions showed that the methylation occur only at sulfur atoms bonded to one nickel ion. The reaction of [Co(Boc-S)2] (1) with an excess of methyl iodide (100-fold excess) in chloroform at 25 °C proceeded according to equation (1):
(1) [Co(Boc-S)2] + 2CH3I → [Co(Boc-SCH3)2]I2
The reaction rates of this complex with methyl iodide have been determined spectrophotometrically as a function of time. The smoothness of the alkylation reactions provide strong evidence that the thiolates are methylated in the cobalt-bound state. The methylation reaction was followed by using the 256-nm absorption of the produced cobalt(II) complex [Co(Boc-SCH3)2]I2 (2). A simple demonstration of the kinetic nature of the methylation reaction can be seen in spectrophotometric scans (Fig. 10) taken over the visible spectral region as a function of time and for different concentrations, while reactions were in process. Thus, the pseudo-first-order rate constants were calculated for five different excess concentrations of methyl iodide (Table 5). The second-order rate constant, obtained using the formula kobs = k[iodo complex], is 10.59 × 10−2 M−1·s−1. The solubility of the complex in a non-coordinating solvent, such as chloroform, and the intense color of the complex have made it an ideal substance to use in studying the reactions of the coordinated sulfur atoms. The mechanism that was proposed for bis(8-mercaptoquinoline)nickel(II) formation [50] is quite similar to the present methylation reaction, as illustrated in Scheme 2.
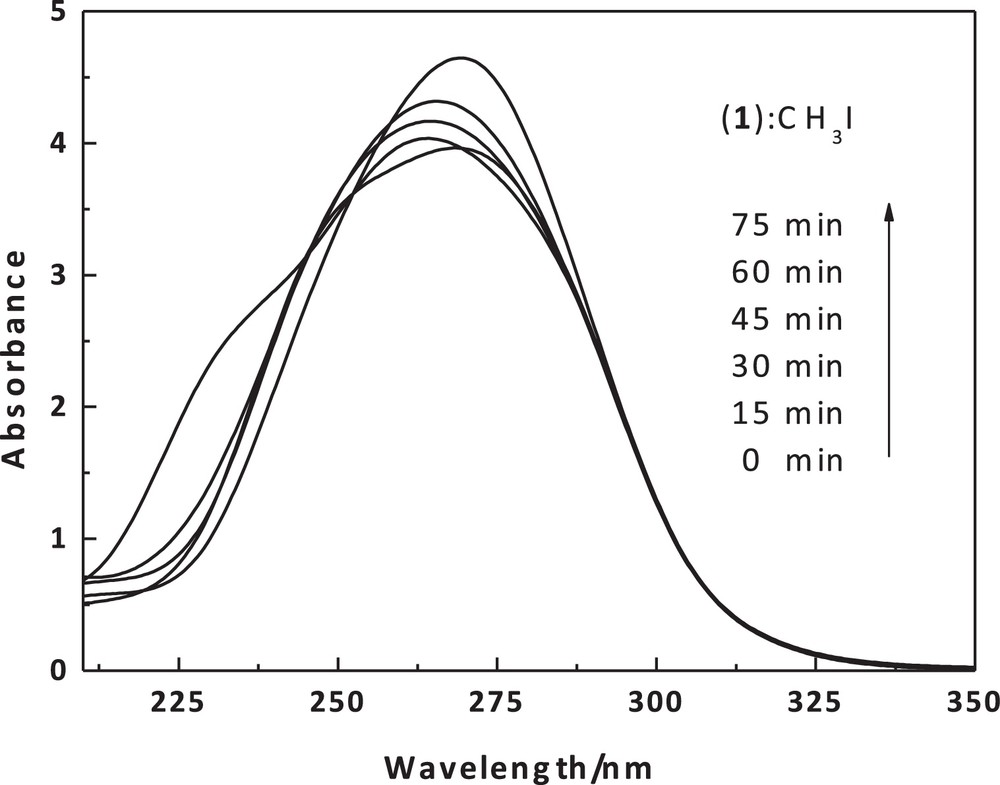
UV–Vis scans of the dithiolate cobalt(II) complex (1) (1 × 10−5) with MeI (0.09 M) in chloroform as a function of time and at 20 °C around 256 nm.
The observed pseudo-first-order rate constants (kobs, s−1) and second-order rate constant (k, M−1·s−1) for the methylation of cobalt(II) complex (1) (1 × 10−5 M) at different concentrations of methyl iodide at 20 °C.
| CH3I [M] | kobs, s−1 | k, M−1·s−1 |
| 0.01 | 0.00274 | |
| 0.03 | 0.00324 | |
| 0.05 | 0.00418 | 0.13245 |
| 0.07 | 0.00939 | |
| 0.09 | 0.0130 |
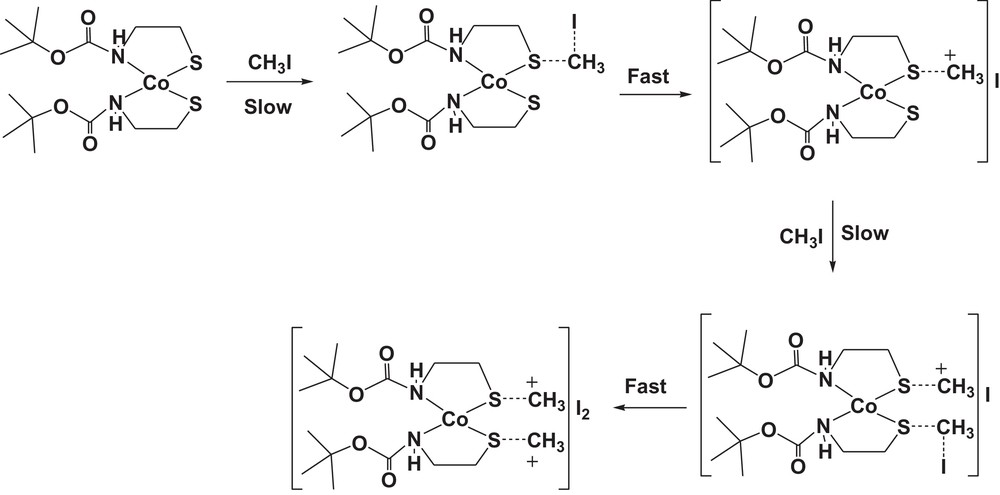
The proposed mechanism for the methylation of cobalt(II) complex (1) by using methyl iodide as methylating agent.
3.9 Computational studies
The geometries of cobalt(II) complexes (1) and (2) were proposed by energy minimization through molecular mechanics calculations. The geometrical optimization was done using force field MM+ and the Polak–Ribière algorithm with RMS gradient 0.01 cal·mol−1, followed by molecular dynamics; the same process was repeated four times. The total energy was noted after each calculation and then the structure with minimum energy was selected as a possible model of the complexes. The result obtained in this calculation was summarized in Table 6. The computational structures of complex (1) were represented in Fig. 11. Quantum chemical parameters such as the energy (EHOMO, ELUMO) of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), the energy gap ΔE = ELUMO – EHOMO between HOMO and LUMO are listed in Table 6. In order to obtain more details, the highest and the lowest values of the two spin degeneracy orbitals for the inhibitor molecules were mapped at the same level and are shown in Fig. 12. Table 5 shows that the highest EHOMO was recorded for the tetrahedral dithiolate cobalt(II) complex [Co(Boc-SCH3)2]I2 (2), whereas the lowest ΔE = ELUMO – EHOMO was observed for the octahedral cobalt(II) complex [Co(Boc-SCH3)2I2]. HOMO–LUMO energy separation (gap) can aid to clarify the chemical reactivity and the kinetic stability of the molecule. The molecular modeling of the obtained dimethylated dithiolate cobalt(II) complex [Co(Boc-SCH3)2]I2 (2) showed that some steric hindrance would be encountered if methyl iodide were attacking in the molecular plane [52–54]. This behavior has been interpreted in terms of steric interaction of the methyl group with the sulfur atom when the latter moiety is in the plane of the molecule.
Energy parameters obtained by using geometry optimization, semi-empirical, PM3.
| [Co(Boc-S)2] (1) | [Co(Boc-S)2I2] | [Co(Boc-S)2]I2 (2) | |
| EHOMO (eV) | –7.54 | –6.608 | –16.135 |
| ELUMO (eV) | –0.791 | –1.20 | –8.67 |
| ΔE (eV) | 6.749 | 5.408 | 7.465 |
| μ (D) | 4.79 | 6.058 | 2.356 |
| Total energy (kcal/mol) | 97399 | –111401 | –110642 |
| Total energy (au) | –155.2 | –177.5 | –176.3 |
| Binding energya kcal/mol) | –3029.0 | –3131.1 | –2974 |
| Isolated atomic energy (kcal/mol) | –94370.0 | –108270.7 | –107667.7 |
| Electronic energy (kcal/mol) | –555689.7 | –671311 | –677741 |
| Core–core interaction (kcal/mol) | 458290.6 | 559909 | 567098 |
| Heat of formation (kcal/mol) | –254.7 | –201 | –149 |
a Binding energy is the energy required to release an electron from its atomic or molecular orbital.
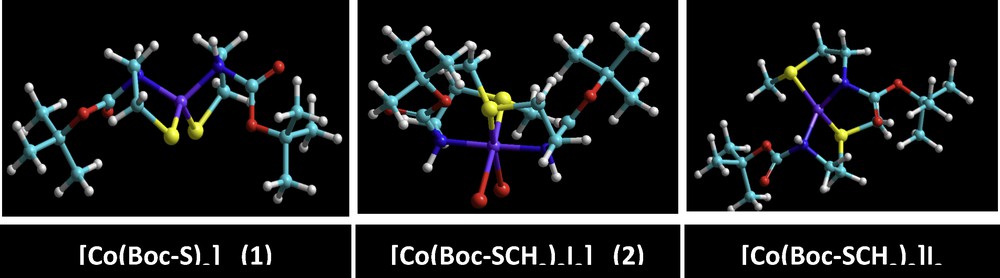
(Color online.) Ball and stick models for the optimized geometries of [Co(Boc-S)2] (1) and its methylated products [Co(Boc-S)2]I2 (2) or [Co(Boc-SCH3)2I2] obtained from the semi-empirical ZINDO/1 method.
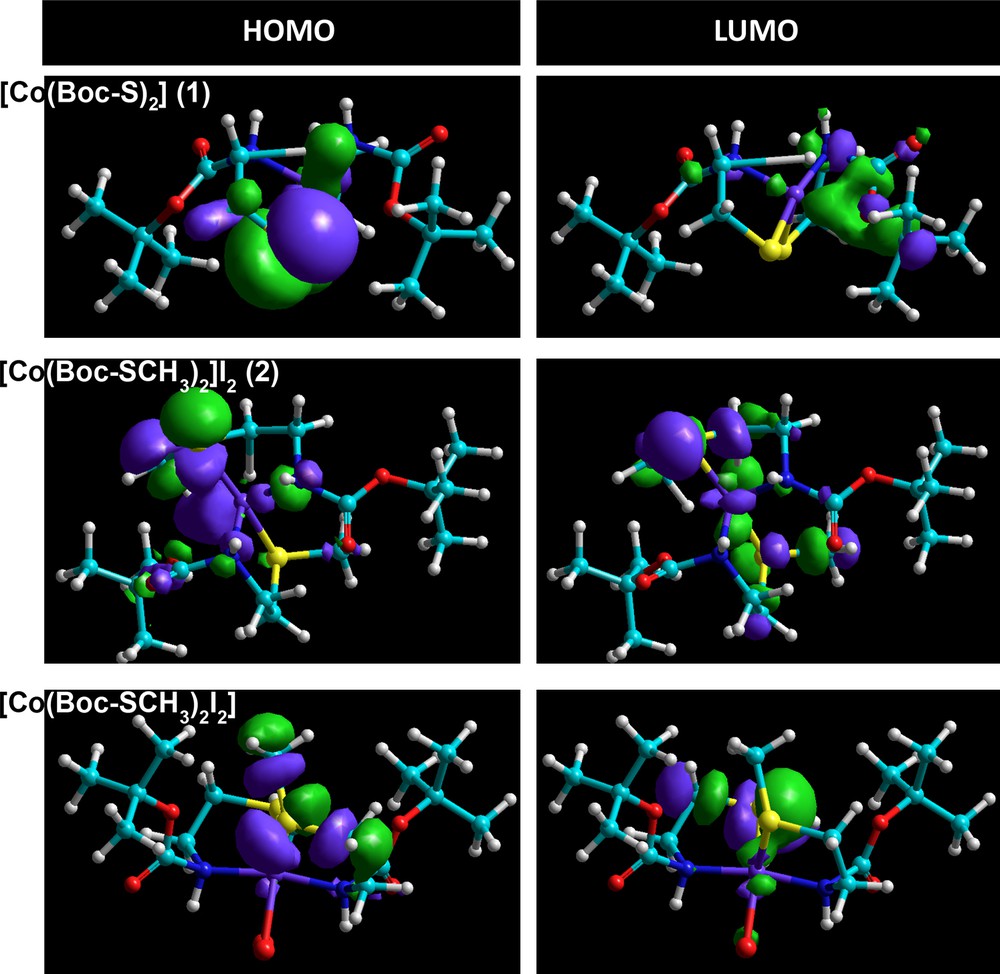
(Color online.) HOMO and LUMO orbitals with the energy of [Co(Boc-S)2] (1) and its methylated products [Co(Boc-S)2]I2 (2) or [Co(Boc-SCH3)2I2] obtained from semi-empirical ZINDO/1 method.
4 Conclusion
Two new thiolate-containing carbamate mononuclear cobalt(II) complexes namely, [Co(Boc-S)2] (1) and [Co(Boc-S)2]I2 (2) (Boc-SH = tert-butyl N-(2-mercaptoethyl)carbamate) were synthesized and characterized. The spectral data showed that the carbamate (Boc-SH) acts as a mono-anionic bidentate ligand coordinated to the cobalt(II) ion through two imine nitrogen and two deprotonated thiolate sulfur donor atoms in a distorted tetrahedral geometry. The TEM image of the oxide residue, resulting from the decomposition of complex (1) shows its nano size clusters, suggesting that the complex (1) may be used as a precursor for nano-oxides. X-ray powder diffraction patterns show an isomorphism among the complex. Its methylation with methyl iodide appears to occur intramolecularly at the cobalt-bound thiolate, forming the tetracoordinated dimethyl dithioether cobalt (II) complex [Co(Boc-S)2]I2, with a clean second-order rate constant of 13.24 × 10−2 M−1·s−1. The homogeneous model catalyst prepared and applied in this study is going to be modified for immobilization on solid support to obtain heterogeneous derivatives, which will be easier to separate from the reaction mixtures and to recycle.
Acknowledgements
This work was financially supported by Taif University, Saudi Arabia, Project No. 1/1433/1817.