

1 Introduction
α-Lipoic acid (α-LA), a widely occurring coenzyme found in prokariotic and eukariotic microorganisms [1] as well as in animals and plants [2], was first isolated by Reed and et al. [3,4].
α-LA is a disulfide derivative of octanoic acid that forms an intramolecular disulfide bond in its oxidized form (Fig. 1b). High electron density resulting from special position of the two sulfur atoms in the 1,2-dithiolane ring confers upon α-LA a high tendency for the reduction of other redox-sensitive molecules according to environmental condition [5,6].

(Color online.) Geometrical structures of β-CD (a) and α-LA (b) optimized at PM6 method.
It has been known for decades to be a crucial prosthetic group of various cellular enzymatic complexes.
α-LA has been characterized as an efficient antioxidant. It has been proposed to be a potential therapeutic agent in the treatment or prevention of different pathologies that may be related to an imbalance of the oxidoreductive cellular status. This occurs in the case of neurodegeneration, ischemia–reperfusion, polyneuropathy, diabetes, AIDS, and hepatic disorder status [7,8].
Although various studies on experimental models, as well as on clinical trials, suggest a beneficial effect of α-LA in the treatment of neurodegenerative conditions, AIDS, and diabetes, the efficacy of α-LA in hepatic and hepatic-associated diseases is controversial.
α-LA is rapidly converted to its metabolite dihydrolipoic acid in most tissues and is in this form against other free radicals.
Recently, Racz et al. [9] have studied the physical–chemical and structural characterization of the encapsulation of α-LA in β-CD.
Cyclodextrins (CDs) are non-toxic cyclic oligosaccharides, consisting of (α-1,4)-linked α-d-glucopyranose units, with a hydrophilic outer surface and hollow hydrophobic interior. The most abundant natural CDs are α-CD, β-CD and γ-CD containing six, seven and eight glucopyranose units, respectively. They have been used in many fields, such as pharmaceutical, food technology, cosmetics and catalysis [10–13]. These compounds have the ability to form host–guest inclusion complexes with a very wide range of guest molecules by molecular complexation, and hence, they can enhance the solubility and stability of the guest molecule [14]. It is well known that different native CDs have the ability to form different inclusion complexes. Thus, the ability of CDs to form inclusion complexes is highly affected by size, shape, hydrophobicity of the guest molecule.
During the course of complexation process, no covalent bond is formed or broken, which strains the physical rather than that the chemical character of process [15]. The complexes improve the characteristics of the drug molecule, such as solubility, the reduction of side effects, chemical stability and bioavailability [15,16]. The driving forces for drug–CD complex formation are hydrogen bonds, van der Waals forces, hydrophobic interactions between the host and guest molecules and entropy released by uncomplexed water molecules from the CD cavity [16].
In the past few years, computational methods in combination with experimental techniques have been mainly focused on the conformational study of the inclusion complex of natural CDs. There are several computational methods used in molecular modelling studies for the complexes of CDs with guest molecules (host–guest systems), such as semi-empirical method, hybrid ONIOM method (our Own N-layer Integrated Orbital Molecular mechanics), Hartree Fock (HF) and density functional theory (DFT) [17–21]. Among these methods, semi-empirical calculations using PM6 level of theory is generally acceptable and frequently used for the structural assignment of CD inclusion complexes, which coincide with the experimental results.
The aim of this work is to give some insights about the location of carboxylic group of α-LA toward primary or secondary hydroxyl of β-CD. We will also determine the driving intermolecular interactions during the formation of such complexes.
2 Computational method
The initial structures of the α-LA was constructed using Hyperchem 7.5 molecular modelling package [22]. The starting geometry of β-CD was taken from Chem-Office 3D ultra (version10, Cambridge Software) [23]. The two structures α-LA and β-CD were then optimized by means of PM6 semi-empirical method prior to using Gaussian09 [24] for all relevant calculations.
The coordinate system used to define the process of complexation is shown in Fig. 2.
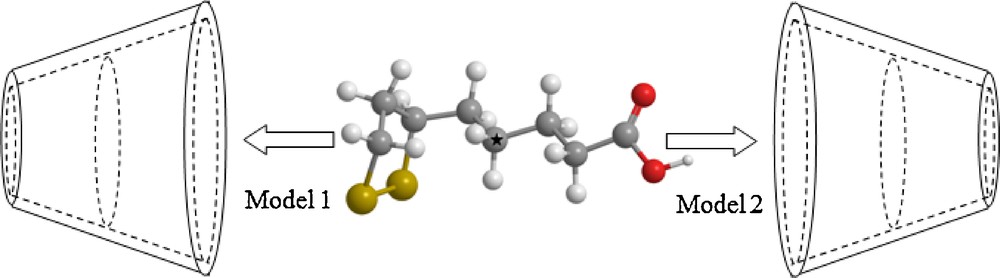
(Color online.) Coordinate systems to describe inclusion processes of α-LA with β-CD.
We followed the method described by Liu and al. to locate the lowest energy minimum of the α-LA/β-CD inclusion complex [20].
Thus, the glycosidic oxygen atoms of β-CD were placed onto the XY plane; their centre was defined as the origin of the coordinate system. The secondary hydroxyl groups of the β-CD were placed pointing toward the positive Z-axis.
The guest molecule was initially placed along the Z-axis. Two possible models of the guest molecule in the complex were considered.
The model in which α-LA entering into the cavity of β-CD from its wide side by cyclic group was called the “model 1”, the other model in which α-LA entering into the cavity of β-CD from its wide side by carboxylic group was called the “model 2” (Fig. 2).
The relative position between the host and the guest was measured by the Z-coordinate of the labeled carbon atom (C*) of the guest (Fig. 2).
Then, the guest was moved into the β-CD cavity along the Z-axis from 10 to −10 Å with 1 Å step. The generated structures at each step were optimized at PM6 methods without imposing any symmetrical restrictions.
In order to find an even more stable structure of the complex, the guest molecule was rotated around Z-axis by 30° from 0 to 360°. Once the preliminary energy minima were determined, the system was re-optimized removing restraint on β-CD and reference atom of α-LA.
To quantify the interaction between host and guest in the optimized geometries, we have evaluated complexation energy (ΔE) using the following formulae [25]:
| (1) |
where Ecomplex, Efree β−CD, and Efree α−LA represent respectively the total energy of the complex, the free optimized β-CD, and the free optimized α-LA energy.
The deformation energy for each component, the guest or the host molecule can be obtained by Eqs. (2) [26]:
| (2) |
where is the single point energy of the component using its geometry in the optimized complex, and is the energy of the optimized geometry of the component.
After that, different levels of calculation were made using HF, DFT and hybrid method (ONIOM2) in vacuo and aqueous solution in the aim to perform a more precise inspection on the geometry and electronic structure of α-LA/β-CD complex.
Then, natural bond orbital (NBO) calculations were carried out to quantify the inter- and intramolecular interactions in particular the establishment of hydrogen bonds between β-CD and α-LA molecules via the determination of the stabilization energy E(2).
At last, based on ONIOM2 optimized geometries, 1H NMR calculations were carried out to quantify the chemical shifts of protons of β-CD, α-LA and their inclusion complex.
3 Results and discussion
In order to look for the optimized structures of α-LA with the lowest energy, the B3LYP level with 6-31G* basis set was employed to perform a conformational search, where dihedral angles were sequentially modified in 12 steps of 30°. As shown in Fig. 3, several stable energy conformers for α-LA were identified and further optimized at the B3LYP/6-31G* level.
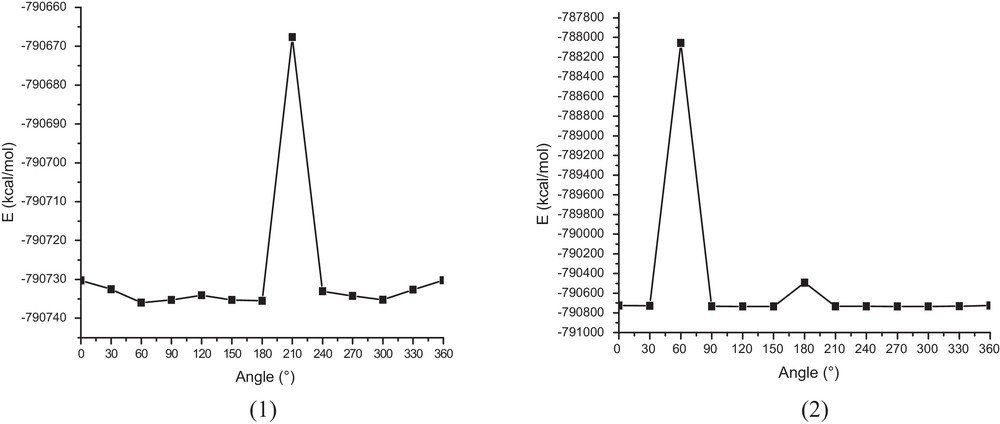
The energies of α-LA calculated at the B3LYP/6-31G* level when scanning dihedral angle. (1) C1–C2–C3–C4, (2) C2–C3–C4–C5.
The most stable conformer was obtained at 60° for dihedral angle C1–C2–C3–C4 (Fig. 3.1). Then, this conformer was used to form the inclusion compound.
The variations of complexation energy in the inclusion process of one molecule of β-CD and one of α-LA at different distance are shown in (Fig. 4a) and at different angle θ are exhibited in (Fig. 4b).
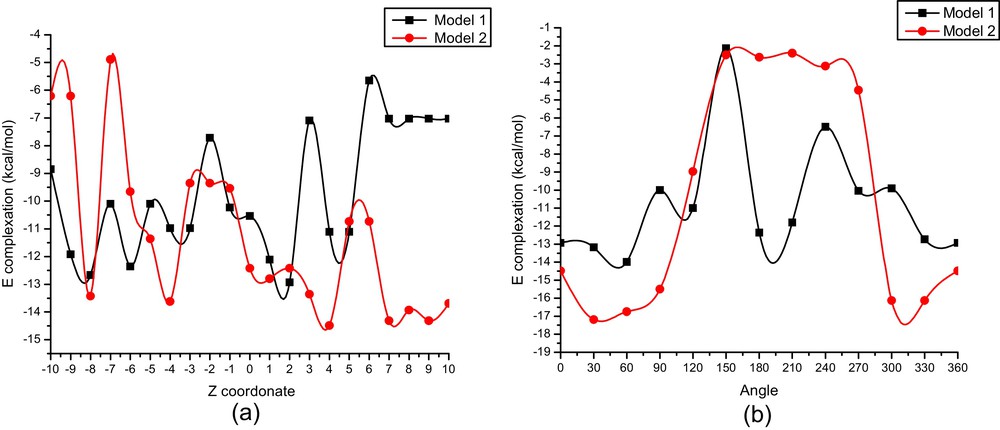
(Color online.) Binding energies of the inclusion complexation of α-LA/β-CD at different positions (Z) (a) and at different angles (θ); (b) for both orientations, PM6 calculations.
The most stable structure of α-LA/β-CD is reached at (2 Å, 60°) for model 1 and at (4 Å, 30°) for model 2.
The calculated energies for the most stable structures obtained by PM6 study in vacuo and aqueous solution are summarized respectively in Table 1.
Complexation energies and thermodynamic parameters of α-LA, β-CD and their inclusion complexes calculated by PM6 method in vacuo and in water.
| α-LA | β-CD | α-LA/β-CD | ||
| Model 1 | Model 2 | |||
| In vacuo | ||||
| PM6 | ||||
| E (kcal/mol) | –114.52 | –1564.63 | 1693.14 | –1696.34 |
| ΔE (kcal/mol) | –13.99 | –17.19 | ||
| Edeformation (α-LA) | – | – | –2.22 | –3.02 |
| Edeformation (β-CD) | – | – | –2.17 | –1.05 |
| H° (kcal/mol) | 18.44 | –941.26 | –835.84 | –838.54 |
| ΔH° (kcal/mol) | –19.70 | –22.78 | ||
| G° (kcal/mol) | –19.64 | –941.26 | –983.68 | –983.68 |
| ΔG° (kcal/mol) | –22.78 | –22.78 | ||
| S° (cal/mol K) | 127.97 | 404.01 | 495.97 | 486.72 |
| ΔS° (cal/mol K) | –36.01 | –45.26 | ||
| In Water | ||||
| PM6 | ||||
| E (kcal/mol) | –125.56 | –1598.20 | –1729.10 | –1736.32 |
| ΔE (kcal/mol) | –5.43 | –12.56 | ||
| Edeformation (α-LA) | – | – | 2.35 | 1.54 |
| Edeformation (β-CD) | – | – | 4.20 | 5.12 |
These results confirm that the complexation energy of model 2 is in favor of 3.20 kcal/mol in vacuo and 7.22 kcal/mol in aqueous solution than model 1.
On the other hand, the results of the investigation of deformation energy reported in Table 1 demonstrate that the α-LA molecule for model 2 requires slightly more energy than that of the model 1 (in vacuo and in aqueous solution) in order to adapt its structure to bind within the cavity of β-CD. This can be supported by the fact that flexibility of the guest structure is one of the important structural requirements for β-CD upon complexation.
The gap energetic between the two orientations obtained with PM6 is increased using single point calculations at B3LYP/6-31G*, M05-2X/6-31G* and HF/6-31G* levels and confirm the preference for the model 2 over the model 1 in vacuo and in aqueous solution (Table 2).
The single point energies and relative energy for the optimized structures of complex α-LA/β-CD in both models in vacuo and aqueous solution.
| α-LA/β-CD | ΔE | ||
| Model 1 | Model 2 | ||
| In vacuo | |||
| B3LYP/6-31G* | |||
| E (kcal/mol) | –3473400.94 | –3473401.51 | |
| ΔE (kcal/mol) | –16.44 | –17.01 | –0.57 |
| M05-2X/6-31G* | |||
| E (kcal/mol) | –3473120.43 | –3473122.38 | |
| ΔE (kcal/mol) | –12.60 | –14.55 | –1.95 |
| HF/6-31G* | |||
| E (kcal/mol) | –3455624.88 | –3455625.93 | |
| ΔE (kcal/mol) | –4.25 | –5.30 | –1.05 |
| E ONIOM2 (B3LYP/6-31G*: PM6) | –792320.85 | –792322.55 | –1.70 |
| In water | |||
| B3LYP/6-31G* | |||
| E (kcal/mol) | –3473425.73 | –3473426.23 | |
| ΔE (kcal/mol) | –7.54 | –8.04 | –0.50 |
| M05-2X/6-31G* | |||
| E (kcal/mol) | –3473143.35 | –3473144.10 | |
| ΔE (kcal/mol) | –8.29 | –9.04 | –0.75 |
| HF/6-31G* | |||
| E (kcal/mol) | –3455662.86 | –34556663.49 | |
| ΔE (kcal/mol) | –3.49 | –4.09 | –0.60 |
| E ONIOM2 (B3LYP/6-31G*: PM6) | –792345.20 | –792359.38 | –14.18 |
3.1 Thermodynamic parameters
To investigate the thermodynamics of the binding process, the statistical thermodynamic calculation were carried out at 1 atm and 298.15 K by PM6. The thermodynamic quantities, the enthalpy change (ΔH), the thermal Gibbs free energy (ΔG) and entropy contribution (ΔS) are given in Table 1. The complex reactions of α-LA with β-CD are exothermic judged from the negative enthalpy changes. And the negative enthalpy changes suggest that both the inclusion processes are enthalpically favorable in nature. In addition, it can be seen that the entropy change (ΔS) of model 1 and 2 are also both negative, this indicates that the formation of the complex becomes an enthalpy-driven process. The two complexation reactions have negative ΔG values and are therefore spontaneous processes, implying that binding interactions are favored. The negative ΔG, ΔH and ΔS values suggest that the formations of β-CD/α-LA inclusion complexes are a spontaneous and enthalpy-driven process. Certainly, since the inclusion reactions happen in aqueous solution, the influence of water molecules on the inclusion process should be very important. However, because of the limitation of our computer, it can hardly calculate the interactions of cyclodextrins systems in aqueous solution. Therefore, the values of thermodynamics calculated have no absolute meaning.
3.2 ONIOM calculations
The weak difference in the complexation energies between the two models obtained with PM6 does not allow determining the nature of the driving forces and their relative contributions. So, we did consider a higher level of calculation.
Thus, starting from the energy minimum structures obtained with PM6 method, we carried out ONIOM2 method fully geometry optimization at level of theory [B3LYP/6-31G*:PM6]. We point out that the high level of calculation was carried out on the α-LA while the low level was applied on the β-CD for the simple reason that β-CD plays only an environment role.
Table 2 it emphasizes the computational results of ONIOM2 study. It is interesting to note that the results indicate that the complexation according to the model 2 is significantly more favorable than model 1 in vacuo and in water which is confirmed to previous results obtained with PM6 method.
Furthermore, it can be seen that the complexation energy of model 2 is in favor than that the model 1 of 1.70 kcal/mol in vacuo and 14.80 kcal/mol in water.
3.3 Natural bond orbital (NBO) analysis
The structures of the energy minimum obtained with ONIOM2 calculations show the presence of several intermolecular hydrogen bond interactions as shown in Fig. 5. In the present study, the hydrogen bond analysis is carried out using NBO approach.
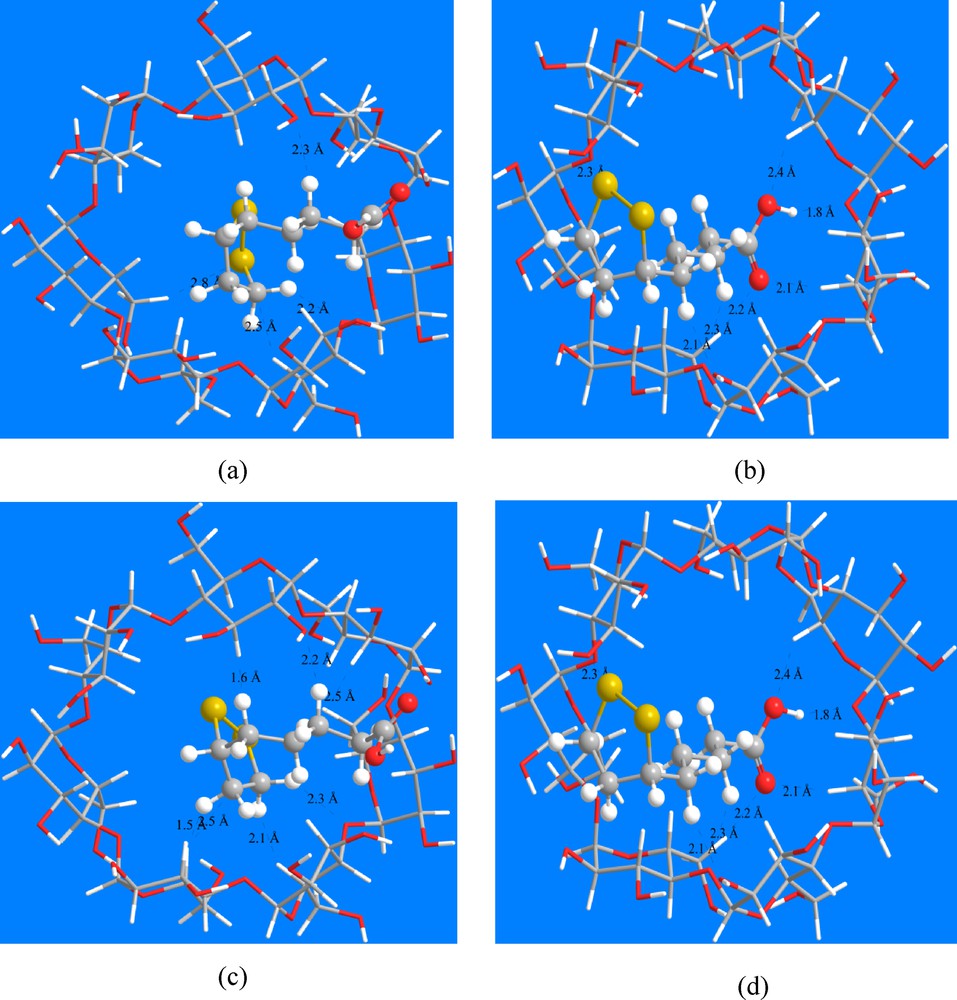
(Color online.) ONIOM2 energy minimized structure of the α-LA/β-CD model 1 (a), model 2 (b) in vacuo, model 1 (c), model 2 (d) in water. Top view. Hydrogen bonds are indicated by dotted lines.
The donor–acceptor interactions of the inclusion complex of α-LA into β-CD cavity were illustrated in Tables 3 and 4. The interaction energies of these contacts are in the range of 1.05–5.93 kcal/mol in vacuo and 0.90–6.75 kcal/mol in water.
Donor–acceptor interactions and stabilization energies E(2) (kcal/mol) of α-LA/β-CD in vacuo.
| Donor | Acceptor | E(2) (kcal/mol) | |
| B3LYP | M05-2X | ||
| Model 1 | |||
| β-CD donor | α-LA acceptor | ||
| δ C 15–H 93 | δ* C 152–H 167 | 3.97 | 3.98 |
| δ C 39–H 121 | δ* C 151–H 165 | 5.35 | 5.32 |
| LP O 55 | δ* C 154–H 171 | 4.33 | 4.99 |
| LP O 56 | δ*C 149–H 163 | 3.31 | 3.74 |
| LP O 74 | δ* C 153–H 169 | 3.25 | 3.83 |
| α-LA donor | β-CD acceptor | ||
| δ C 148–H 161 | δ* C 23–H 102 | 4.82 | 4.94 |
| δ C 151–H 165 | δ* C 39–H 121 | 5.53 | 5.59 |
| δ C 152–H 167 | δ* C 15–H 93 | 4.41 | 4.51 |
| δ C 153–H 169 | δ* C 3–H 81 | 3.64 | 3.71 |
| LP O 157 | δ* O 43–H 126 | 5.79 | 6.70 |
| Model 2 | |||
| β-CD donor | α-LA acceptor | ||
| δ C 21–H 100 | δ*C 153–H 170 | 4.21 | 4.21 |
| δ C 27–H 107 | δ* C 152–H 167 | 4.05 | 4.05 |
| δ C 29–H 109 | δ* C 154–H 171 | 3.41 | 3.41 |
| δ O 56 | δ* O 156–H 173 | 4.94 | 4.94 |
| α-LA donor | β-CD acceptor | ||
| δ C 152–H 167 | δ* C 27–H 107 | 5.93 | 5.93 |
| δ C 153–H 170 | δ* C 21–H 100 | 5.46 | 5.46 |
| δ C 154–H 171 | δ* C 29–H 109 | 5.23 | 5.23 |
| LP O 157 | δ*C 30–H 110 | 2.96 | 2.96 |
Donor–acceptor interactions and stabilization energies E(2) (kcal/mol) of α-LA/β-CD in water.
| Donor | Acceptor | E(2) (kcal/mol) | |
| B3LYP | M05-2X | ||
| Model 1 | |||
| β-CD donor | α-LA acceptor | ||
| δ C 15–H 93 | δ* C 152–H 167 | 4.81 | 4.83 |
| δ C 23–H 102 | δ* C 148–H 161 | 3.62 | 3.63 |
| δ C 39–H 121 | δ* C 151–H 165 | 4.89 | 4.88 |
| LP O 55 | δ* C 154–H 171 | 5.02 | 5.73 |
| LP O 61 | δ* C 148–H 160 | 2.83 | 3.22 |
| LP O 7 4 | δ* C 153–H 169 | 4.90 | 5.52 |
| α-LA donor | β-CD acceptor | ||
| δ C 148–H 161 | δ* C 23–H 102 | 5.77 | 5.85 |
| δ C 149–H 163 | δ* C 11–H 90 | 2.43 | 2.45 |
| δ C 151–H 165 | δ* C 39–H 121 | 5.21 | 5.26 |
| δ C 152–H 167 | δ* C 15–H 93 | 5.07 | 5.19 |
| δ C 153–H 169 | δ* C 3–H 81 | 3.30 | 3.36 |
| LP O 157 | δ* O 43–H 126 | 6.04 | 6.99 |
| Model 2 | |||
| β-CD donor | α-LA acceptor | ||
| δ C 21–H 100 | δ* C 153–H 170 | 3.07 | 3.11 |
| δ C 29–H 109 | δ* C 154–H 171 | 3.33 | 3.33 |
| δ C 33–H 114 | δ* C 149–H 163 | 2.52 | 2.55 |
| LP O 56 | δ* O 156–H 173 | 4.00 | 4.13 |
| LP O 59 | δ* C 151–H 166 | 6.75 | 7.49 |
| LP O 75 | δ*S 158–S 159 | 2.27 | 2.63 |
| α-LA donor | β-CD acceptor | ||
| δ C 152–H 167 | δ* C 27–H 107 | 3.38 | 3.50 |
| δ C 153–H 170 | δ* C 21–H 100 | 4.02 | 4.07 |
| δ C 154–H 171 | δ* C 29–H 109 | 4.76 | 4.85 |
| LP O 157 | δ* C 30–H 110 | 2.21 | 2.54 |
The interactions are in detail:
- • in vacuo: when α-LA plays the role of donor, the important intermolecular hydrogen bond is observed between δ C 151–H 165 and δ* C 39–H 121 with energy equal to 5.53 kcal/mol in model 1 and between δ C 152–H 167 and δ* C 27–H 107 with energy of 5.93 kcal/mol in model 2. On the other side, when the α-LA is an acceptor, the important H-bond is formed between δ C 39–H 121 and δ* C 151–H 165 with energy 5.35 kcal/mol in model 1 and between δ O 56 and δ* O 156–H 173 with energy 4.94 in model 2;
- • in water: when α-LA plays the role of donor, the important intermolecular hydrogen bond is observed between LP O 157and δ* O 43–H 126 with energy equal to 6.04 kcal/mol in model 1 and between δ C 154–H 171 and δ* C 29–H 109 with energy of 4.76 kcal/mol in model 2.
On the other side, when the α-LA is an acceptor, the important H-bond is formed between LP O55 and δ*C 154–H 171 with energy 5.02 kcal/mol in model 1 and between LP O59 and δ* C 151–H 166 with energy 6.75 in model 2.
3.4 GIAO/DFT calculation
Based on ONIOM2 optimized geometries, the Gauge-Including Atomic Orbital (GIAO) method as implemented in Gaussian 09 was employed for 1H NMR calculations and by employing the density functional theory B3LYP at 6-31G* basis set with using corresponding TMS shielding calculated at the same theoretical level as the reference. The solvent effects have been investigated using the PCM method for water as a solvent (ɛ = 78.35) [27–29].
1H NMR calculations of isolated species of α-LA, β-CD and their more stable inclusion complex are presented in Table 5 and Fig. 6.
Comparison of experimental and theoretical 1H chemical shifts of the inclusion complex of α-LA/β-CD calculated at the B3LYP/6-31G* level of the theory.
| α-LA | β-CD | α-LA/β-CD | Δδ | ||
| Model 2 | Exp [9] | ||||
| Ha | 2.63 | 2.33 | 2.30 | 0.03 | |
| Hb | 1.70 | 2.66 | – | – | |
| Hc | 1.13 | 1.77 | 1.46 | 0.31 | |
| Hd | 1.92 | 3.50 | 1.63 | 1.87 | |
| He | 4.43 | 3.41 | – | – | |
| Hf | 2.48 | 2.77 | 2.47 | 0.30 | |
| Hg | 2.93 | 4.01 | 3.22 | 0.79 | |
| H1 | 5.37 | 5.15 | – | – | |
| H2 | 3.99 | 3.87 | 3.64 | 0.23 | |
| H3 | 4.23 | 4.04 | 3.99 | 0.05 | |
| H4 | 3.14 | 3.26 | 3.59 | –0.33 | |
| H5 | 4.34 | 4.22 | 3.83 | 0.39 | |
| H6 | 3.74 | 3.63 | 3.87 | –0.24 |

(Color online.) 1H NMR chemical shifts (ppm) of α-LA (a) and β-CD (b) before and after complexation calculated by GIAO method at B3LYP/6-31G*.
As can be seen from Table 5, the protons signals of Hb, d, e, g of α-LA after complexation shifted to a large field. Whereas the signals of the other protons of α-LA and all protons of β-CD have lowest chemical shifts changes.
In addition, it is commonly accepted that this changes in chemical shifts are based on hydrogen bond interactions.
Also, the calculated NMR data shows very detailed information about the chemical shifts allowing us to identify each hydrogen atom.
The comparison of the computed shielding obtained for the optimized structures with the experimental chemical shifts [9] is illustrated in Table 5 and Fig. 7.
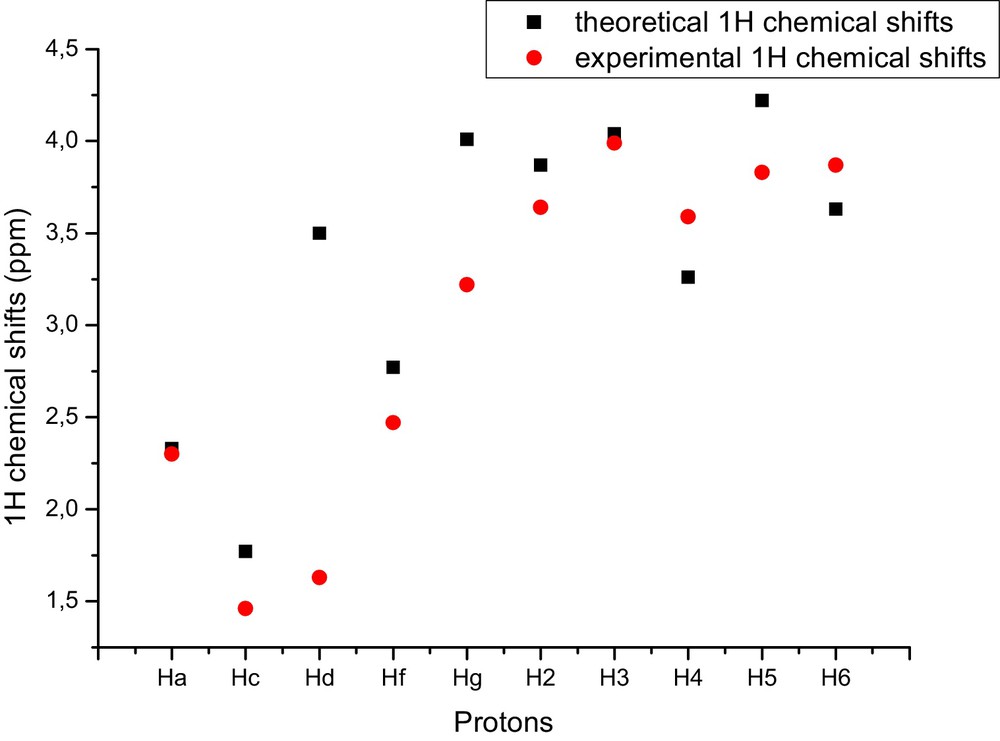
(Color online.) Comparison of experimental and theoretical 1H chemical shifts of the inclusion complex of α-LA/β-CD calculated at the B3LYP/6-31G* level of the theory.
From Table 5, the largest difference between the theoretical chemical shifts and experimental chemical shifts is seen in protons Hd, g.
4 Conclusion
The inclusion process for α-lipoic acid with β-CD was studied according two models using quantum mechanics PM6, B3LYP level theory with 6-31G* basis set and ONIOM2 (B3LYP/6-31G*: PM6) hybrid calculations. The minimum energy structure for each model was localized with PM6 method. The affinity of these minimum energies structures was carried out with DFT and ONIOM methods. The DFT and ONIOM results show that the model 2 is preferred according to complexation energy, in which the carboxylic group is near to primary hydroxyls of β-CD. The analyses of the thermodynamic calculations indicate that the negative ΔG, ΔH and ΔS values suggest that the formations of β-CD/α-LA inclusion complexes in vacuo are a spontaneous and enthalpy-driven process. The NBO analysis shows that the driving forces for the complexes formation was due to the intermolecular hydrogen bonds. Finally, 1H NMR computational methods can be used as a support to indicate the structures for supramolecular arrangements.
Acknowledgments
This study was supported by the Algerian minister of higher education and scientific research through project CNEPRU (No. B01520090002) and PNR (8/u24/4814).