

1 Introduction
Free radicals are ubiquitous compounds in nature and can be produced from external sources, such as exposure to X-rays, ozone, cigarette smoking, air pollution, and industrial chemicals [1]. Free radicals are the reactive oxygen species (ROS) that include alkoxyl (RO•), hydroxyl (HO•), peroxyl (ROO•) and superoxide (O2−), they are capable of attacking proteins in tissues, lipids in cell membranes, carbohydrates, amino acids and DNA, resulting in oxidative damage [2,3]. Antioxidants can prevent the damage caused by free radicals [4,5]. Numerous reports suggest that a fruit-and-vegetable-rich diet reduces cancer risk in humans, and therefore using dietary antioxidants has become a fascinating strategy for cancer chemoprevention [6]. Resveratrol (3,5,4′-trihydroxy-trans-stilbene) (A1 in Fig. 1), a naturally occurring phytoalexin derived from more than 72 plant species, including a wide variety of fruits and vegetables, such as grapes, berries, and peanuts, is one such dietary antioxidant and cancer chemopreventive agent [7,8]. Resveratrol has been reported to be a potent antioxidant against the peroxidation of low-density lipoproteins (LDL) [9], and liposomes [10], a powerful inhibitor of lipoxygenase [11], and to be able to protect the rat's heart from ischemia reperfusion injury [12]. The structural simplicity of this molecule has prompted interest in designing novel analogs with improved antioxidant potency [13–15]. Theoretical studies of the antioxidant activity of trans-resveratrol have also been undertaken by many authors, like Cao et al. [16]. Their results indicate that this compound is a strong antioxidant because its radicals have a semiquinone structure in which the unpaired electron is mainly localized on the O atoms in para- and ortho-positions. On the other hand, it has been proved that all trans-resveratrol oligomers [17], trans-resveratrol-3-O-glucuronide and glucosides exhibit remarkably higher antioxidant activity than trans-resveratrol and that trans stereoisomers of epsilon-viniferin and piceid are more efficient antioxidants than their cis stereoisomers. From the results obtained, the dimer of trans-4,4′-DHS and trans-resveratrol-3-O-glucuronide are predicted to be potential antioxidants in living organisms. Queiroz et al. [18] studied the antioxidant activity of resveratrol and of some analogs; the results revealed that the antioxidant pharmacophore of resveratrol is 4-hydroxystilbene. Very recently, in the same field, we demonstrated that the antioxidant potency of molecules bearing para-di-hydroxyl groups depends on the para-quinone structure [19]. The calculations revealed that 2-hydroxystilbene is a novel pharmacophore in the stelbenic skeleton [19]. The elongation of the conjugated links is an important strategy to improve the antioxidant activity of resveratrol analogs: for that, 15 compounds were synthesized by the insertion of additional double bonds between two aromatic rings (diene and triene compounds of series B and C in Fig. 1); the electron donors (ED) groups (methoxy and hydroxyl) were introduced in ortho or para position to 4-OH or 4′-OH [20].

(Color online.) Molecular structures of trans-resveratrol and of its analogs.
On the other hand, we studied the antioxidant activity of resveratrol and of some phenanthrenes analogues in vacuo, in water and in benzene [21]. Our results revealed that the 4′-OH group is the most favoured site for homolytic and heterolytic O–H breaking in trans-resveratrol, in all the environments studied. On the other hand, the 3-OH site is the most preferred in all phenanthrenes studied, with the exception of the B molecule, the phenanthrene analogue of RSV. The introduction of the single bond between the two rings A and B in resveratrol creates a novel pharmacophore on the 4-OH (3-OH in RSV) site, suggesting its importance for the construction of a phenanthrene ring [21]. However, Caruso et al. [22] proved that SPLET is the dominant mechanism in the reaction of free radicals with various antioxidants and that the para 4′-OH group is more acidic than the other hydroxyl groups; this observation is consistent with its free radical-scavenging ability. In the paper by Caruso et al. [22], the influence of the solvent was not taken into consideration. The mechanism by which the naturally occurring polyphenolic compound resveratrol and its metabolite piceatannol scavenge free radicals was studied using experimental and density functional theory methods [23]. Piceatannol was found to be more efficient than resveratrol because:
- • piceatannol can share its 3′-OH hydrogen atom with its adjacent neighbor, O-4′, so the abstraction and transfer of the 4′-H atom to the free radical becomes easier;
- • the resulting piceatannol semiquinone radical is more stable.
In this work, and as part of our ongoing theoretical research project on antioxidants molecules, we studied the antioxidant activity of the above compounds by means of a DFT method; this investigation includes the BDE, AIP, PDE, PA, and ETE. Therefore, BDE, AIP and PA may be used to determine the most thermodynamically preferred reaction pathway [24,25]. On the other hand, the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) distributions, and the spin density in free radicals were also studied. The effects of both the elongation of the conjugated links and the introduction of the hydroxyl and methoxy groups as EDG (electro-donors groups) were studied. The contributions of the ortho- and para-uinone structures on the antioxidant activity were also investigated.
2 Computational methods
All calculations were performed using GAUSSIAN 09 program package [26]. Firstly, the original structures of molecules studied and their corresponding free radicals, cations and anions were optimized by AM1 [27], then a full optimization to these structures was performed at 6-31G** basis set by B3LYP method (Fig. 2). For the species having several conformers, all of them were investigated. The conformer with the lowest electronic energy was used in this work.
(Color online.) B3LYP/6-31G** fully optimized geometries of trans-resveratrol (A1) and of its analogs.
The two main working mechanisms of phenols (denoted as ArOH) antioxidant action [28,29] are:
(1) the hydrogen atom transfer (HAT) pathway;
| ArOH → ArO• + H• | (1) |
(2) the single-electron transfer followed by proton transfer (SET–PT);
| ArOH → ArOH+• + e− | (2.1) |
| ArOH+• → ArO• + H+ | (2.2) |
A third mechanism has been discovered and confirmed on the basis of kinetics experiments:(3) the sequential proton loss electron transfer (SPLET) [30,31],
| ArOH → ArO− + H+ | (3.1) |
| ArO− → ArO• + e− | (3.2) |
The net results of all three mechanisms are the same, i.e. the formation of phenoxy radical ArO•. Kinetics measurements showed that the reaction enthalpies related to individual steps of the above-described mechanisms are usually denoted as follows:
- • BDE: O–H bond dissociation enthalpy related to eq. (1);
- • AIP: ionization potential, enthalpy of electron transfer from the antioxidant (eq. (2.1));
- • PDE: proton dissociation enthalpy (eq. (2.2));
- • PA: proton affinity of phenoxide anion (eq. (3.1));
- • ETE: electron transfer enthalpy (eq. (3.2)).
In this work, the following antioxidant descriptors were considered:
- • ;
- • ;
- • ;
- • ;
- • .
The frequency calculation revealed that the B3LYP-optimized structures were real minima (no imaginary frequency). The absence of spin contamination was noted with the S2 value, which is about 0.750 in all cases. The enthalpy of the parent molecule is then corrected for the translational, rotational, and vibrational terms to obtain the thermal corrections including the zero-point energy (ZPE). The gas phase enthalpy of hydrogen atom at 298.15 K, including the translation and PV corrections (–0.49764 hartree), was used. The total enthalpies of the chemical systems studied were computed from the following formula:
| Htot = E0 + ZPE + ΔHtrans + ΔHrot + ΔHvib + RT | (1) |
The calculated gas phase total enthalpies of proton and electron are 6.197 kJ/mol and 3.145 kJ/mol, respectively [32]. AIP and ETE calculations require the value of the electron-solvation enthalpy, ΔsolvH(e−); therefore, we used a published value: –105 (in water). For PA and PDE calculations, we need the proton solvation enthalpy, ΔsolvH(H+) in the studied solvents, we utilized the value –1090 (in water) [24,25].
3 Results and discussion
From the optimized values of the dihedral angles α (C6′-C1′-C8-C7) and θ (C8-C7-C1-C2) at the B3LYP/6-31G** level, it is evident that all resveratrol analogs studied in this work are strictly planar. It must be pointed out that the strictly planar geometry of these compounds determines the energetically favourable delocalization of the π-electrons, and the stacking interaction of the planarity of the stilbene system may also facilitate electron transfer.
3.1 HOMO
The HOMO energy, characterizing the ability to donate electrons, is one of the most appropriate parameters representing the free radical-scavenging efficiency of phenolic compounds, because the process to inhibit auto-oxidation includes, not only the abstraction of the H atom, but also electron transfer [33]. The molecule is considered a better electron donor when it exhibits a higher ɛHOMO. In contrast, ɛLUMO presents the aptitude of a molecule to receive electrons [34]. The HOMO disposition of a phenolic compound can indicate qualitatively its active site of scavenging free radicals. The wide range of delocalization of this orbital indicates that the compound investigated has many active redox sites, specially the hydroxyl groups of 4 and 4′-positions, which are easily attacked by either the electrophilic or nucleophilic agents, such as radicals, metal ions and O2−. As shown in Fig. 3, the HOMO orbitals are delocalized over the whole molecules studied, particularly in 4′-O-position. There is also a significant HOMO density contribution from all the double bonds between A- and B-rings of resveratrol and its analogs. These double bonds contribute to the antioxidant activity, as they ensure π-electron delocalization between the A- and B-rings, which contributes to the stabilization of RO• after H abstraction.

(Color online.) The HOMO orbital distribution in trans-resveratrol (A1) and of its analogs.
We can also see from Fig. 3 that the HOMO is distributed on all hydroxyl sites, except for 3- and 5-OH sites in A1, B1 and C1, whereas the HOMO density is higher in 4- and 4′-OH than the other hydroxyl positions.
Based on the results of our calculations, the HOMO values vary in the opposite direction to AIP and BDE and followed the sequence: triene > diene > ene. For example, A1 showed a HOMO value of –5.21 eV, while B1 and C1 showed HOMO values of –5 and –4.88 eV, respectively. The elongation of the double bond link of resveratrol and its hydroxylated derivatives increased the HOMO values. In the diene group, the HOMO value of B2 was –5.02 eV, the addition of hydroxyl groups increased the HOMO values as shown in B1, B3, and B4 molecules (–5, –4.83 and –4.97 eV), respectively. The same result was found with the introduction of methoxyl groups in B5 (–4.80 eV) and B6 (–4.91 eV). The most nucleophilic compound was compound C3 with HOMO and AIP values of –4.72 eV and 137.2 kcal/mol, respectively.
3.2 HAT mechanism
3.2.1 Bond dissociation energy (BDE)
Stilbenes can protect cell damage by either donating an H atom or an electron. Molecules with low BDE values are expected to exhibit strong antioxidant activity. In particular, a low BDE value indicates that the antioxidant is able to donate a hydrogen atom during a reaction with free radicals. The calculated BDE values are often attributed to π-electron delocalization, leading to the stabilization of the radicals obtained after H abstraction. Therefore, molecules with the lowest BDE are more active.
It has been stressed that the gas phase and solution BDE values do not always follow the same trend. Some compounds can easily transfer the H atom or electron into the gas phase, but become inactive in the presence of solvents. This was rather expected, since the solvents employed in the calculations tend to form intermolecular hydrogen bonds with solutes affecting hydrogen donation [35]. Generally, the BDE values computed in the condensed phase are lower than those obtained in the gas phase.
As shown in Table 1, the BDE values of the studied compounds follow the sequence: ene > diene > triene, in both water and gas phase, except for A4 and B4, for which the lowest BDE values have been recorded. On the grounds of computed BDE values for the parent compounds, the most efficient radical scavengers are predicted to be those bearing a catechol moiety in all studied media. The ortho-hydroxy group is reported to be able to promote the H atom donating ability by the intramolecular hydroxyl bond, which can decrease the BDE of the parent molecule and stabilize the radical formed after the H atom has been abstracted.
The computed values of BDE (kcal/mol) obtained at the B3LYP/6-31G** level of theory.
| Compounds | BDE (vacuum) | BDE (water medium) | k2a |
| A1 | 77.7 | 76.9 | 15.7 |
| A2 | 78.2 | 76.7 | 9.7 |
| A3 | 77.4 | 75.6 | 109 |
| A4 | 68.8 | 69.1 | 1.47 × 103 |
| A5 | 76.6 | 75.5 | 33 |
| A6 | 77.4 | Not favored | 101 |
| B1 | 76.3 | 75.3 | 90.8 ± 3.3 |
| B2 | 76.3 | 75.1 | 68.0 ± 4.2 |
| B3 | 75.6 | 74.1 | 274 ± 5 |
| B4 | 67.6 | 68.4 | (3.36 ± 0.12) × 103 |
| B5 | 75.5 | 74.1 | 171 ± 7 |
| B6 | 75.5 | 73.8 | 282 ± 4 |
| C1 | 75.3 | 74.0 | 142 ± 1 |
| C2 | 75.3 | 73.9 | Not favored |
| C3 | 74.8 | 73.1 | 441 ± 13 |
a Tang et al., 2011 [20].
The BDE value for A2 is 78.2, the introduction of hydroxyl groups decreases this value, such as in A1 (77.7 kcal/mol), A3 (77.4 kcal/mol), and A4 (68.8 kcal/mol). The same change was observed with the addition of a methoxy group on the 4′-para-position of A5 (76.6 kcal/mol), whereas this value increases in A6 due to an H-bond formation between the hydrogen in the 4-OH group and the oxygen atom of the methoxy group on C3. As a consequence, the BDE on this site is higher, because H-removal also implies the breaking of the H-bond.
Intramolecular hydrogen bond formation between the hydroxyl and the O-methoxy compounds in compound A6 and B6 (2.0757 and 2.0836 Ǻ), respectively, explains the higher BDE values. We should note that the homolytic cleavage of O–H in A6 is not favoured in aqueous media. The BDE values of compounds studied follow the sequence (with the exception of A6 in water):
4 < 5< 6 ≈ 3 < 1 < 2.
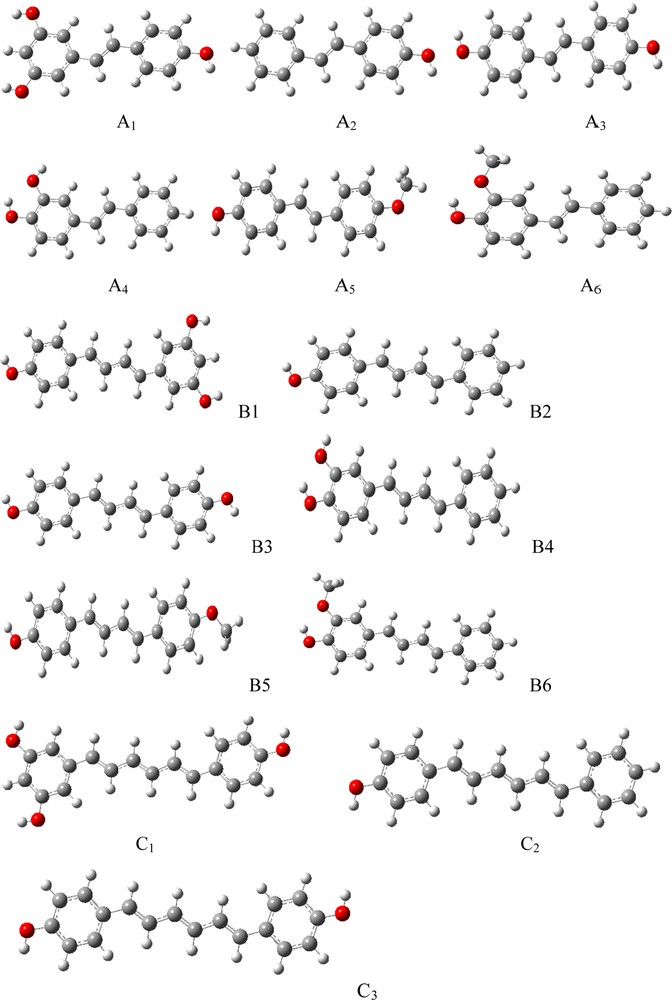
On the other hand, the experimental results revealed that 6 and 3 present a glavinoxyl radical (GO•) scavenging activity higher than that of 5 [20]. Moreover, BDE values are characterized by experimental second-order rate constants (k2). We should also point out that each one of the compounds A3 and B3 could donate a second hydrogen atom, forming a quinone (Fig. 4). The BDE values of the para-quinone form of compounds A3, B3 and C1 are lower than the sum of BDEs, corresponding to a simultaneous 4-H and 4′-H abstraction [19].
Mechanism of 4,4′-DHS inhibited peroxidation.
The trend of the BDE value became 3 < 5, in accordance with that obtained with the k2 values.
The elongation of the conjugated links decreased the BDE values and increased the number of resonance structures for the semiquinone radical and the antioxidant activity.
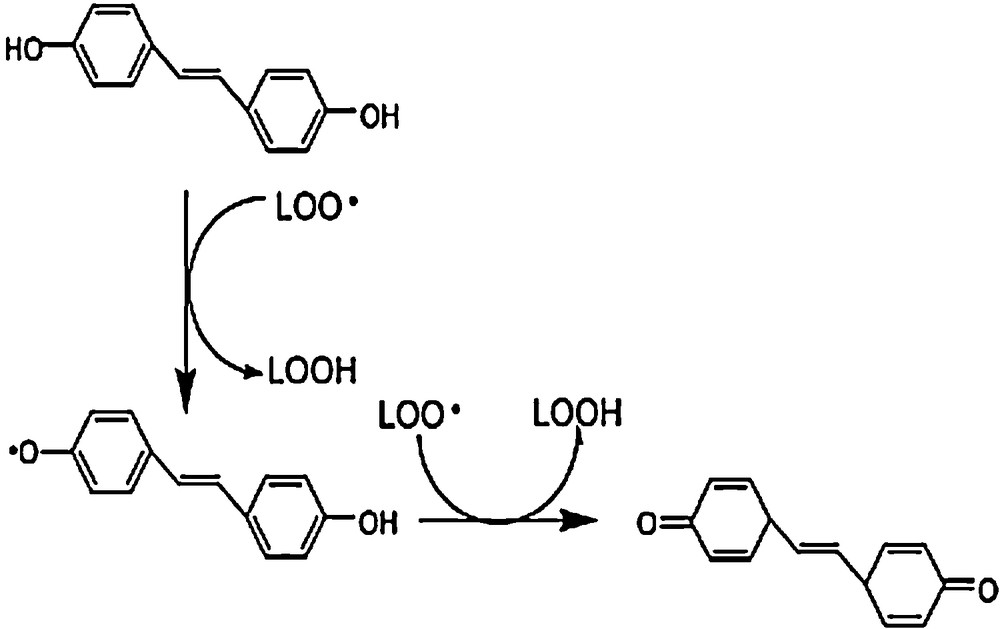
On the other hand, compounds bearing 3,4-ortho-di-hydroxyl groups, such as A4 and B4, show a strong antioxidant activity due to the abstraction of hydrogen atoms from the ortho-position hydroxyls. This abstraction can occur continuously to form a semiquinone or even a quinone structure (Fig. 5).
Mechanism of 3,4-DHS inhibited peroxidation.
We calculated the BDEs of the quinone structure formed by losing the two hydrogen atoms in A4 and B4; they were 157.1 and155.0 kcal/mol, respectively. The difference between the latter values and the sum of BDEs corresponding to the simultaneous loss of 4-H and 3-H is only 2.7 kcal/mol. On the other hand, the HOMO and LUMO values of the ground-state molecules, semiquinone and quinone structures of A4, B4, and C4 indicate that the semiquinone form would provide an electron again to continue the H abstraction in order to form the quinone structure. The BDEs of para 4,4′-quinone structures of A3, B3 and C3 were 159.1, 158 and 157.1 kcal/mol, respectively. The halves of these values are lower than those obtained for the semiquinone radicals BDEs (Table 1). According to the discussion above, it would be inferred that the semiquinone free radical and both 3,4-ortho and 4,4′-para-quinone structures are the proper explanations for the higher antioxidant activity of these compounds. This reaffirms the results of our previous work [19]. No significant difference between the BDE values has been evidenced in all the studied media. It must be stressed that the BDE values in water were lower than those in vacuo, with the exception of A4 and B4. The results indicate that the BDE values for a given molecule are closer in all studied environments. Based on the presented analysis, it may be concluded that the HAT is the most preferable antioxidant mechanism in vacuo for all molecules studied.
3.3 Spin density
Another molecular parameter correlated with the free radical-scavenging activity is spin density, which was also calculated. Spin density characterizes the distribution of electron spins in the free radicals and is responsible for their stability. It must be stressed that the more delocalized the spin density in the radical, the easier is the radical formed, and thus, the lower is the BDE [36]. The spin density is delocalized through the whole radicals in all studied molecules (Fig. 6). This is due to the presence of vinyl bonds. The lowest spin density values on the oxygen in phenoxy radicals, enhancing the stability of the radical formed after H abstraction, were found in the triene group. The results obtained from Table 2 show that the spin density values of the compounds with the same substituents decrease with both elongation of the conjugated links and introduction of EDGs. We should point out that the spin density values in water were lower than those in vacuo, with the exception of A4 and B4. This is in excellent agreement with the computed values of BDEs.

(Color online.) Spin density distribution in the 4-O-radical of trans-resveratrol (A1) and of its analogs.
Spin density on the oxygen atoms in phenoxy radicals of trans-resveratrol and its analogs.
| Compounds | Spin density (water) | Spin density (vacuum) |
| A1 | 0.28 | 0.32 |
| A2 | 0.28 | 0.32 |
| A3 | 0.27 | 0.31 |
| A4 | 0.26 | 0.28 |
| A5 | 0.26 | 0.31 |
| A6 | 0.54 | 0.32 |
| B1 | 0.24 | 0.28 |
| B2 | 0.24 | 0.28 |
| B3 | 0.23 | 0.27 |
| B4 | 0.24 | 0.26 |
| B5 | 0.23 | 0.27 |
| B6 | 0.25 | 0.27 |
| C1 | 0.22 | 0.26 |
| C2 | 0.21 | 0.25 |
| C3 | 0.20 | 0.25 |
3.4 SET–PT mechanism
3.4.1 Adiabatic ionization potentials (AIP)
The AIP represents the ease of electron donation of a molecule. Electron abstraction is the first antioxidant mechanism. Therefore, molecules with the lowest AIP are more active [18]. Nevertheless, low values of AIP do not guarantee a high antioxidant action of molecules. On the other hand, it should be pointed out that very low AIP values will result in air instability of electron-rich antioxidants, which limits their antioxidant efficacy, because they might react with molecular oxygen to generate a superoxide [20]. As seen from Table 3, it must be stressed that the compounds studied have AIP values following the sequence ene > diene > triene in all studied media. As an example, the AIP values of A1, B1 and C1 were 153.4, 146.4 and 140.7 kcal/mol, respectively. In the same group of molecules, the AIP values decreased in molecules with an additional hydroxyl in ortho position to form a catechol group such as A4 (151.2 kcal/mol) or para position, like A3 (148.1 kcal/mol). The same change was obtained with the introduction of methoxyl groups on these sites to obtain A6 (151.1 kcal/mol) and A5 (140.8 kcal/mol), respectively. With the absence of the two meta-hydroxyl groups, A2 showed the highest AIP values (154.6 kcal/mol). However, electron donor (ED) groups are of great importance for the electron-donating capacity of antioxydants. Therefore, the addition of EDs in both ortho and para positions decreased AIP values and increased the electron-donating capacity. The inductive and resonance effects are mainly responsible for the cation free radical stabilization. Compounds with more resonance structures are more stable and showed the lowest AIP values (for instance, for C3, AIP = 137.2 kcal/mol). The electron-donating ability of the studied compounds is related to an extended electronic delocalization over the entire molecule and systems having a high degree of π-delocalization are more active among the systems in which one electron transfer mechanism takes place.
Computed values of AIP and PDE (kcal/mol) obtained at the B3LYP/6-31G** level of theory.
| Compounds | AIP (vacuum) | PDE (vacuum) | AIP (water medium) | PDE (water medium) | Reducing capacitya |
| A1 | 153.4 | 239.2 | 93.8 | 9.7 | 1.53 ± 0.05 |
| A2 | 154.6 | 237.7 | 93.5 | 9.9 | 1.29 ± 0.02 |
| A3 | 148.1 | 243.3 | 88.4 | 13.5 | 2.15 ± 0.11 |
| A4 | 153.2 | 230.8 | 92.6 | 4.5 | 4.07 ± 0.30 |
| A5 | 146.9 | 245.0 | 88.2 | 13.9 | 1.79 ± 0.12 |
| A6 | 151.1 | 241.5 | 91.2 | Not favored | 1.51 ± 0.11 |
| B1 | 146.4 | 245.0 | 88.8 | 13.2 | 1.67 ± 0.05 |
| B2 | 147.5 | 243.9 | 88.4 | 13.4 | 1.78 ± 0.03 |
| B3 | 142.3 | 248.6 | 84.5 | 16.3 | 2.56 ± 0.02 |
| B4 | 145.9 | 236.9 | 87.3 | 7.7 | 4.65 ± 0.15 |
| B5 | 140.8 | 249.9 | 84.3 | 16.4 | 2.25 ± 0.17 |
| B6 | 144.2 | 247.9 | 86.8 | 13.6 | 2.03 ± 0.11 |
| C1 | 140.7 | 249.7 | 84.9 | 15.7 | 1.70 ± 0.04 |
| C2 | 141.6 | 248.9 | 84.6 | 15.9 | Not favored |
| C3 | 137.2 | 252.8 | 81.6 | 18.2 | 2.82 ± 0.11 |
a Tang et al., 2011 [20].
The ionization potentials of studied molecules in the gas phase reached higher values in comparison to water values. Note that, with the increase in the solvent polarity, ionization potentials decrease. Again, more polar solvents cause larger attenuation of solvent effect than non-polar solvents [25].
The solvents induce large changes in ionization potentials, because these include solvated charged species (radical and cations). The AIP depends on the HOMO energy of the neutral compound, and for a conjugated system, the abstraction of electrons becomes very easy. These results show a good correspondence with both BDE and HOMO values.
A significant influence of the solvent on AIP values was observed comparatively to the solvent effect on BDE. Hence, highest ionization potentials were found in the gas phase. Average differences between AIPs in the gas phase and water reached 60, 57.8 and 56.1 kcal/mol in ene, dine, and triene group, respectively. Note that the solvent effect decreases with the elongation of the ethylene bridge. Comparatively with BDE values, compounds with a catechol moiety have not the lowest AIP values, as shown in Table 3. This discrepancy can be attributed to the fact that BDE is affected by the local environment induced by the substituents, whereas the AIP value is affected by the structure of the whole molecule (the extended delocalization and conjugation of the π-electrons) [28]. Very high AIP values in vacuo show that SET–PT is not favoured in the gas phase. Unlike BDE values, those of AIP seem to be more influenced by the polarity of the solvent, since the latter may affect charge separation in a molecule [37].
3.4.2 Proton dissociation enthalpies (PDE)
Proton dissociation enthalpies related to the second step of SET–PT mechanism (Eq. (2.2)) show the most thermodynamically preferred OH group for the deprotonation of radical cations formed in the first step, Eq. (2.1). Nevertheless, it is important to study the effect of substituents, the elongation of the double bonds and solvents on PDE. In our work, we need the proton solvation enthalpies ΔsolvH(H+) in the studied solvent.
The calculated PDEs for the studied molecules are summarized in Table 3. The highest PDE values were found in the gas phase. The PDE values decrease significantly from the gas phase to water, due to the high solvation enthalpies of the proton. The average difference between PDE values in the gas phase and water reached 229.2, 231.9 and 233.8 kcal/mol in ene, diene and triene groups, respectively. Our results are in agreement with those of other authors [24,38,39].
Unlike AIP, the solvent effect increases with the elongation of the ethylene bridge in PDE values.
As shown in Table 3, the PDEs for resveratrol analogs studied in both water and in vacuo can be arranged in the following order: triene > diene > ene. Compared to BDE values, the highest PDE values were found in the triene group. This indicates that the elongation of the conjugated links of resveratrol and its analogs decreases the deprotonation potency in the cation form.
The compounds bearing a catechol moiety present the lowest PDE values. Electron-donating substituents cause an increase in PDE values. The deprotonation of the radical cation in A5 is not favored in water due to the intermolecular hydrogen bond.
By comparison of the values obtained in both studied media, we can note that BDE values are always lower than those of AIP. This indicates that the SET–PT mechanism is not the probable process in the studied environments.
3.5 SPLET mechanism
3.5.1 Proton affinities
PA represents the reaction enthalpy of the first step in SPLET mechanism (eq. (3.1)). The reaction enthalpy of the first step corresponds to the proton affinity (PA) of the phenoxide anion, ArŌ. Several experimental works have shown that phenoxide anions formed from flavonoids are better electron donors and radical scavengers than neutral molecules [40]. The deprotonation of the phenolic OH group in the gas phase and solvents was studied by calculating the proton affinity PA. The calculated PA values for the studied molecules are summarized in Table 4.
Computed values of PA and ETE (kcal/mol) obtained at the B3LYP/6-31G** level of theory.
| Compounds | PA (vacuum) | ETE (vacuum) | PA (water medium) | ETE (water medium) |
| A1 | 342.1 | 45.1 | 40.2 | 63.3 |
| A2 | 342.4 | 45.3 | 40.4 | 62.9 |
| A3 | 344.3 | 42.6 | 40.8 | 61.4 |
| A4 | 332.6 | 45.8 | 34.4 | 62.8 |
| A5 | 343.7 | 42.5 | 41.3 | 60.8 |
| A6 | 343.2 | 43.6 | 42.5 | Not favored |
| B1 | 338.6 | 47.2 | 38.9 | 63.0 |
| B2 | 338.3 | 47.5 | 39.7 | 62.1 |
| B3 | 340.0 | 45.1 | 40.4 | 60.4 |
| B4 | 330.0 | 47.2 | 33.6 | 61.4 |
| B5 | 340.0 | 45.0 | 40.3 | 60.4 |
| B6 | 342.5 | 43.8 | 41.8 | 58.6 |
| C1 | 335.8 | 49.0 | 39.2 | 61.4 |
| C2 | 335.5 | 49.3 | 39.1 | 61.4 |
| C3 | 337.1 | 47.2 | 39.9 | 59.9 |
Among the studied compounds, A4 and B4 have always the lowest PAs in all the studied environments. The PAs of the molecules in the studied environments can be arranged in the following order:
ene > diene > triene.
Similar to PDE, the PA values decrease significantly from the gas phase to water, due to the high solvation enthalpies of the proton. The average differences between PA in the gas phase and water were 301.4, 299.4, and 296.6 kcal/mol. This finding suggested that the solvation effect on the PA parameter was higher than on PDE by an average value of 67.5 kcal/mol. Confronted with PDE, the elongation of the conjugated links increases the deprotonation potency in the neutral system. Note that the PDE values in the present study are lower than the PA values; this is due to the ease of proton release in cations compared to neutral systems. Computed results also indicate that SPLET mechanism should be thermodynamically favoured in water, because PA values in water are lower than the AIPs and BDEs of all the studied molecules.
3.5.2 Electron transfer enthalpies (ETE)
ETE represents the reaction enthalpy of the second step in SPLET mechanism (Eq. (3.2)). The single-electron transfer processes in the gas phase and in water were studied by calculating electron transfer enthalpies (ETE). Calculated B3LYP/6-31G** ETEs are presented in Table 4. The ETEs of the molecules in the studied environments can be arranged in the following order:
ene > diene > triene.
Confronted with AIP values, the results revealed that the ETEs calculated in solvent are higher than those in vacuo. This indicates that the anionic form is more inclined to donate an electron than its neutral form. Note that the electron-donating groups decrease ETE [41], which explains the lower ETE value for A5 and B5.
3.6 Thermodynamically preferred mechanism
Our results demonstrate that the studied environments reaction enthalpies follow the sequence:
- • gas phase: BDE < AIP < PA;
- • water: PA < BDE < AIP.
It is clear that the BDE values are significantly lower than those of AIP and PA; hence, HAT mechanism is dominant in vacuo. On the other hand, PAs of OH groups of studied compounds are significantly lower in water than the corresponding BDEs and AIPs. Therefore, the SPLET mechanism represents the most thermodynamically probable reaction pathway in aqueous media. The SET–PT mechanism is not the preferred one both in water and in vacuo, because AIPs are always higher than BDEs and PAs. Due to the large enthalpy of proton solvation in all the studied solvents, PAs are significantly lower than gas-phase values. There is no pronounced difference between BDEs in the studied environments. On the other hand, Iuga et al. [42] found that the main reaction mechanism for the global reactivity of trans-resveratrol toward radicals, in water, at physiological pH, is proposed to be the sequential electron–proton transfer (SE–PT).
4 Conclusion
The results of our DFT computations revealed that resveratrol and its studied analogs were excellent antioxidants. BDE, AIP, PA and spin density values decrease in compounds with the same substituents with the elongation of the conjugated links. The calculations demonstrate that the antioxidant potency depends on the geometry of the neutral compounds, their phenoxy radicals, cation and anion structure, the number and position of the hydroxyl and methoxy groups, semiquinone and quinone structures. The elongation of the conjugated links, the induced effect of both hydroxyl and methoxy groups, the intramolecular hydrogen bond effect after H abstraction contribute strongly to antioxidant activity. The elongation of the conjugated links is an important strategy to improve the antioxidant activity of resveratrol analogs, including hydrogen atom or electron-donating ability. The H atom transfer mechanism is indicated as the most important one for the antioxidants in vacuo, while the SPLET process was the most probable mechanism in water. The SET–PT mechanism is not the most preferred one both in water and in vacuo. The PDE values revealed that the elongation of the conjugated links of resveratrol and its analogs decrease the deprotonation potency in the cation form, while the PA values indicated that the elongation of the ethylene bridge increases the deprotonation potency in the neutral system.
The experimental observations are satisfactorily explained by the results obtained. More importantly, the compounds bearing 4,4′-dihydroxy groups (A3, B3, and C3) exhibited remarkably higher cytotoxicity on HL-60 and HepG2 cells than resveratrol [20]. Compound C3 is the most cytotoxic among the compounds examined. In our opinion, these results are related to the contribution of the para-quinone structure in the compounds bearing 4,4′-dihydroxy groups.