

1 Introduction
Reactive oxygen species (ROS), as products of cellular metabolism, can have both beneficial and harmful roles [1]. Oxidative stress occurs as a consequence of overproduction of reactive species and may cause significant damage to molecules of high biological importance, thus contributing to various health problems such as cancer [2], aging [3], heart diseases [4], and neurodegenerative disorders [5]. Antioxidants are substances that, in low concentration, can prevent or slow down damage to cells caused by free radicals and inhibit oxidative stress, DNA mutations, malignant changes, and other forms of cell damage [6]. Among them, phenolic compounds (including phenolic acids) have attracted much attention because of their ubiquitous occurrence in nature and pronounced free radical–scavenging activity attributed to the radical-scavenging function of their phenolic hydroxyl groups [7]. o-Dihydroxyl configuration (i.e., catecholic group) is commonly considered to be a substantial characteristic which governs the antiradical activity of phenolic antioxidants [8]. Protocatechuic acid (3,4-dihydroxybenzoic acid) is a naturally occurring catechol-type phenolic acid found in many fruits and vegetables and is reported to have antibacterial [9], antimutagenic [10], antiinflammatory [11], and antihyperglycemic [12] activities. It is also identified as a potent antioxidant agent acting as a good protector against oxidative stress, thus demonstrating the preventive effect on different malignant diseases [13,14].
1,3,4-Thiadiazole ring, as a nitrogen- and sulfur-containing heterocyclic scaffold, is well known for its synthetic application as a useful synthon for the construction of bioactive molecules with a wide range of biological activities [15,16]. A series of 2,5-disubstituted-1,3,4-thiadiazoles have been synthesized by Khan et al. [17] and evaluated for their antioxidant activities, pointing out one compound with higher superoxide anion–scavenging activity than the standard drug n-propyl gallate. The carboxamide and carbothioamide derivatives of 1,3,4-thiadiazole have been found to exhibit promising antioxidant effects, which are determined by their high efficacy in trapping the reactive species generated during the oxidative damage [18].
In our previous report, we have carried out the synthesis of novel conjugates combining 1,3,4-thiadiazole ring and a phenolic acid moiety [19]. The obtained derivatives exerted good antioxidant and antiproliferative activity, and these results prompted us to proceed with the synthesis and examination of a series of 1,3,4-thiadiazole–chalcone hybrids containing a phenolic moiety [20]. This series of compounds also exhibited a strong antioxidant potential. In continuation of this research, herein we present the synthesis of new 1,3,4-thiadiazole amides obtained from protocatechuic acid and evaluation of their antioxidant potential by 2,2-diphenyl-1-picrylhydrazyl (DPPH) and 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) methods, supported by density functional theory (DFT) study and electrochemistry.
2 Results and discussion
2.1 Chemistry
The synthesis of a series of novel 1,3,4-thiadiazole amides (3a–o) is performed in two steps presented in Scheme 1. Initially, heterocyclic 1,3,4-thiadiazole precursors (2a–o) were prepared by the reaction of corresponding carboxylic acid (1a–o) with thiosemicarbazide using phosphoryl chloride as the reagent and solvent [19]. In the next step, the obtained 5-substituted-1,3,4-thiadiazole-2-amines (2a–o) were coupled with 3,4-dihydroxybenzoyl chloride (protocatechuic acid chloride) in dry dioxane, resulting in the formation of the final amide derivatives 3a–o in moderate to good yields (51–75%). The coupling reaction was performed in the presence of sodium hydrogen carbonate to neutralize liberated hydrogen chloride. In accordance with previously published synthesis of 1,3,4-thiadiazole derivatives [20], the preparation of an amide requires long time and high temperature because of poor nucleophilicity of the amino group of 1,3,4-thiadiazole. In addition, an excess of protocatechuic acid chloride was necessary to complete the reaction. The final compounds were prepared with satisfactory purity, but to obtain compounds with very high purity, they can be further purified by recrystallization from a hot aqueous solution of EtOH. All compounds were prepared for the first time, and their structural elucidation was performed by means of 1H and 13C nuclear magnetic resonance (NMR) spectroscopy, infrared (IR) spectroscopy, and elemental analysis (see Supplementary Material). Spectral data obtained from 1H NMR spectra showed that aromatic protons corresponding to the phenolic group appeared in the expected range of ppm scale (6.82–7.97 ppm), giving a singlet for H-2 and two doublets corresponding to H-5 and H-6, for most of the compounds. The proton belonging to the amide functional group, as anticipated, appeared as a singlet at the highest δ-values (12.12–12.88 ppm) for all derivatives, except for 3e, where a broad singlet was formed combining signals from two OH and one NH proton.

Reagents and conditions: (a) POCl3, rt, H2NHNC(=S)NH2, 1 h, reflux; (b) 3,4-dihydroxybenzoyl chloride, dioxane, 24 h, reflux.
To determine the contribution of the amide functional group to the antioxidative activity of the synthesized derivatives, we have performed the synthesis of the corresponding imino (4) and amino (5) analogues of the 1,3,4-thiadiazole amide derivative 3a. The synthesis of these two derivatives is presented in Scheme 2. In the first step, the desired 1,3,4-thiadiazole derivative containing an imine fragment (4) was prepared by condensation of 3,4-dihydroxybenzaldehyde (protocatechualdehyde) and 5-phenyl-1,3,4-thiadiazol-2-amine 2a, in the presence of the catalytic amount of glacial acetic acid. Owing to the decreased reactivity of the amino group of the 1,3,4-thiadiazole derivative 2a, an excess of aldehyde and prolonged reaction time were necessary for completing this reaction too. Finally, amine analogue 5 was obtained via simple reduction of the 1,3,4-thiadiazole imine derivative 4, using NaBH4 as a reduction agent in methanol at room temperature. Final compounds, 4 and 5, were also prepared for the first time, and their structure was determined by previously mentioned methods (1H and 13C NMR, IR, and elemental analysis, see Supplementary Material). The presence of a singlet at 8.79 ppm, corresponding to the proton from the CH=N group, in 1H NMR spectrum of derivative 4 confirmed the proposed structure. On the other hand, 1H NMR spectrum of compound 5 showed a doublet at 4.35 ppm corresponding to the methylene group protons. These protons are coupled with the adjacent NH proton, which appeared as a triplet at 8.34 ppm.

Reagents and conditions: (a) 3,4-dihydroxybenzaldehyde, AcOH, EtOH, 18 h, reflux; (b) NaBH4, MeOH, 1 h, rt.
2.2 Antioxidant activity
To determine the antioxidant potential of the synthesized compounds, as well as to compare them with previously reported results [19,20], we have used two of the most widely used antioxidant methods—DPPH and ABTS assays. The results of DPPH radical– and ABTS radical cation–scavenging activities of the tested compounds, expressed as a concentration of tested compound that reduces 50% (IC50) of the DPPH and ABTS species, are presented in Table 1. As can be seen, all examined derivatives showed high potential to scavenging DPPH radical and ABTS radical cation compared with the well-known antioxidants ascorbic acid and nordihydroguaiaretic acid (NDGA). Owing to the very poor solubility of most derivatives from the 3 series in methanol, determination of radical-scavenging activity of amides was performed in diluted dimethyl sulfoxide (DMSO). However, the solubility of compounds 3a, 4, and 5 was sufficient to allow the execution of DPPH assay in methanol. Because these three compounds were selected to examine and compare the influence of the amido, imino, and amino fragments of the molecule on the antioxidative activity, we wanted to avoid the possible solvent effects on the rate of H-abstraction from phenolic compounds [21].
DPPH- and ABTS-scavenging activities of the 1,3,4-thiadiazoles 3a–o, 4, and 5.a
| Compounds | IC50 (μM) | |
| DPPH• | ABTS•+ | |
| 3a | 4.34 ± 0.33 | 31.22 ± 0.72 |
| 4.48 ± 0.06b | ||
| 3b | 4.31 ± 0.24 | 27.67 ± 0.05 |
| 3c | 3.53 ± 0.25 | 30.31 ± 0.27 |
| 3d | 3.82 ± 0.11 | 27.21 ± 2.08 |
| 3e | 5.44 ± 0.22 | n.d. |
| 3f | 4.93 ± 0.01 | n.d. |
| 3g | 5.08 ± 0.35 | n.d. |
| 3h | 4.57 ± 0.21 | 27.73 ± 0.03 |
| 3i | 3.97 ± 0.13 | 27.13 ± 0.46 |
| 3j | 4.54 ± 0.16 | 33.00 ± 0.56 |
| 3k | 4.50 ± 0.47 | n.d. |
| 3l | 3.73 ± 0.04 | 27.86 ± 0.02 |
| 3m | 4.05 ± 0.03 | n.d. |
| 3n | 3.85 ± 0.04 | n.d. |
| 3o | 4.15 ± 0.08 | n.d. |
| 4 | 5.20 ± 0.17b | 25.90 ± 0.11 |
| 5 | 6.08 ± 0.07b | 21.69 ± 0.20 |
| PCA | 7.81 ± 0.29 | 99.72 ± 0.88 |
| Ascorbic acid | 14.26 ± 0.36 | 125.44 ± 1.86 |
| NDGA | 3.57 ± 0.04 | 45.78 ± 0.11 |
a Results are mean values ± SD from three measurements.
b Tested compound dissolved in methanol.
In DPPH assay, all examined compounds showed significantly stronger antioxidant potential than referent ascorbic acid and parent protocatechuic acid, whereas NDGA showed higher or similar DPPH-scavenging activity compared with the synthesized compounds. The nature of the R-substituents within the amide series was selected in such a manner to cover electron-donating, electron-withdrawing, and steric properties. Electron-donating groups are found to increase the electron density on the radical center formed after abstraction of a hydrogen atom from oxygen or nitrogen and consequently stabilize free radical intermediates. In accordance with this statement, the highest DPPH-scavenging activity was found for derivatives containing electron-donating alkyl groups in their structure (3c, 3d, 3l, and 3n). Among them, compound 3c, with the IC50 value of 3.53 μM, possessed slightly better activity than standard antioxidant NDGA (IC50 3.57 μM). Based on the results obtained from the DPPH assay, derivatives containing an electron-withdrawing chloro-substituent (3h–j) showed a lower antioxidant activity. The compounds 3e–g containing a methoxy group, which acts as an electron-donating group in ortho and para positions but shows an electron-withdrawing effect in the meta position, also exerted weaker activity because of the negative inductive effect of oxygen. Furthermore, it can be concluded that the substituent position at the phenyl ring also influenced the measured activity of the molecule, with m-substituted derivatives having the highest DPPH-scavenging potential. To examine the influence of the amido group on the antioxidative potential of the tested series, the compounds 4 and 5 were prepared and tested. Both of them possessed a low DPPH-scavenging activity, which is still higher than that of protocatechuic acid and ascorbic acid.
For further examination of the antioxidative potential, 10 compounds were selected (3a–d, 3h–j, 3l, 4, and 5) based on their prominent DPPH radical–scavenging activity. ABTS-scavenging assay showed that all tested compounds had better ABTS radical cation–scavenging ability than ascorbic acid, NDGA, and 3,4-dihydroxybenzoic acid. In this method, in contrast to the results obtained from the DPPH assay, the highest activity was observed for compounds 4 and 5, with IC50 values 25.90 and 21.69 μM, respectively. This is approximately 2–5 times stronger than the activities of referent antioxidant compounds. The compounds of the amide series were found to be less effective against ABTS, with IC50 values between 27 and 33 μM, but still more active than the most powerful examined referent antioxidant NDGA (IC50 45.78 μM). Obtained IC50 values show that compounds 4 and 5 with the best ABTS-scavenging property possessed the lowest activities in the scavenging of DPPH radicals among synthesized compounds. These differences may be attributed to the different steric accessibility of antioxidants to the radical site of DPPH and ABTS [22].
DPPH- and ABTS-scavenging activities of compounds 3c and 3i, which showed high antioxidant potential in both applied methods, were measured during 90 min and compared with the activities of protocatechuic acid and NDGA. The changes in the percentage of inhibition of DPPH and ABTS for these compounds applied at a concentration of 100 μM are presented in Fig. 1. In both applied methods, referent antioxidant NDGA showed the highest percentage of inhibition (about 60%) at the beginning of the reaction. The percentage of inhibition of ABTS for 3c and 3i was approximately 60% after 5 min of incubation, whereas in the DPPH method, percentages of inhibition for these compounds at the same time were 76.45 and 80.36, respectively. NDGA demonstrated the maximum activity in the neutralization of DPPH radicals after 15 min of reaction, whereas 3c and 3i displayed maximum activity after 25 min of reaction. Regarding ABTS radical cation–scavenging activity, the percentage of inhibition for 3c and 3i greatly increased up to 20 min with a slight increase of inhibition until the end of incubation. The percentage of inhibition for NDGA in the ABTS method was almost the same from 0 min to 90 min. Protocatechuic acid showed different behavior in inhibitions of DPPH and ABTS compared with compounds 3c, 3i, and NDGA. In reactions with DPPH and ABTS, 3,4-dihydroxybenzoic acid demonstrated an almost linear increase in the percentage of inhibition of these radicals from 0 min to 90 min of reaction. This suggests that protocatechuic acid reacts much slower with the tested radicals than referent antioxidant NDGA and synthesized compounds 3c and 3i, indicating considerably better antioxidant properties of the synthesized compounds in comparison with protocatechuic acid.

Percentage of inhibition of DPPH (A) and ABTS (B) measured during 90 min of reaction time for compounds 3c, 3i, protocatechuic acid (PCA), and NDGA. ABTS, 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid); DPPH, 2,2-diphenyl-1-picrylhydrazyl; NDGA, nordihydroguaiaretic acid.
2.3 Conformational analysis
The most stable rotamers of the compound 3a are examined, with different orientations of the protocatechuic acid part of the molecule. The most stable conformers obtained by the conformational analysis (3a and 3a1) are presented in Fig. 2. The energy difference between the two most stable structures in methanol is not negligible (22.19 kJ/mol). The interconversion barrier between the two minima is 25.9 kJ/mol, which indicates that conformational interconversion takes place easily at room temperature. In the following text, we will focus only on the most stable conformer 3a, as well as on the structures of other compounds derived from this rotamer. The structures of 3a are nonplanar and have one internal hydrogen bond (IHB) between two hydroxyl groups bonded to aromatic ring A (O3H3···O4). The natural bond orbital (NBO) analysis of investigated compounds reveals that the lone pair–antibonding orbital interactions between the oxygens and adjacent OH bond are responsible for the IHB formation. Namely, it is found that the charge transfer from the lone pair on O4 (2p orbital) to the σ* antibonding O3H3 orbital is a favorable donor–acceptor interaction. The conformations without IHBs are less stable with respect to the absolute minimum.
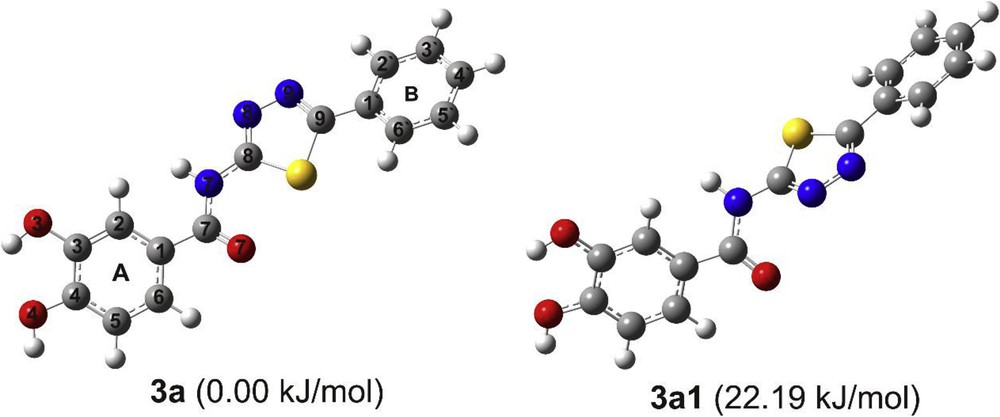
Two most stable structures of the compound 3a.
Unlike examined compounds of the 3 series, compounds 4 and 5 are not amides; the amide group which links ring A with the rest of the molecule is replaced by imino (4) and amino (5) groups. These two compounds are also not planar, having one IHB between two adjacent hydroxyl groups on the ring A. It means that similar antioxidant properties can be expected for compounds of the 3 series. The most stable conformers of all the investigated compounds are presented in Fig. S35 in the Supplementary Material.
2.4 Radicals, radical cations, and anions
Homolytic cleavage of the OH and NH bonds, for each of the investigated compounds, leads to the formation of three free radicals (Fig. 3). The exception is compound 4, which can form only two free radicals, because of the lack of NH bond. The corresponding bond dissociation enthalpy (BDE) values are presented in Table 2. The BDE values for both of the OH groups are mutually similar and significantly lower than those for the NH groups. Both oxygen radicals (Fig. 3) are stabilized with one hydrogen bond, unlike the nitrogen radical. Because no experimental values are available for investigated compounds, the obtained BDE values are compared with theoretical values for protocatechuic acid in water. The BDE values for all the investigated compounds given in this article are similar to those obtained for protocatechuic acid (339 and 341 kJ/mol, for 3-OH and 4-OH groups in water) [23]. It should also be noted that calculated BDE values of these compounds are significantly lower than BDE values calculated in benzene and water for chrysin (392.5, 390.8 kJ/mol), pinocembrin (396.1, 391.4 kJ/mol), apigenin (372.2, 368.1 kJ/mol), and naringenin (368.3361.8 kJ/mol) [24]. As can be seen from Scheme 3 and Fig. 3, additional delocalization of the spin density for compound 4 is carried out via the nitrogen of imino group. On the other hand, no additional delocalization of spin density over nitrogen was noticed for all other investigated compounds. In the case of the compound 5, there is no additional delocalization of the spin density.

Spin density distribution in all radicals obtained from 3, 4, and 5 in methanol.
Calculated thermodynamic parameters (kJ/mol) for the antioxidative activity of compounds 3a–d, 3h–j, 3l, 4, and 5.
| Compound | BDE | IP | PDE | PA | ETE | |
| 3a | 554.1 | |||||
| 3-OH | 337.1 | −48.0 | 122.3 | 383.8 | ||
| 4-OH | 339.4 | −45.7 | 111.3 | 397.2 | ||
| NH | 388.2 | 3.1 | 116.3 | 440.9 | ||
| 3b | 550.2 | |||||
| 3-OH | 337.1 | −44.0 | 122.2 | 383.9 | ||
| 4-OH | 338.8 | −42.4 | 110.9 | 396.9 | ||
| NH | 389.1 | 7.9 | 117.5 | 440.6 | ||
| 3c | 549.4 | |||||
| 3-OH | 337.2 | −43.3 | 122.1 | 384.1 | ||
| 4-OH | 338.8 | −41.6 | 111.0 | 396.9 | ||
| NH | 387.2 | 6.7 | 116.2 | 440.0 | ||
| 3d | 541.5 | |||||
| 3-OH | 336.8 | −35.7 | 122.5 | 383.4 | ||
| 4-OH | 338.8 | −33.7 | 111.6 | 396.1 | ||
| NH | 384.4 | 11.9 | 117.2 | 436.3 | ||
| 3h | 549.9 | |||||
| 3-OH | 340.5 | −40.3 | 122.3 | 387.2 | ||
| 4-OH | 339.1 | −41.8 | 111.1 | 397.0 | ||
| NH | 394.4 | 13.6 | 115.1 | 448.3 | ||
| 3i | 550.4 | |||||
| 3-OH | 328.3 | −44.3 | 121.7 | 384.4 | ||
| 4-OH | 330.0 | −42.6 | 109.9 | 397.8 | ||
| NH | 383.0 | 10.4 | 112.4 | 448.4 | ||
| 3j | 555.0 | |||||
| 3-OH | 337.0 | −49.0 | 122.0 | 384.0 | ||
| 4-OH | 338.9 | −47.1 | 110.5 | 397.4 | ||
| NH | 388.6 | 2.6 | 113.2 | 444.4 | ||
| 3l | 550.1 | |||||
| 3-OH | 337.3 | −43.8 | 122.9 | 383.4 | ||
| 4-OH | 338.6 | −42.4 | 111.5 | 396.2 | ||
| NH | 400.0 | 18.9 | 119.5 | 449.5 | ||
| 4 | 540.4 | |||||
| 3-OH | 339.2 | −32.2 | 121.5 | 386.7 | ||
| 4-OH | 333.0 | −38.5 | 103.9 | 398.1 | ||
| 5 | 517.9 | |||||
| 3-OH | 328.6 | −20.3 | 136.2 | 361.4 | ||
| 4-OH | 325.8 | −23.1 | 135.5 | 359.3 | ||
| NH | 369.3 | 20.4 | 172.1 | 366.2 |
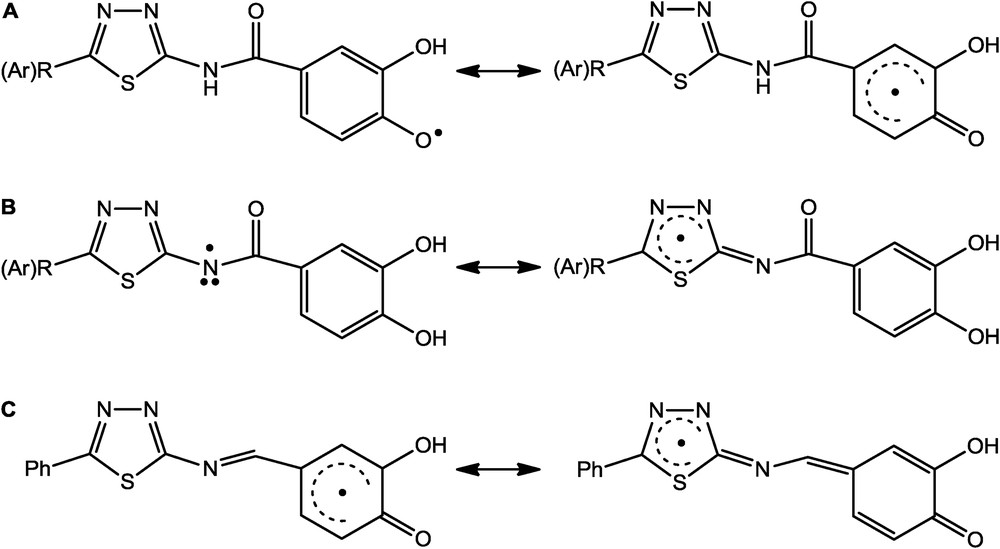
Resonance stabilization of radicals formed from amides (3a–o) after heterolytic (A) OH and (B) NH cleavage and radicals formed from imine 4 after heterolytic (C) OH cleavage and loss of one electron according to the SPLET mechanism. SPLET, sequential proton loss electron transfer.
When a single electron is removed from a molecule, a radical cation is formed. Ionization potential (IP) is a measure for ease of the formation of a radical cation. The IP values for the investigated compounds in methanol as a solvent are presented in Table 2. A careful analysis of the obtained results shows that the IP values are significantly higher than the other thermodynamic parameters. Even more, the values of the IP of these compounds are significantly higher than the IP value for the protocatechuic acid [23]. For these reasons, the single electron transfer followed by proton transfer (SET-PT) mechanism can be excluded in case of results obtained from the DPPH test. On the other hand, if we compare the IP values for compounds 3a, 4, and 5 with experimental IC50 values for the ABTS-scavenging assay, it is clear that these values are in good correlation with IP values. This means that SET-PT can be a likely mechanistic pathway in the case of an ABTS radical cation. The fact that the IC50 values for ABTS radical cation are significantly higher than those for the DPPH radical, i.e., that the reactions free radical neutralization with ABTS radical cation are taking place much more slower than the corresponding ones with the DPPH radical, is supporting this assumption. The confirmation of this assumption will be the subject of our future investigations that will address at examining the kinetics of these reactions.
The calculated proton affinity (PA) values for both OH groups and NH groups of all compounds are given in Table 2. The PA value is always the lowest for the 4-OH group, indicating that proton transfer from the 4-OH group is easier than from the NH and another OH group. Significantly lower PA values in methanol are the consequence of the interactions of OH and NH groups and anionic oxygen with the solvent molecules. Distribution of the negative charge for all anions is presented in Fig. S36. As already mentioned, the most stable anions are obtained by deprotonation of the 4-OH group. The negative charge in these anions is delocalized over O4, C5, and C6 atoms of the aromatic ring A and oxygen atom of the amide group, which contributes to the additional stability of the anion. The neighboring of OH group additionally contributes to the stability of the anion by formation of hydrogen bond with the adjacent hydroxyl group.
Owing to lack of experimental PA values, the obtained values are compared with the calculated ones for protocatechuic acid in water. The PA values for all the investigated compounds are slightly lower than those obtained for protocatechuic acid (130 and 119 kJ/mol, for 3-OH and 4-OH groups in water) [23]. It should also be noted that calculated PA values of these compounds are significantly lower than PA values calculated in water for ortho-, meta-, and para-hydroxybenzoic acids (191, 158, 150 kJ/mol, respectively) [25]. On the other hand, they are almost the same as those calculated for gallic acid (129 and 109 kJ/mol) [26].
The exception is compound 4. Only in the case of this compound, additional delocalization of the negative charge is noticed via the imino group, whereas for other compounds, this effect was not observed. The consequence of charge delocalization is also reflected on PA values (Table 2). It is obvious that the obtained theoretical results are in good agreement with the experimental values for DPPH (Table 1).
2.5 DPPH radical–scavenging mechanisms of investigated compounds
As mentioned previously, at least three mechanisms of the antioxidant activity are operative. To evaluate which mechanism is dominant under given reaction conditions, it is necessary to calculate the enthalpies associated with these three mechanisms [hydrogen atom transfer (HAT), SET-PT, and sequential proton loss electron transfer (SPLET)]. It is possible to evaluate the preferred reaction pathway by simple comparison of BDE, IP, and PA values [27–29]. The lowest value of these thermodynamic parameters indicates a more probable mechanism.
It is clear, based on the values presented in Table 2, that IP values are significantly greater than other thermodynamic parameters. This means that SET-PT is a nonoperating mechanism for these compounds.
On the other hand, PA values for 3-OH, 4-OH, and NH groups are significantly lower than BDE and IP values, for all investigated compounds. It means that the SPLET mechanism represents the most probable reaction path in polar solvents such as methanol. Despite the fact that the 4-OH group is the most active reaction center, it should be noted that all three reaction centers have mutually similar corresponding PA values. Moreover, unlike the HAT mechanism, the NH group plays a more important role in the SPLET mechanism. The PA values for all NH groups are very close to that of the 4-OH group, especially in the case of compounds 3i and 3j.
In our previous research, we have determined that better radical-scavenging activity of phenolic acid derivatives containing 1,2,4-triazole [30] or 1,3,4-oxadiazole [31] ring in comparison with parent acids can be attributed to the participation of the heterocyclic scaffold in the delocalization of the unpaired electron after homolytic cleavage of the OH and NH bonds by the DPPH radical. This was also supported by DFT calculations for 1,2,4-triazole derivatives [30]. Similarly, in case of previously reported conjugates combining 1,3,4-thiadiazole ring and a phenolic acid moiety [19], as well as 1,3,4-thiadiazole–chalcone hybrids containing a phenolic moiety [20], the resonance stabilization of the resulting phenoxyl or nitrogen radical was achieved by the participation of the 1,3,4-thiadiazole ring. In this study, as we can see from Table 1, the presence of 1,3,4-thiadiazole moiety significantly increases the radical-scavenging activity of the compounds 3a–o in comparison with parent protocatechuic acid and referent antioxidants. As a result of the SPLET mechanism, the phenoxyl radical formed from amide derivatives can be stabilized through resonance which includes a phenyl ring and neighboring carbonyl group (Scheme 3A). Herein, the heterocyclic scaffold does not participate in the stabilization of the radical formed after homolytic cleavage of the OH bond. In the case of homolytic cleavage of the NH bond, the resonance stabilization includes only 1,3,4-thiadiazole ring (Scheme 3B). The imino analogue (4) was found to have the lowest calculated PA value for the 4-OH bond (Table 2), whereas amine 5 had generally the highest calculated PA values for 3-OH, 4-OH, and NH bonds. This is in agreement with the high resonance stabilization of the phenoxyl radical formed after homolytic cleavage of the 4-OH bond of imino derivative, where the unpaired electron can be delocalized across the whole molecule (Scheme 3C).
2.6 Electrochemistry
Selected compounds (3a, 3d, 3j, 3l, 4, and 5) were investigated in N,N-dimethylformamide (DMF) solutions containing tetrabutylammonium perchlorate in the potential amplitude of +1.6 V to −2.1 V.
In the applicable positive potential range, all compounds exhibit one (3l and 4) or two (3a, 3d, 3j, and 5) oxidation peaks. For the sake of comparison, the oxidation process for 3a is presented together with a voltammogram of PCA, the parent phenolic acid, protocatechuic acid (Fig. 4). In this range, as reported previously [30], this acid displays a two-step one-electron oxidation process at peaks located at close potentials. Here, the shape and the current function of the first oxidation peak of 3a are quite similar to those of PCA. Because this is observed at all new compounds, it seems reasonable to attribute the first oxidation process to the already described oxidation located at hydroxyl groups [30]. Shifting of the peak potential toward less-positive values and better “reversibility” of its counter peak must be understood as a result of an easier oxidation process at a conjugated molecule which is in accordance with the aforementioned discussion.
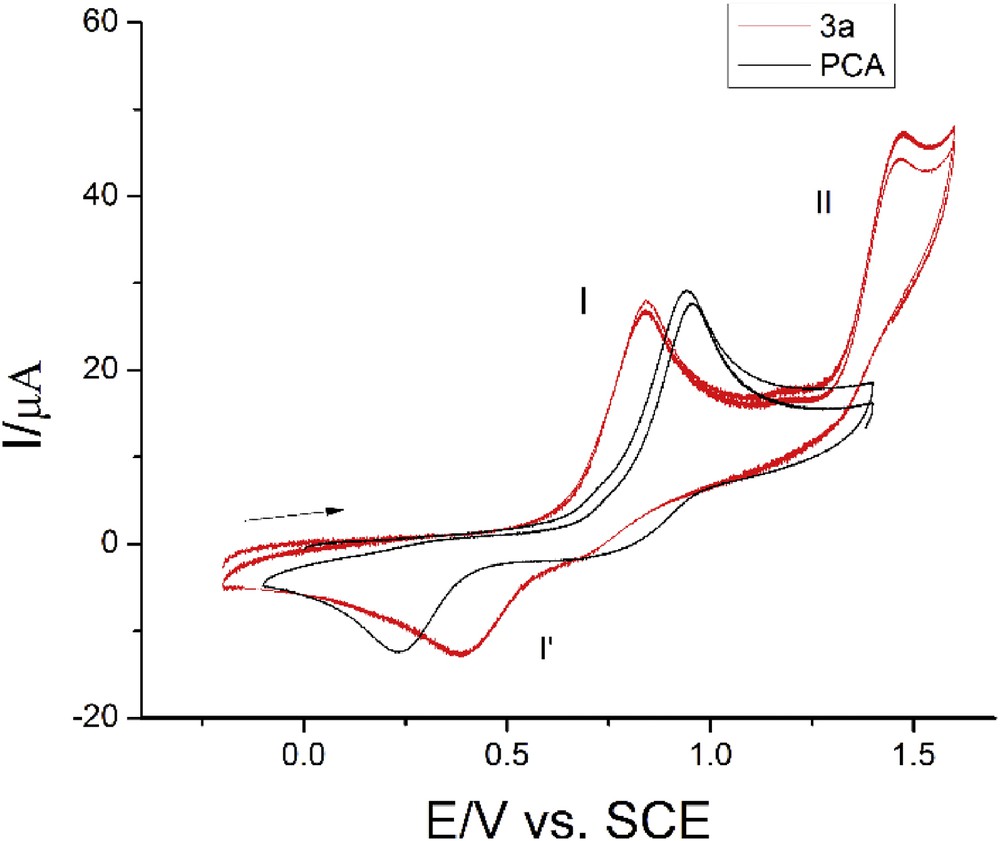
Cyclic voltammograms for 3a (c = 1.14 mM) and PCA (c = 1.15 mM); GC electrode, v = 0.10 Vs−1. PCA, protocatechuic acid (3,4-dihydroxybenzoic acid); GC, glassy carbon; SCE, saturated calomel electrode.
The electrochemical characteristics of the first process are given as follows: ΔEpa/c for peaks I/I' for all the compounds at moderate scan rates lie between 300 and 500 mV, while cathodic parts significantly deform at higher scan rates showing irreversibility of the electron transfer process. In addition, kinetic parameters of the peak I such as half-peak width which amounts from 80 mV (at 0.02 Vs−1) up to about 150 mV for higher scan rates together with ΔEp/Δ log v ranging from 80 mV (v = 0.02–0.20 Vs−1) to 130 mV (0.2–2 Vs−1) are evidence of an overall complex process. However, if the amplitude of potential is limited behind the peak I, excellent repeatability of the peaks’ I/I' shapes and potentials was recorded just as presented in Fig. 4, suggesting reversible chemical steps after the electron release.
The second oxidation process for all 3-type compounds (a, d, and j) is shifted to the positive limit of potential and has the characteristics of irreversible electron transfer. In these voltammograms, the current function of the peak II is somewhat higher than for the peak I, which suggests a possible two-electron process (Fig. 4). Assessment of these peaks is not easy partly because the aforementioned compounds display some adsorptive features at these potentials. This oxidation process, however, is much better defined for compound 5 where the second process appears at the least positive potentials of all compounds (Fig. 5). These peaks have apparently similar characteristics as the peaks I/I'. Because by cycling the potential in the amplitude −0.2 to +1.5 V both oxidation peaks are fully recovered, it means that in the cited potential range, no irreversible chemical reaction is present in either step. The second oxidation process may be ascribed to oxidation of NH; that is why no peak was observed at 4. On the other hand, the process in 3l voltammograms is shifted behind the limit of positive potentials; the reason for that might lie in the highest BDE value for the NH bond, which makes the oxidation more difficult (vide infra, Table 2).
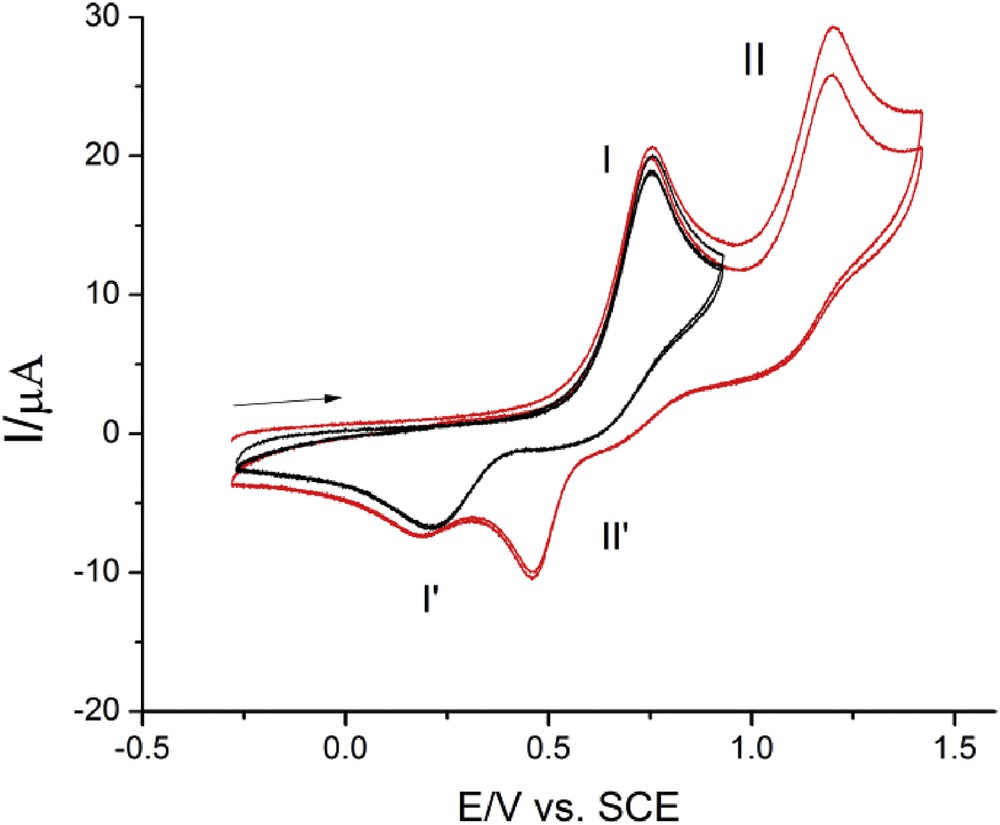
Cyclic voltammograms for 5 (c = 0.74 mM); GC electrode, v = 0.10 V s−1. GC, glassy carbon; SCE, saturated calomel electrode.
The potentials of the characteristic peaks on the voltammograms of the selected compounds are given in Table 3.
Potentials of characteristic peaks; GC electrode (v = 0.10 V s−1).
| Compounds | Ep(I)a | Ep(I') | Ep(II) | Ep(Ir) |
| 3a | +0.81 | +0.39 | +1.44 | −1.82 |
| 3d | +0.84 | +0.41 | +1.44 | −1.86 |
| 3j | +0.83 | +0.44 | +1.47 | −1.75 |
| 3l | +0.86 | +0.34 | >1.5 | <−2.1 |
| 4 | +0.75 | +0.41 | – | −1.80 |
| 5 | +0.76 | +0.46 | +1.20 | −2.06 |
a In V versus SCE.
It is common that the compounds containing amino, imino, or amido nitrogen display reduction processes with a variety of chemical reactions coupled with electron transfer [32]. The chemical steps usually involve either radical dimerization or addition of a proton to stabilize the obtained anion from the medium or through self-protonation if there is such a possibility [33–36]. As mentioned previously, in our cases, three nitrogen forms are present: amido in compounds denoted by 3, imino in 4, and amino in 5. Among all these, only 4 displayed well-defined peaks, whereas the voltammograms of other compounds have the ill-shaped irreversible peaks shifted to potentials around −2.0 V (Fig. 6). These processes are situated near the adsorptive multielectron peaks at the negative potential limit and therefore appear to be unsuitable for a detailed characterization. However, judging by the current functions, all the peaks correspond to a one-electron reduction. In contrast to that, reduction of the imine group in 4 is carried out in a one-electron quasireversible pair of peaks: ΔEp(Ir'/Ir) amounts from 65 mV (0.02 Vs−1) to 130 mV (2 Vs−1). At scan rates higher than 0.02 Vs−1, a new small oxidation peak at ∼+0.10 V appears which in voltammetric time scale apparently does not affect either oxidation or reduction processes. This peak is most pronounced at 4 but only slightly visible in voltammograms of all other compounds. Obviously, it belongs to a reactive intermediate formed after reduction, the composition and nature of which could be established by application of other methods such as controlled potential electrolysis. However, it was beyond the scope of the present study.
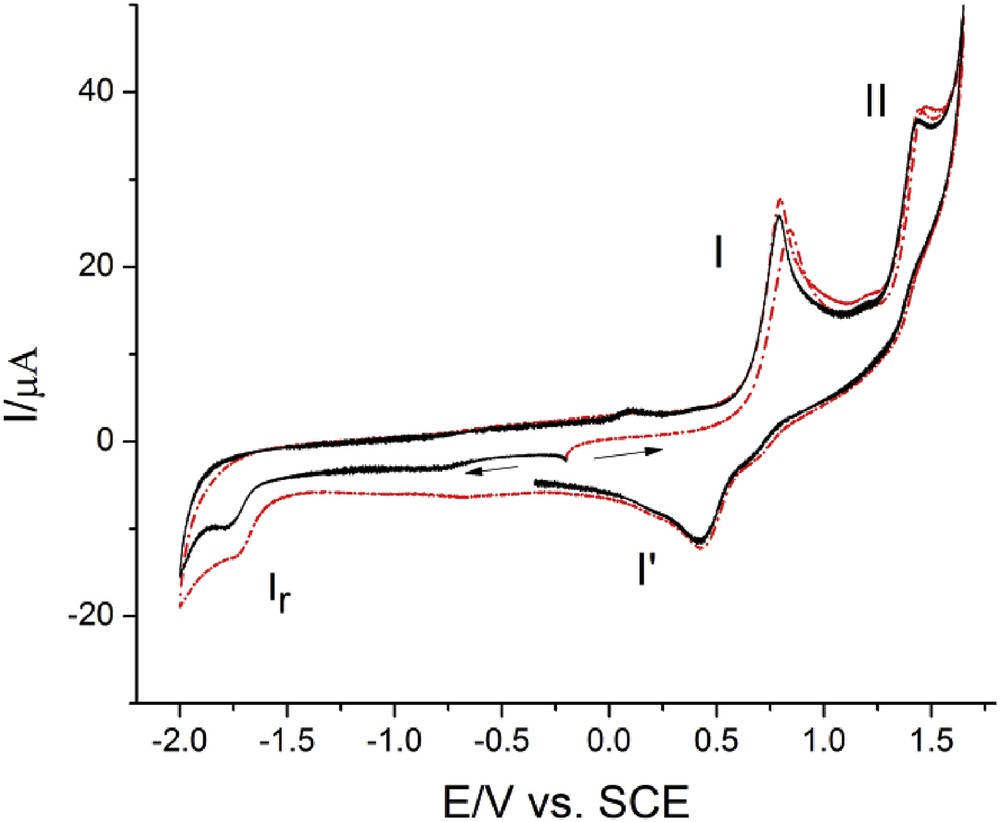
Cyclic voltammograms for 3j (c = 0.87 mM); GC electrode, v = 0.10 V s−1. GC, glassy carbon; SCE, saturated calomel electrode.
As can be seen in Table 3, the most easily oxidizable compounds are 4 and 5. In general, the first oxidation process is similar for all six compounds and easier than oxidation of the parent protocatechuic acid recorded under the same experimental conditions (Ep = +0.96 V at v = 0.10 Vs−1) [30]. The second oxidation process ascribed to oxidation of amido (3), imino (4), and amino (5) nitrogen proceeds most easily at the latter one. On the other hand, reduction, which takes place also at these nitrogen atoms, is irreversible for all compounds except for 4; this can be ascribed to the possibility of additional stabilization of the transferred electron by charge delocalization in the imino derivative.
The obtained electrochemical data served for calculation of highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energies (Table 4), according to the procedure described in the study by Filipović et al. [37]. The obtained HOMO values are compared with that for protocatechuic acid, which appears to be the lowest of all. Because the acid does not show any reduction process in the applied potential range, its LUMO cannot be calculated. The highest value of HOMO in 5 is related with the lowest IP (Table 2), whereas its LUMO energy correlates with the lowest electron affinity, i.e., value of electron transfer enthalpy (ETE) (vide infra).
Calculated values of HOMO and LUMO orbitals.
| Compounds | EOxonset/[V]a | HOMO/[eV]b | ERedonset/[V] | LUMO/[eV]c |
| 3a | +0.128 | −4.93 | −2.140 | −2.66 |
| 3d | +0.163 | −4.96 | −2.093 | −2.71 |
| 3j | +0.148 | −4.95 | −2.082 | −2.72 |
| 3l | +0.175 | −4.98 | – | – |
| 4 | +0.032 | −4.83 | −2.103 | −2.70 |
| 5 | +0.008 | −4.81 | −2.377 | −2.42 |
| PCA | +0.213 | −5.01 | – | – |
a V versus Fc/Fc+ in DMF containing 0.1 M TBAP as the supporting electrolyte at a scan rate of 0.10 V s−1, GC electrode.
b EHOMO = −4.8 − (EOxonset − E1/2Fc/Fc+) [37].
c ELUMO= −4.8 − (ERedonset − E1/2Fc/Fc+); E1/2Fc/Fc+ = +0.487 V versus SCE.
3 Conclusion
In conclusion, 15 novel 1,3,4-thiadiazole amide derivatives containing a protocatechuic acid moiety (3a–o), as well as the corresponding imino (4) and amino (5) analogues of 3a, were prepared and examined for their antioxidative potential. All tested compounds exerted better DPPH radical–scavenging activity than parent 3,4-dihydroxybenzoic acid and referent ascorbic acid, with the highest activity found for derivatives containing electron-donating alkyl groups (3c, 3d, 3l, and 3n). The ABTS assay showed that all examined derivatives had better radical cation–scavenging ability than referent ascorbic acid, NDGA, and 3,4-dihydroxybenzoic acid. In this method, in contrast to the results obtained from the DPPH assay, the highest activity was observed for compounds 4 and 5. To evaluate which radical-scavenging mechanism is dominant under given reaction conditions, the values of thermodynamic parameters associated with these mechanisms were calculated and compared. The obtained theoretical results are in good agreement with the experimental values for the DPPH assay. The PA values are significantly lower than BDE and IP values, for all investigated compounds, which means that the SPLET mechanism represents the most probable reaction path in polar solvents. Furthermore, the IP values for compounds 3a, 4, and 5 are in good correlation with experimental IC50 values for the ABTS-scavenging assay. This indicates that SET-PT can be a likely mechanistic pathway in the case of an ABTS radical cation. The obtained electrochemical data showed that the most easily oxidizable compounds are 4 and 5. On the other hand, reduction, taking place at amido (3), imino (4), and amino (5) nitrogen atoms, is irreversible for all compounds except for 4. This can be ascribed to the possibility of additional stabilization in the imino derivative, which also justifies the lowest calculated PA value for this compound.
4 Experimental
4.1 Chemistry
4.1.1 Physical measurements and methods
Melting points were determined on a Mel-Temp capillary melting points apparatus, model 1001 and are uncorrected. Elemental (C, H, N, S) analysis of the samples was carried out in the Center for Instrumental Analysis, Faculty of Chemistry, Belgrade. UV spectra were recorded using an Agilent Technologies, Cary 300 Series UV-Vis Spectrophotometer. IR spectra were obtained using a PerkinElmer Spectrum One FT-IR spectrometer with a KBr disc. 1H and 13C NMR spectra were recorded using a Varian Gemini 200 MHz spectrometer.
4.1.2 Procedure for the preparation of 3a–o, 4, and 5
4.1.2.1 Procedure for the synthesis of 2a–o
A mixture of corresponding acid, 1 (2.00 mmol), and POCl3 (0.80 ml) was stirred for 20 min at room temperature. Then, thiosemicarbazide (2.0 mmol, 0.182 g) was added, and the resulting suspension was refluxed for 1 h. After cooling the flask in an ice bath, 2.4 ml of distilled water was added carefully, and reflux was continued for 1 h. The mixture was then cooled to the room temperature, saturated aqueous solution of NaOH was added until pH 8.5 was reached, and the suspension was stirred for 1 h at the room temperature. The formed precipitate of the corresponding 2-amino-1,3,4-thiadiazole derivative (2a–o) was then filtrated, dried over CaCl2, and recrystallized from hot 50% aqueous EtOH.
4.1.2.2 Procedure for the synthesis of 3a–o
To the mixture of 3,4-dihydroxybenzoic acid (2.00 mmol, 0.308 g) in dry dichloromethane (8.0 ml), SOCl2 (20.00 mmol, 1.5 mL) was slowly added, followed by two drops of DMF. The resulting mixture was then stirred for 2 h at room temperature. Afterward, the solvent was evaporated under reduced pressure, and the excess of SOCl2 was removed by azeotropic distillation with toluene. Corresponding substituted 2-amino-1,3,4-thiadiazole, 2a–o (1.00 mmol), NaHCO3 (2.00 mmol, 0.168 g), and dry dioxane (10.0 ml) were added to the formed acid chloride, without previous isolation, and the mixture was then refluxed for 24 h. Afterward, the solvent was evaporated under reduced pressure, 15.0 mL of H2O was added to the residue, as well as solid Na2CO3, until pH 9 was reached, and the formed suspension was stirred for 30 min at the room temperature, filtrated, washed with water, and dried over CaCl2. The final compounds were obtained with satisfactory purity, but to obtain compounds with very high purity, they can be subjected to further purification by recrystallization from a hot aqueous solution of 50% EtOH, for 3l, 65% EtOH, for 3b and 3o, 70% EtOH, for 3a, 3c, 3f, and 3n, 75% EtOH, for 3m, 80% EtOH, for 3k, 85% EtOH, for 3d and 3e, 90% EtOH, for 3j and 3g, and 96% EtOH, for 3h and 3i.
4.1.2.3 Procedure for the synthesis of 4
To the mixture of 3,4-dihydroxybenzaldehyde (1.87 mmol, 0.259 g) and 2-amino-5-phenyl-1,3,4-thiadiazole (1.50 mmol, 0.266 g) in absolute ethanol (5.0 ml), three drops of glacial acetic acid were added, and the mixture was refluxed for 18 h. The mixture was then cooled to the room temperature, 10.0 ml of distilled water was added, and the suspension was then stirred for 30 min at the room temperature. The precipitate was then filtrated, dried over CaCl2, and recrystallized from hot acetone.
4.1.2.4 Procedure for the synthesis of 5
Solid NaBH4 (15.67 mmol, 0.593 g) was slowly added to the stirring suspension of formed imine, 4 (1.00 mmol, 0.297 g) in methanol (10.0 ml), and stirring of the resulting mixture was then continued for 1 h at the room temperature. Afterward, 20.0 ml of distilled water was added, as well as 2 M HCl, until pH 2 was reached, and the formed precipitate was stirred for 30 more minutes at the room temperature. After cooling the flask in a refrigerator for 1.5 h, the precipitate was filtrated, dried over CaCl2, and recrystallized from hot 50% aqueous EtOH.
4.1.2.5 3,4-Dihydroxy-N-(5-phenyl-1,3,4-thiadiazol-2-yl)benzamide × H2O (3а)
Beige powder; yield: 0.20 g (64%); mp > 250 °C; 1H NMR (200 MHz, DMSO-d6): 6.86 (d, 1H, J = 8.2 Hz, Ar–H); 7.53–7.62 (m, 5H, Ar–H); 7.97 (s, 1H, Ar–H); 7.97 (d, 1H, J = 8.2 Hz, Ar–H); 9.44 (s, 1H, OH); 9.93 (s, 1H, OH); 12.81 (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): 115.22, 116.09, 120.94, 122.37, 126.89 (2C), 129.33 (2C), 130.39, 130.47, 145.26, 150.48, 159.56, 161.75, 164.73; IR (KBr, cm−1): 3524, 3414, 3107, 1632, 1614, 1592, 1520, 1291, 754; anal. calcd. for C15H11N3O3S × H2O (313.33 g/mol): C, 57.50; H, 3.54; N, 13.41; S, 10.23; found: C, 57.48; H, 3.55; N, 13.39; S, 10.25.
4.1.2.6 3,4-Dihydroxy-N-(5-(o-tolyl)-1,3,4-thiadiazol-2-yl)benzamide (3b)
Beige powder; yield: 0.20 g (61%); mp = 246–247 °C (Dec.); 1H NMR (200 MHz, DMSO-d6): 2.53 (s, 3H, CH3); 6.86 (d, 1H, J = 8.0 Hz, Ar–H); 7.31–7.42 (m, 3H, Ar–H); 7.55–7.62 (m, 2H, Ar–H); 7.70 (d, 1H, J = 7.0 Hz, Ar–H); 9.77 (bs, 2H, OH); 12.43 (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): 21.27, 115.28, 116.11, 121.01, 122.46, 126.47, 129.57, 129.93, 130.12, 131.52, 136.41, 145.34, 150.55, 160.06, 161.31, 164.85; IR (KBr, cm−1): 3404, 3154, 2955, 1685, 1604, 1527, 1300, 1204, 753; anal. calcd. for C16H13N3O3S (327.36 g/mol): C, 58.70; H, 4.00; N, 12.84; S, 9.80; found: C, 58.72; H, 4.01; N, 12.82; S, 9.82.
4.1.2.7 3,4-Dihydroxy-N-(5-(m-tolyl)-1,3,4-thiadiazol-2-yl)benzamide × 0.5H2O (3c)
Beige powder; yield: 0,24 g (74%); mp > 250 °C; 1H NMR (200 MHz, DMSO-d6): 2.39 (s, 3H, CH3); 6.86 (d, 1H, J = 8.2 Hz, Ar–H); 7.32 (d, 1H, J = 7.6 Hz, Ar–H); 7.41 (t, 1H, J = 7.6 Hz, Ar–H); 7.56 (s, 1H, Ar–H); 7.61 (d, 1H, J = 8.2 Hz, Ar–H); 7.73–7.79 (m, 2H, Ar–H); 9.72 (bs, 2H, OH); 12.60 (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): 21.05, 115.29, 116.14, 121.02, 122.45, 124.12, 127.41, 129.26, 130.38, 131.19, 138.80, 145.35, 150.56, 159.59, 161.92, 164.78; IR (KBr, cm−1): 3407, 3281, 3187, 1685, 1608, 1542, 1521, 1320, 1298, 1107, 749; anal. calcd. for C16H13N3O3S × 0.5H2O (327.36 g/mol): C, 58.70; H, 4,00; N, 12.84; S, 9.80; found: C, 58.71; H, 3.99; N, 12.87; S, 9.79.
4.1.2.8 3,4-Dihydroxy-N-(5-(p-tolyl)-1,3,4-thiadiazol-2-yl)benzamide × 0.5H2O (3d)
White powder; yield: 0.23 g (70%); mp > 250 °C; 1H NMR (200 MHz, DMSO-d6): 2.36 (s, 3H, CH3); 6.86 (d, 1H, J = 8.2 Hz, Ar–H); 7.34 (d, 2H, JAB = 8.0 Hz, Ar–H); 7.55 (s, 1H, Ar–H); 7.59 (d, 1H, J = 8.2 Hz, Ar–H); 7.85 (d, 2H, JBA = 8.0 Hz, Ar–H); 9.54 (bs, 1H, OH); 9.74 (bs, 1H, OH); 12.75 (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): 19.04, 113.17, 114.03, 118.89, 120.34, 124.76 (2C), 125.63, 127.84 (2C), 138.31, 143.23, 148.42, 157.22, 159.77, 162.61; IR (KBr, cm−1): 3397, 3168, 2956, 1682, 1609, 1532, 1459, 1309, 1298, 1206, 810; anal. calcd. for C16H13N3O3S×0.5H2O (327,36 g/mol): C, 58.70; H, 4.00; N, 12.84; S, 9.80; found: C, 58.74; H, 4.02; N, 12.85; S, 9.81.
4.1.2.9 3,4-Dihydroxy-N-(5-(2-methoxyphenyl)-1,3,4-thiadiazol-2-yl)benzamide × 0.5H2O (3e)
Beige powder; yield: 0.23 g (68%); mp > 250 °C; 1H NMR (200 MHz, DMSO-d6): 4.03 (s, 3H, OCH3); 6.86 (d, 1H, J = 8.0 Hz, Ar–H); 7.13 (t, 1H, J = 8.0 Hz, Ar–H); 7.27 (d, 1H, J = 8.0 Hz, Ar–H); 7.50 (dd, 1H, J = 8.0 and 1.4 Hz, Ar–H); 7.55 (s, 1H, Ar–H); 7.70 (d, 1H, J = 8.0 Hz, Ar–H); 8.30 (dd, 1H, J = 8.0 and 1.4 Hz, Ar–H); 10.41 (bs, 2H, OH and 1H, NH); 13C NMR (50 MHz, DMSO-d6): 54.14, 110.37, 113.16, 113.99, 117.09, 118.85, 19.14, 120.46, 125.32, 129.60, 143.26, 148.41, 153.32, 154.45, 159.27, 162.60; IR (KBr, cm−1): 3398, 3153, 2944, 1681, 1601, 1545, 1526, 1314, 1299, 1260, 1018, 747; anal. calcd. for C16H13N3O4S × 0.5H2O (343.36 g/mol): C, 55.97; H, 3.82; N, 12.24; S, 9.34; found: C, 55.99; H, 3.81; N, 12.26; S, 9.35.
4.1.2.10 3,4-Dihydroxy-N-(5-(3-methoxyphenyl)-1,3,4-thiadiazol-2-yl)benzamide × 0.5H2O (3f)
Light brown powder; yield: 0.24 g (70%); mp > 250 °C (Dec.); 1H NMR (200 MHz, DMSO-d6): 3.85 (s, 3H, OCH3); 6.86 (d, 1H, J = 8.0 Hz, Ar–H); 7.09 (d, 1H, J = 8.0 Hz, Ar–H); 7.40–7.62 (m, 5H, Ar–H); 9.68 (bs, 2H, OH); 12.72 (bs, 1H, NH); 13C NMR (50 MHz, DMSO-d6): 55.49, 111.47, 115.27, 116.12, 116.65, 119.55, 121.00, 122.40, 130.60, 131.69, 145.33, 150.55, 159.78 (2C), 161.62, 164.79; IR (KBr, cm−1): 3386, 3169, 2940, 1676, 1605, 1526, 1457, 1304, 1217, 1217, 1112, 745; anal. calcd. for C16H13N3O4S × 0.5H2O (343,36 g/mol): C, 55.97; H, 3.82; N, 12.24; S, 9.34; found: C, 55.94; H, 3.83; N, 12.23; S, 9.31.
4.1.2.11 3,4-Dihydroxy-N-(5-(4-methoxyphenyl)-1,3,4-thiadiazol-2-yl)benzamide × H2O (3g)
Beige powder; yield: 0.18 g (51%); mp > 250 °C; 1H NMR (200 MHz, DMSO-d6): 3.83 (s, 3H, OCH3); 6.85 (d, 1H, J = 8.4 Hz, Ar–H); 7.08 (d, 2H, JAB = 8.8 Hz, Ar–H); 7.54 (s, 1H, Ar–H); 7.59 (d, 1H, J = 8.2 Hz, Ar–H); 7.90 (d, 2H, JBA = 8.8 Hz, Ar–H); 9.66 (bs, 2H, OH); 12.65 (bs, 1H, NH); 13C NMR (50 MHz, DMSO-d6): 55.56, 114.84 (2C), 115.27, 116.11, 120.96, 122.49, 123.00, 128.51 (2C), 145.33, 150.51, 159.04, 161.06, 161.61, 164.72; IR (KBr, cm−1): 3397, 3181, 2937, 1663, 1607, 1521, 1462, 1305, 1258, 1178, 829; anal. calcd. for C16H13N3O4S × H2O (343.36 g/mol): C, 55.97; H, 3.82; N, 12.24; S, 9.34; found: C, 55.98; H, 3.80; N, 12.27; S, 9.36.
4.1.2.12 N-(5-(2-Chlorophenyl)-1,3,4-thiadiazol-2-yl)-3,4-dihydroxybenzamide × H2O (3h)
Beige powder; yield: 0.21 g (61%); mp > 250 °C; 1H NMR (200 MHz, DMSO-d6): 6.86 (d, 1H, J = 8.2 Hz, Ar–H); 7.49–7.62 (m, 4H, Ar–H); 7.65–7.72 (m, 1H, Ar–H); 8.12–8.17 (m, 1H, Ar–H); 9.59 (bs, 1H, OH); 9.80 (bs, 1H, OH); 12.85 (bs, 1H, NH); 13C NMR (50 MHz, DMSO-d6): 115.28, 116.14, 121.05, 122.27, 127.90, 129.25, 130.64, 130.93, 131.16, 131.75, 145.34, 150.60, 157.86, 161.25, 164.84; IR (KBr, cm−1): 3420, 3160, 2969, 1684, 1606, 1527, 1451, 1300, 1203, 753; anal. calcd. for C15H10ClN3O3S × H2O (347.78 g/mol): C, 51.80; H, 2.90; N, 12.08; S, 9.22; found: C, 51.82; H, 2.91; N, 12.10; S, 9.20.
4.1.2.13 N-(5-(3-Chlorophenyl)-1,3,4-thiadiazol-2-yl)-3,4-dihydroxybenzamide (3i)
White powder; yield: 0.19 g (54%); mp > 250 °C; 1H NMR (200 MHz, DMSO-d6): 6.86 (d, 1H, J = 8.2 Hz, Ar–H); 7.53–7.63 (m, 4H, Ar–H); 7.87–7.95 (m, 1H, Ar–H); 8.02 (d, 1H, J = 1.8 Hz, Ar–H); 9.42 (s, 1H, OH); 9.95 (s, 1H, OH); 12.88 (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): 115.26, 116.11, 121.01, 122.22, 125.78, 126.16, 130.24, 131.30, 132.35, 134.08, 145.32, 150.59, 160.08, 160.33, 164.74; IR (KBr, cm−1): 3421, 3365, 3114, 1659, 1617, 1601, 1518, 1300, 1256, 1115, 753; anal. calcd. for C15H10ClN3O3S (347.78 g/mol): C, 51.80; H, 2.90; N, 12.08; S, 9.22; found: C, 51.78; H, 2.89; N, 12.09; S, 9.23.
4.1.2.14 N-(5-(4-Chlorophenyl)-1,3,4-thiadiazol-2-yl)-3,4-dihydroxybenzamide (3j)
White powder; yield: 0.22 g (64%); mp > 250 °C; 1H NMR (200 MHz, DMSO-d6): 6.86 (d, 1H, J = 8.2 Hz, Ar–H); 7.55–7.62 (m, 2H, Ar–H); 7.60 (d, 2H, JAB = 8.4 Hz, Ar–H); 7.99 (d, 2H, JBA = 8.4 Hz, Ar–H); 9.47 (bs, 1H, OH); 9.91 (bs, 1H, OH); 12.84 (bs, 1H, NH); 13C NMR (50 MHz, DMSO-d6): 115.19, 116.06, 120.96, 122.22, 128.50 (2C), 129.21, 129.35 (2C), 135.04, 145.25, 150.50, 159.80, 160.58, 164.66; IR (KBr, cm−1): 3426, 3182, 1681, 1657, 1614, 1603, 1523, 1455, 1298, 1234, 829; anal. calcd. for C15H10ClN3O3S (347.78 g/mol): C, 51.80; H, 2.90; N, 12.08; S, 9.22; found: C, 51.84; H, 2.90; N, 12.06; S, 9.19.
4.1.2.15 N-(5-(4-Bromophenyl)-1,3,4-thiadiazol-2-yl)-3,4-dihydroxybenzamide × 0.5H2O (3k)
Light yellow powder; yield: 0.21 g (54%); mp > 250 °C; 1H NMR (200 MHz, DMSO-d6): 6.85 (d, 1H, J = 8.2 Hz, Ar–H); 7.54–7.61 (m, 2H, Ar–H); 7.73 (d, 2H, JAB = 8.4 Hz, Ar–H); 7.92 (d, 2H, JBA = 8.4 Hz, Ar–H); 9.48 (bs, 1H, OH); 9.89 (bs, 1H, OH); 12.84 (bs, 1H, NH); 13C NMR (50 MHz, DMSO-d6): 115.26, 116.12, 121.01, 122.29, 123.83, 128.78 (2C), 129.63, 132.34 (2C), 145.32, 150.57, 159.90, 160.72, 164.76; IR (KBr, cm−1): 3391, 3199, 1675, 1605, 1521, 1455, 1299, 1209, 1071, 749; anal. calcd. for C15H10BrN3O3S × 0.5H2O (392.23 g/mol): C, 45.93; H, 2.57; N, 10.71; S, 8.18; found: C, 45.92; H, 2.58; N, 10.73; S, 8.19.
4.1.2.16 N-(5-Benzyl-1,3,4-thiadiazol-2-yl)-3,4-dihydroxybenzamide (3l)
Gray powder; yield: 0.19 g (57%); mp > 250 °C (Dec.); 1H NMR (200 MHz, DMSO-d6): 4.36 (s, 2H, CH2); 6.82 (d, 1H, J = 8.2 Hz, Ar–H); 7.24–7.37 (m, 5H, Ar–H); 7.48 (s, 1H, Ar–H); 7.52 (d, 1H, J = 8.2 Hz, Ar–H); 9.38 (s, 1H, OH); 9.88 (s, 1H, OH); 12.57 (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): 35.20, 115.23, 116.04, 120.85, 122.48, 127.06, 128.84 (2C), 128.91 (2C), 137.92, 145.27, 150.40, 159.90, 163.53, 164.67; IR (KBr, cm−1): 3433, 3347, 3063, 1673, 1612, 1527, 1454, 1304, 1218, 750; anal. calcd. for C16H13N3O3S (327.36 g/mol): C, 58.70; H, 4.00; N, 12.84; S, 9.80; found: C, 58.65; H, 3.98; N, 12.80; S, 9.83.
4.1.2.17 3,4-Dihydroxy-N-(5-(phenoxymethyl)-1,3,4-thiadiazol-2-yl)benzamide (3m)
Beige powder; yield: 0.26 g (75%); mp > 250 °C; 1H NMR (200 MHz, DMSO-d6): 5.51 (s, 2H, CH2); 6.84 (d, 1H, J = 8.2 Hz, Ar–H); 7.00 (t, 1H, J = 7.4 Hz, Ar–H); 7.08 (d, 2H, J = 8.2 Hz, Ar–H); 7.33 (t, 2H, J = 8.2 Hz, Ar–H); 7.51 (s, 1H, Ar–H); 7.56 (d, 1H, J = 8.2 Hz, Ar–H); 9.43 (bs, 1H, OH); 9.89 (s, 1H, OH); 12.73 (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): 64.22, 115.08 (2C), 115.26, 116.09, 120.97, 121.65, 122.31, 129.72 (2C), 145.32, 150.54, 157.48, 160.42, 160.73, 164.82; IR (KBr, cm−1): 3391, 3178, 1684, 1601, 1531, 1497, 1455, 1299, 1231, 746; anal. calcd. for C16H13N3O4S (343.36 g/mol): C, 55.97; H, 3.82; N, 12.24; S, 9.34; found: C, 55.99; H, 3.83; N, 12.21; S, 9.35.
4.1.2.18 N-(5-Cyclohexyl-1,3,4-thiadiazol-2-yl)-3,4-dihydroxybenzamide (3n)
Beige powder; yield: 0.20 g (64%); mp > 250 °C; 1H NMR (200 MHz, DMSO-d6): 1.17–1.55 (m, 5H, CH2cyclohexyl); 1.62–1.79 (m, 3H, CH2cyclohexyl); 2.01–2.07 (m, 2H, CH2cyclohexyl); 2.97–3.10 (m, 1H, CH2cyclohexyl); 6.83 (d, 1H, J = 8.2 Hz, Ar–H); 7.50 (s, 1H, Ar–H); 7.54 (d, 1H, J = 8.2 Hz, Ar–H); 9.59 (bs, 2H, OH); 12.52 (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): 25.49 (2C), 25.58, 33.15 (2C), 38.84, 115.20, 116.05, 120.82, 122.65, 145.25, 150.32, 158.92, 164.66, 169.01; IR (KBr, cm−1): 3391, 3294, 2932, 2855, 1686, 1614, 1552, 1524, 1312, 1293, 1193, 656; anal. calcd. for C15H17N3O3S (319.38 g/mol): C, 56.41; H, 5.37; N, 13.16; S, 10.04; found: C, 56.37; H, 5.38; N, 13.15; S, 10.05.
4.1.2.19 N-(5-(Adamantan-1-yl)-1,3,4-thiadiazol-2-yl)-3,4-dihydroxybenzamide (3o)
White powder; yield: 0.25 g (67%); mp > 250 °C; 1H NMR (200 MHz, DMSO-d6): 1.75 (s, 6H, Hadamantyl); 2.02 (s, 9H, Hadamantyl); 6.82 (d, 1H, J = 8.2 Hz, Ar–H); 7.50 (s, 1H, Ar–H); 7.54 (d, 1H, J = 8.2 Hz, Ar–H); 9.74 (bs, 2H, OH); 12.12 (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): 28.09 (3C), 36.07 (3C), 37.37, 42.90 (3C), 115.19, 116.03, 120.80, 122.65, 145.26, 150.35, 158.84, 164.68, 173.44; IR (KBr, cm−1): 3381, 3220, 2906, 2851, 1637, 1607, 1525, 1448, 1296, 1226, 744; anal. calcd. for C19H21N3O3S (371.45 g/mol): C, 61.44; H, 5.70; N, 11.31; S, 8.63; found: C, 61.47; H, 5.69; N, 11.33; S, 8.65.
4.1.2.20 4-(((5-Phenyl-1,3,4-thiadiazol-2-yl)imino)methyl)benzene-1,2-diol (4)
Yellow powder; yield: 0.24 g (53%); mp = 228–229 °C (Dec.); 1H NMR (200 MHz, DMSO-d6): 6.92 (d, 1H, J = 8.2 Hz, Ar–H); 7.39 (dd, 1H, J = 8.2 and 1.8 Hz, Ar–H); 7.51 (d, 1H, J = 1.8 Hz, Ar–H); 7.54–7.58 (m, 3H, Ar–H); 7.93–7.98 (m, 2H, Ar–H); 8.79 (s, 1H, N=CH); 9.65 (s, 1H, OH); 10.18 (s, 1H, OH); 13C NMR (50 MHz, DMSO-d6): 115.02, 115.84, 125.44, 126.23, 127.20 (2C), 129.38 (2C), 130.14, 131.11, 146.01, 151.96, 165.02, 168.56, 174.15; IR (KBr, cm−1): 3533, 2924, 1603, 1585, 1516, 1297, 1291, 1192, 762; anal. calcd. for C15H11N3O2S (297.33 g/mol): C, 60.59; H, 3.73; N, 14.13; S, 10.78; found: C, 60.56; H, 3.74; N, 14.12; S, 10.76.
4.1.2.21 4-(((5-Phenyl-1,3,4-thiadiazol-2-yl)amino)methyl)benzene-1,2-diol (5)
Yellow powder; yield: 0.24 g (80%); mp = 217–218 °C (Dec.); 1H NMR (200 MHz, DMSO-d6): 4.35 (d, 2H, J = 5.4 Hz, CH2); 6.64 (dd, 1H, J = 8.0 and 1.6 Hz, Ar–H); 6.69 (t, 1H, J = 8.0 Hz, Ar–H); 6.79 (d, 1H, J = 1.4 Hz, Ar–H); 7.44–7.52 (m, 3H, Ar–H); 7.73–7.77 (m, 2H, Ar–H); 8.34 (t, 1H, J = 5.4 Hz, NH); 8.85 (s, 1H, OH); 8.94 (s, 1H, OH); 13C NMR (50 MHz, DMSO-d6): 48.21, 115.33, 115.52, 118.84, 126.38 (2C), 129.21 (2C), 129.28, 129.67, 130.99, 144.65, 145.28, 156.00, 168.56; IR (KBr, cm−1): 3431, 3205, 3108, 2955, 1607, 1580, 1573, 1516, 1425, 1362, 1289, 1195, 761; anal. calcd. for C15H12N3O2S (299.35 g/mol): C, 60.19; H, 4.38; N, 14.04; S, 10.72; found: C, 60.17; H, 4.39; N, 14.05; S, 10.71.
4.2 Biology
4.2.1 DPPH free radical–scavenging assay
The DPPH free radical–scavenging activity of the studied compounds was determined according to the method described by Kontogiorgis and Hadjipavlou-Litina [38]. Briefly, 1 mL (0.05 mM) of DPPH solution in methanol was mixed with an equivalent volume of the tested compound [20 μL of the compound solution (in DMSO for 3a–o or in methanol for 3a, 4, and 5), and 980 μL of methanol]. The sample was stored in dark at room temperature. The absorbance was acquired at 517 nm after an incubation period of 30 min. Methanol was taken as the control. IC50 values represent the concentration necessary to obtain 50% of a maximum scavenging activity. Ascorbic acid was used as the positive control. The results are presented as mean ± standard deviation (SD) calculated from the independent triplicate experiments using Microsoft Excel software.
4.2.2 ABTS-scavenging assay
The radical-scavenging activity of synthesized compounds using stable ABTS radical cation was determined according to the method of Re et al. [39] with slight modifications. ABTS•+ was generated by addition of Na2S2O8 (2.45 mM) to 7 mM ABTS solution during 16 h in the dark. For experimental purposes, the obtained solution was diluted with methanol to reach the absorbance of 0.70 ± 0.02 at 734 nm. Tested compounds were dissolved in DMSO to obtain a concentration of 10 mM and then diluted with methanol to prepare a concentration range between 20 and 40 μM. The ABTS•+ scavenging test was carried out by mixing 100 μL of examined compounds solutions prepared at different concentrations with 900 μL of ABTS•+ solution and incubated at ambient temperature for 30 min. Then, the absorbance at 734 nm was measured. The percentage of ABTS•+-scavenging activity was expressed as % radical-scavenging activity = [(Ac − As)/Ac] × 100, where Ac is the absorbance of ABTS•+ solution without the test sample (the blank control) and As is the absorbance of the test sample. The concentration of the tested compound that reduces 50% of the free-radical concentration (IC50) was calculated using the equation obtained from the linear regression analysis.
4.3 Computational details
Theoretical calculations of the investigated compounds were carried out using Gaussian 09 software program [40]. Geometry optimizations of all investigated compounds and their corresponding reactive species such as radicals, radical cations, and anions have been performed using M062X hybrid meta functional [41]. To find the most stable geometries, all possible rotamers are calculated. The geometries of investigated compounds were fully optimized by using the DFT method using the 6–311++g(d,p) basis set. This theoretical model provides satisfactory results in the analysis of a range of organic molecules [42–44]. All geometries were optimized without any symmetry constraints. In addition, vibrational frequency calculations were performed to confirm the minima on the potential surface. The solvent effect of methanol (ε = 32.67) on the calculated structures was investigated by the solvation model based on the quantum mechanical charge Density of a solute molecule interacting with a continuum (SMD) [45]. To obtain thermodynamic descriptors of the antioxidant activity, such as BDE, IP, PDE, structures of not only the neutral molecules but also the corresponding radical cations, radicals, and anions were optimized.
4.4 Antioxidant capacity
Antioxidant activity of phenols has been explained via at least three possible mechanisms [46–55]. These mechanisms will be discussed here in short.
- 1. Hydrogen atom transfer (HAT):
| ArOH → ArO• + H• |
| BDE = H(ArO•) + H(H•) |
The HAT mechanism is presenting the ability of a different phenolic compound to exist as stable radical species after the abstraction of a hydrogen atom from the OH group. The stability of the resulting radical intermediates (ArO·) formed during the abstraction of hydrogen atoms is determined by the fact whether the reaction is going to terminate or continue. It means that if the radical is better stabilized by the resonance delocalization of the unpaired electron within the aromatic ring, its antiradical activity is better. This mechanism is governed by the OH bond dissociation enthalpy (BDE).
- 2. Single electron transfer followed by proton transfer (SET-PT):
| ArOH → ArOH •+ + e− |
| ArOH •+ → ArO• + H+ |
| IP = H(ArOH •+) + H(e−) – H(ArOH) |
| PDE = H(ArO•) + H(H+) – H(ArOH •+) |
The first step of this mechanism is governed by an electron transfer from the antioxidant to the free radical. The second step of this two-step mechanism is abstraction of a proton from the radical cation, which is formed during the first step. The main product in this case is the same as in the HAT mechanism, a new phenoxy radical. The IP and proton dissociation enthalpy (PDE) are thermodynamic parameters responsible for the first and the second step of the SET-PT mechanism.
- 3. Sequential proton loss electron transfer (SPLET)
| ArOH → ArO− + H+ |
| ArO− → ArO• + e− |
| PA = H(ArO−) + H(H+) – H(ArOH) |
| ETE = H(ArO•) + H(e−) – H(ArO−) |
This is also a two-step reaction mechanism. In the first step, phenoxide anion is formed by a heterolytic cleavage of OH bond of the investigated antioxidant. In the second step, this anion loses an electron that leads to formation of the stable phenoxide radical. The thermodynamic descriptors derived from this mechanism are the PA and ETE for the first and the second step, respectively.
4.5 Cyclic voltammetry
The cyclic voltammetric experiments were performed on a PST050 Voltalab instrument. The three-electrode cell consisted of a glassy carbon (GC) disk electrode (3 mm) polished with fine alumina suspension before each curve, with Pt wire counter electrode and a saturated calomel electrode (SCE) as a reference one. Voltammetric measurements were carried out in the potential range +1.6 to −2.1 V, applying scan rates from 0.01 to 2 Vs−1. The solutions of the compounds (0.6–1.5 mM) were prepared in doubly distilled DMF previously dried over molecular sieves for two days; the solutions contained tetrabutylammonium perchlorate (TBAP) as the supporting electrolyte. The solutions were carefully deaerated by purging nitrogen before an experiment, and a strong stream of the gas was maintained during the measurements.
Acknowledgments
The authors are grateful to the Ministry of Education, Science and Technological Development of the Republic of Serbia for financial support (Grant Nos 172016, 172014, 172015, and III43004).
Appendix A Supplementary data
The following is the supplementary data to this article:
Supplementary data (copies of 1H and 13C NMR spectra for 3a–o, 4, and 5 and structures of the most stable conformers of all the investigated compounds) related to this article can be found at https://doi.org/10.1016/j.crci.2019.06.001.