

1 Introduction
During a synthetic program aimed at generating biologically active novel neurological agents, we sought to carry out synthetic transformations from structurally complex bridge-head heterocyclic indoline fused benzazepine class of compound 1 to compound 2 (Fig. 1).
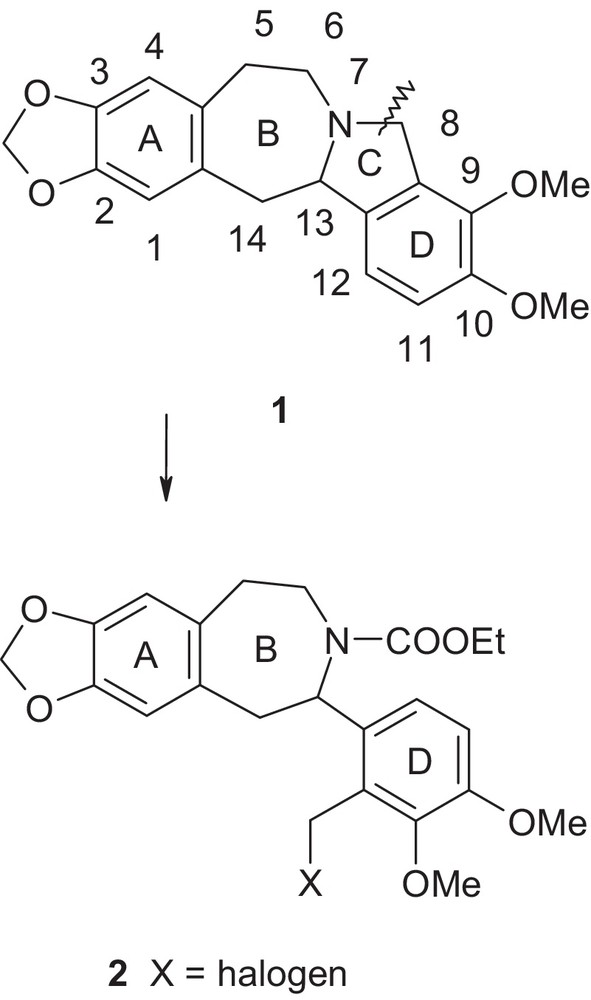
Anticipated transformations from compound 1 to compound 2.
Our anticipation was based on a literature reported similar N–C8 bond cleavage reaction. For example, refluxing biologically active natural product tetrahydroprotoberberine class of compounds 3 in ethyl chloroformate generated compound 4 (method a, Fig. 2; note the numbering systems in compounds 1 and 3 and afterwards maintain the same pattern for the uniformity in the discussion) [1–4]. A modified procedure of treating compound 3 in acetone with ethyl chloroformate in the presence of sodium iodide at room temperature and in dark to generate compound 5 (method b, Fig. 2) had been also reported [5,6].

Reported ring fragmentation procedures.
2 Chemistry
Compound 1 was accessed by following a literature reported procedure [7]. Once in hand, refluxing the compound 1 with excess ethyl chloroformate over a period of 24 h resulted in no appreciable change of the starting material. On the other hand, treatment of compound 1 in acetone with ethyl chloroformate in the presence of sodium iodide resulted in exclusive cleavage, not at expected C8–N bond but at C6–N bond generating compound 6, instead of the anticipated compound 2 (Scheme 1).

Reagents and conditions. (a) Ethyl chloroformate, NaI, acetone, dark, room temperature, 72 h, 70%; (b) LAH, ether, heat, 1 h, 65%.
The mode of cleavage was proven by reduction of compound 6, with lithium aluminum hydride, to the corresponding tertiary base 7. The 1H NMR spectrum of compound 7 displayed a three-proton triplet at δ 1.14 with a coupling constant of 7.5 Hz and a two-proton quartet at δ 2.57 with a coupling constant of 7.5 Hz representing the aromatic –CH2CH3 group. The spectrum also exhibited three-proton singlet at δ 2.49 for the N–CH3 group.
Concurrently, we were also developing a total synthesis of the novel tetrahydrohomoprotoberberine compound 8 (Scheme 2). Commercially available compound 9 was converted to compound 10 [5]. Compound 10, on treatment with sodium cyanide, produced compound 11 that underwent basic hydrolysis (sodium hydroxide) to generate compound 12 without affecting the N-ethoxycarbonyl group. Its removal needed a harsher condition with careful monitoring by heating compound 12 at a high temperature in a sealed tube in the presence of potassium hydroxide to generate compound 13. Subsequent internal cyclization of the amino acid 13 to lactam 14 was carried out in hot decalin. Compound 14 was reduced by lithium aluminum hydride to generate the free base 8.

Reagents and conditions. (a) Ethyl chloroformate, NaI, acetone, dark, room temperature, 72 h, quantitative; (b) NaCN, DMSO, room temp., 1 h, quantitative; (c) NaOH, EtOH–H2O, reflux, overnight, 80%; (d) KOH, EtOH–H2O, sealed tube, 140 °C, overnight, 70%; (e) decalin, 180 °C, 2 h, 90%; (f) LAH, ether, reflux, 1 h, 90%.
The difference in behavior of compounds 1 and 3 in the bond cleavage reactions as well as the availability of compound 8 at this stage prompted us to explore its behavior in the same paradigm. Treatment of compound 8 in refluxing ethyl chloroformate [Scheme 3, step (a) (i)] overnight resulted in the production of a compound (50% yield), displaying a deep blue fluorescence under short UV light. The same product was also generated in 90% yield when Rönsch protocol [Scheme 3, step (a) (ii)] was followed. The 1H NMR spectrum of the compound revealed two one-proton doublets at δ 6.81 and δ 6.85, respectively, with a coupling constant of 13.5 Hz, indicating two vicinal protons of an olefinic moiety of a trans-stilbene system indicating the product might be the compound 15. This was further proven by reduction of compound 15, with lithium aluminum hydride, to corresponding tertiary base 16. In the 1H NMR spectrum of compound 16, the vicinal protons around the olefinic moiety appeared as two doublets at δ 6.82 and 6.89, respectively with a coupling constant of 16.2 Hz. The spectrum also displayed a singlet at δ 2.37 for the N–CH3 group.
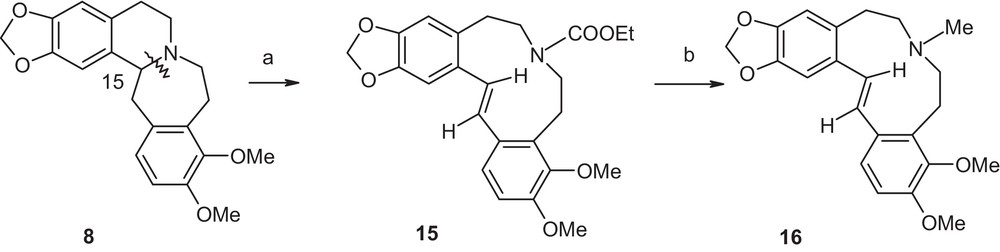
Reagents and conditions. (a) (i) Ethyl chloroformate, reflux, overnight, 50%; or (ii) Ethyl chloroformate, NaI, acetone, dark, room tempearture, overnight, 90%; (b) LAH, ether, heat, 1 h, 90%.
3 Discussion
It had been proposed in the literature [4] that for ring cleavage in compound 3, the reaction is initiated by the nucleophilic attack by the lone pair of electrons on nitrogen in compound 3 at the electrophilic carbethoxy group generating the reactive acyl ammonium intermediate A (Fig. 3).
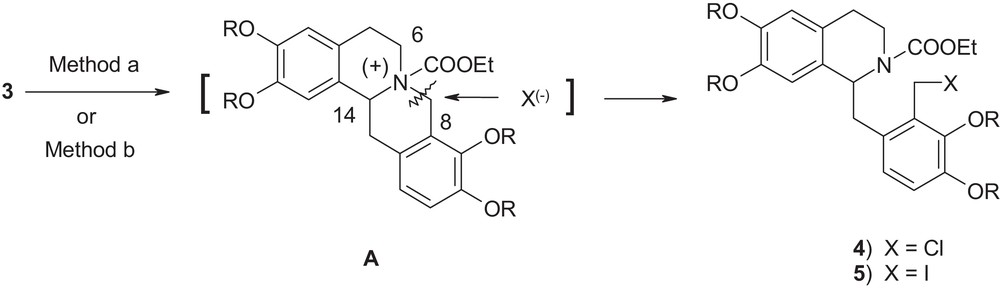
Proposed mechanism for compound 3.
Nucleophilic halide anion (X−) then attacks the sterically less hindered primary benzylic position 8 over sterically congested tertiary benzylic position 14, thus, cleaving the N–C8 bond in an SN2 fashion producing compounds 4 or 5. Thus, the course of the reaction is guided by both electronic and steric factors.
For compound 1, it appears that the initial step also involves the nucleophilic attack by the lone pair of electrons on nitrogen in compound 1 at the electrophilic carbethoxy group generating the reactive acyl ammonium intermediate B (Fig. 4). However, due to the constrained nature of the five-member indoline ring fused to a seven-member azepine ring with a bridge-head nitrogen atom, primary benzylic position 8 assumes an envelop form getting steric resistance from the newly-attached carbethoxy group. Thus, this position becomes less accessible to a nucleophilic attack. Same is true for the sterically congested tertiary benzylic position 13. However, in order to relieve the steric congestion in the newly formed cationic species, the iodide anion, a superior nucleophile then finds the remaining available position 6, though it is not benzylic in nature. Note the fragmentation did not take place in neat ethyl chloroformate where counter anion was less nucleophilic chloride anion. Thus, the course of the reaction in this case is guided by steric factor.
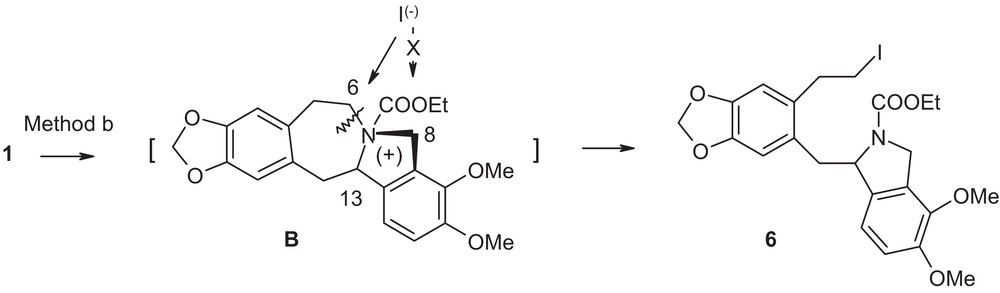
Suggested mechanism for compound 1.
For compound 8, the similar initial step provides the intermediate C (Fig. 5). Note in the intermediate C, position 8 is no longer benzylic in nature, but position 15 remains so.
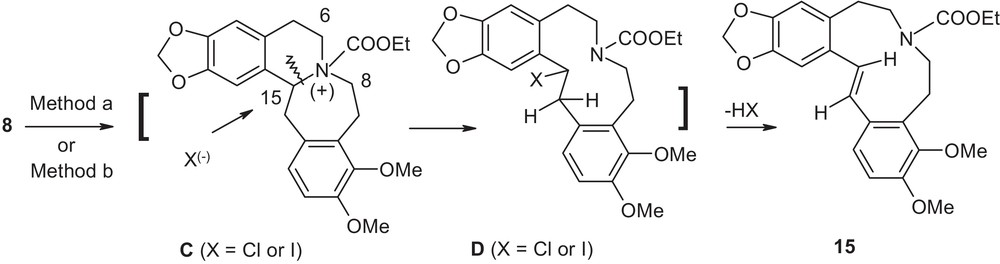
Suggested mechanism for compound 8.
The inherent flexibility of the seven-member ring accommodates the anion (X(−)) to interact with sterically congested position 15 over non-benzylic but sterically accessible position 6 (cf. intermediate B in Fig. 4) affording the second intermediate D. Intermediate D then undergoes concomitant in situ trans-dehydrohalogenation reaction generating the final compound 15. Thus, the course of the reaction in this case is guided mainly by electronic factor with accommodation from steric factor. Interestingly, the fragmentation pattern also allowed an entry to generate an 11-membered flexible cyclic system, that itself might be a driving force in the outcome of the cleavage.
After completion of our previous efforts, we located compound 17 from our chemical library [8]. Interestingly, the treatment of compound 17 in acetone with ethyl chloroformate in the presence of sodium iodide generated both compounds 18 and 19 in 5:1 ratio, in a total yield of 90% (Scheme 4). Both of them were additionally characterized by reducing them to corresponding bases 20 and 21, respectively. As can be seen from the ratio of the initial cleavage products (18 and 19), though both available benzylic sites took part in the ultimate outcome, the attack by the nucleophile at sterically favored primary benzylic position 8 predominated over sterically congested benzylic position 15. The same ratio of products, but with a lower total yield (50%) was obtained when compound 17 was heated in neat ethyl chloroformate.2
Acknowledgements
Authors wish to acknowledge Mr. Greg Hostetler for his help with literature search.
2 Compound 16. A solution of compound 15 (0.043 g, 0.1 × 10−3 mmol) in anhydrous Et2O (10 mL) was slowly added to a refluxing suspension of lithium aluminum hydride (0.015 g, 0.4 × 10−3 mmol) in anhydrous Et2O (100 mL). Heating was continued for another 1 h. After cooling to 0 °C, the reaction mixture was carefully quenched with the slow addition of a saturated aq. Na2SO4 solution. The supernatant was decanted and the inorganic residue was well-washed with Et2O (3 × 10 mL). The combined organic layer was washed successively with water and brine, dried (MgSO4), concentrated and purified via preparative chromatography (eluant: 90% CH2Cl2 10% 7 N methanolic ammonia) to generate compound 17 as a colorless oil (0.030 g, 80%). 1H NMR (200 MHz; CDCl3) δ 2.37 (s, 3H, –NCH3), 3.83 (s, 3H, –OCH3), 3.88 (s, 3H, –OCH3), 5.93 (s, 2H, –OCH2O-), 6.67 (s, 1H, ArH), 6.79 (d, J 8.4 Hz, 1H, ArH), 6.82 (d, J 16.2 Hz, 1H, –CHCH–), 6.88 (s, 1H, ArH), 6.89 (d, J 16.2 Hz, 1H, –CH = CH–), 7.05 (d, J 8.4 Hz, 1H, ArH). MS. m/e 368 (M+H).