

1 Introduction
On account of the ever-increasing concern for environment protection, the safe treatment and detoxification of tetrachloromethane and other organo-chlorinated pollutants has acquired great importance. Several methods, including incineration, biological treatment, photo-catalytic decomposition, and catalytic hydrodechlorination [1–5], have been proposed for the treatment of chlorinated organic wastes. Among these methods, used for disposal or recycling of chlorinated hydrocarbons, catalytic hydrogenolysis of carbon–chlorine bond (referred to as hydrodechlorination, HdCl) is regarded as the most universal and promising method. HdCl is emerging as a simple, safe and non-destructive alternate technology whereby the chlorinated waste can be converted into hydrocarbons, products of commercial value. Additionally, HdCl operates at low temperatures, so the process is economically viable.
Even though, all Group VIII metals are known for their CCl bond cleavage and hydrogenation ability [6], palladium occupies a unique position in this group by virtue of its superior activity and high selectivity towards the desired hydrocarbon, i.e. non-chlorine containing, products. However, there are some problems such as the improvement of the stability of Pd catalysts and selectivities to target products, i.e., C1C5 hydrocarbons, remaining to be met. The main drawback for practical application is rapid deactivation of palladium catalysts during HdCl of CCl4 reaction, although they show a very high initial catalytic activity [7–15]. Therefore, the development of new catalysts with highly selective dechlorinating activity and high stability becomes the strategy for the hydrodechlorination of chlorinated hydrocarbons.
As commonly observed in the hydro-treating catalytic reactions, catalytic performance such as activity, selectivity, and resistance towards catalyst deactivation are strongly dependent on the nature of the catalyst. Therefore, various catalyst systems such as noble metals, non-noble metals, and alloy catalysts have been investigated for HdCl reactions. It is known that the HdCl activity of supported palladium catalysts is affected by many factors: the morphology of metal particles, the nature of the support [16–18] and the presence of a second metal in active phase [19–25].
Bimetallic catalysts are very promising as they feature an interesting catalytic behaviour with respect to monometallic systems [26]. Many bimetallic systems demonstrate enhanced properties in terms of selectivity and activity as well as resistance to poisoning and/or metal sintering. Bimetallic palladium–gold catalysts find industrial application, for instance in the selective hydrogenation of various organic compounds [27], production of vinylacetate [28], hydrodechlorination of chlorofluorocarbons [29] and trichloroethene [30].
The ultimate size of the metal particles and good homogeneity of active metal phase (namely Pd–Au alloy) are essential for improving the catalytic properties of palladium. Our previous results show that for supported Pd–Au catalysts prepared by impregnation techniques a satisfactory extent of Pd–Au mixing was not achieved [23,31]. The reductive deposition of gold to palladium catalysts resulted in a significantly higher, although still not perfect, degree of Pd–Au alloying [32]. Our recent paper [33] deals with similarly prepared carbon-supported Pd–Au catalysts investigated in CCl4 hydrodechlorination, where attention was focused on the negative effect of alloy inhomogeneity on the catalyst's stability. Although this defect could be largely reduced by removing unalloyed palladium species (by treatment with nitric acid [33]), nevertheless further progress in this research could be made by application of another catalyst's preparation technique which would lead to a still better Pd–Au alloying.
In the present work we decided to prepare palladium and palladium–gold catalysts supported on mesoporous active carbons by the “water-in-oil” reverse microemulsion method [34,35]. In this special method, nanoparticles with precisely defined size are obtained. Nanoparticles are formed by reduction/co-reduction of metal ions present inside nanodroplets of aqueous solution of metal salts stabilized by a surfactant in a non-polar solvent. Proper adjusting of metals ions concentration, concentration and type of reducing agent as well as composition of microemulsion (especially water to surfactant molar ratio) allow to obtain metal particles precisely predicted in size in a very narrow range (which is difficult to obtain in traditional synthesis routes, e.g., by impregnation). Our recent studies showed that such prepared Pd–Au catalysts are homogeneous and active in the reaction of hydrogenation of cinnamaldehyde [36]. The present work is aimed at the performance of similarly prepared carbon-supported Pd–Au catalysts in the hydrodechlorination of tetrachloromethane. High degree of alloy homogeneity attainable by using the “water-in-oil” reverse microemulsion method should allow us to establish more precisely the activity pattern as a function of Pd–Au composition. Our previous work [33] indicated a number of synergistic effects associated with palladium alloying with Au in HdCl of CCl4, however uncertainty as to the degree of Pd–Au homogeneity did not allow us to render more rigorous relations.
2 Experimental
2.1 Catalyst preparation characterization
Two types of commercial active carbons were used as catalyst's supports: activated pyrocarbon Sibunit (Novosibirsk, Russia) [37] and furnace black Vulcan XC-72 (Cabot Corporation) [38]. Before preparation of catalysts, the active carbon Sibunit was washed with a boiling mixture of concentrated HCl and HF, rinsed with large amounts of redistilled water and dried at 253 K in an air oven for 12 hours. Carbon Vulcan XC-72 was used without any purification. The properties of the supports are given in Table 1. It is evident that the Vulcan carbon is characterized by a larger average size of pores.
Characteristics of active carbons used in this worka.
| Measure | Sibunit | Vulcan XC72 |
| Grain size, μm | ∼10 | 0.1–0.3 |
| Surface area (BET), m2/g | 308 | 228 |
| t-plot micropore area, m2/g | 8 | 24 |
| Total pore volume (BJH desorption), cm3/g | 0.890 | 1.770 |
| Mezopore volume, cm3/g | 0.841 | 1.722 |
| H.-K. micropore volume, cm3/g | 0.029 | 0.048 |
| H.-K. micropore width, nm | 0.97 | 0.74 |
| Average pore width (BJH desorption), nm | 5 | 14 |
a The surface areas of the supports, their pore volumes and diameters were determined from desorption isotherms of nitrogen adsorbed at 75 K after evacuation at 623 K for 5 h on ASAP 2020 (Micromeritics, USA).
The catalysts consisting of the same palladium loading equal to 2 wt% Pd and the growing Au content were prepared by means of the reverse “water-in-oil” microemulsion method according to the procedure described previously [39,40]. In the PdAu/carbon catalysts the molar ratio of Pd:Au ranges were from 95:5 up to 70:30. The monometallic 2 wt%Pd/C (Pd100) and 2 wt%Au/C (Au100) catalysts (for catalyst notation, Table 2) were also prepared using the same preparation procedure. The reverse micellar solutions were prepared using surfactant dioctyl sulfosuccinate sodium salt (AOT, pure 98%, Fluka) and heptane (pure, Aldrich) as the oil phase. The aqueous solutions of PdCl2 (99.9% Grade, Johnson Matthey) and HAuCl4 (pure, Avantor) were used as the precursors. Hydrazine hydrate (reagent grade 98%, Aldrich) was applied as a reducing agent. Taking into account our previous studies [39,40] the value of molar ratio of aqueous solution (PdCl2 and/or HAuCl4) to AOT surfactant W = 5 was applied.
Characteristics of Pd–Au/Sibunit carbon and Pd–Au/Vulcan carbon catalysts.
| Catalysta | wt% | Metal particle size, nm | ||
| Pd | Au | SEM | XRDb | |
| Sibunit series | ||||
| 2 wt%Pd100/Sibunit | 2.00 | – | 4.7 (257)c | ∼5 |
| 2.19 wt%Pd95Au5/Sibunit | 2.00 | 0.19 | 4.6 (203)c | ∼5 |
| 2.41 wt%Pd90Au10/Sibunit | 2.00 | 0.41 | 4.5 (391)c | ∼7 |
| 2.65 wt%Pd85Au15/Sibunit | 2.00 | 0.65 | 4.4 (425)c | ∼5 |
| 3.59 wt%Pd70Au30/Sibunit | 2.00 | 1.59 | 4.2 (121)c | ∼5 |
| 2 wt%Au100/Sibunit | – | 2.00 | n.m.d | ∼9 |
| Vulcan XC72 seriese | ||||
| 2 wt%Pd100/Vulcan | 2.00 | – | 6.7 | 5.5–6.0 |
| 2.41 wt%Pd91Au9/Vulcan | 2.00 | 0.41 | 5.5 | 5.5 |
| 3.59 wt%Pd71Au29/Vulcan | 2.00 | 1.59 | 5.2 | 4.0–4.5 |
| 2 wt%Au100/Vulcan | – | 2.00 | 8.2 | 8.0–9.0 |
a In the notation W wt% PdXAuY, W represents overall metal (Pd + Au) weight loading. X and Y denote atomic percentages of Pd and Au in the metal phase supported on carbons.
b From the broadening of XRD profiles (111 and 200).
c Numbers in the parentheses indicate quantities of measured metal particles.
d Not measured.
e Characterization taken from [36].
In the first stage of the preparation of the catalysts, AOT was dissolved in heptane, then an aqueous solution of precursor(s) was introduced to heptane-surfactant mixture and stirred to obtain a transparent yellow/orange suspension. To this microemulsion, hydrazine was added directly, the hydrazine to metal(s) molar ratio was equal to 20. The reduction of metal ions proceeded quickly and the colour of the liquid changed into black. Then, carbon support (Vulcan XC72 or Sibunit) was introduced into microemulsion and the system was kept under stirring for 1 h. Then, destabilization of microemulsion was carried out by introducing the THF solvent at a very slow rate, 0.25 cm3/min using an automatic syringe pump with the solution vigorously stirred. In the preparation of 1 g of catalyst with 2 wt % metal loading on carbon support it takes ca. 1–1.5 h. Upon addition of THF, the colour of liquid gradually changed from black to grey and finally the liquid phase was colourless, showing complete deposition of metal particles on the support. The obtained catalyst was separated by filtration, dried in air and washed with acetone, then with methanol to remove surfactant and heptane. Finally, the catalyst was washed with water to remove chloride ions and then dried at 393 K in air.
The list of the catalysts used in this work, their metal loadings, Pd–Au alloy composition, metal particle/crystallite size from SEM and XRD, respectively, are reported in Table 2. Characteristics of the Vulcan-supported Pd–Au catalysts were taken from [36].
Reduced (in a flow of 20%H2/Ar mixture, 50 cm3/min, at 433 K) and post-reaction Pd–Au/Sibunit samples were investigated by X-ray diffractometry (Rigaku Denki, Ni-filtered Cu Kα radiation). The obtained XRD profiles were fitted to an analytical function of the PEARSON-VII type using commercial PEAKFIT software. For phase identification and calculation of the phase composition, centroids of the fitted profiles were used, and for the calculation of the crystallite size, the half-widths of the fitted profiles were taken.
Electron Microscopy (SEM) studies were performed by means of Field Emission Scanning Electron Microscope JEOL JSM-7500 F. Two detectors were used and the images were recorded in two modes. The secondary electron detector provided SEI images, and the backscattered electron detector provided BSE (COMPO) micrographs. To prepare the particle size distribution diagram and estimation of the average particle size the particles (100 or more) were manually counted using micrographs registered in BSE (COMPO) mode. The counting was carried out using several electron micrographs for each catalyst taken at 200,000 × magnification, where metal particles very well contrasted with the support and they were clearly observable. On these micrographs registered in BSE (COMPO) mode the accuracy of the particle size scale was 0.5 nm (the particle size 3 nm corresponds to 1.5 mm on the micrograph).
Temperature-programmed hydride decomposition (TPHD) experiments were performed in a flow system, with data acquisition by a microcomputer. After reduction in 10% H2/Ar, the catalyst sample (∼0.25 g) was cooled to ∼273 K and subjected to subsequent temperature-programmed study in 10% H2/Ar flow, ramping the temperature from ∼273 K to 393 K, at 8 K/min. Since the samples had already been reduced, the aim of such experiments was to monitor hydrogen evolution in the process of β-hydride decomposition [41].
TPH-MS (temperature-programmed hydrogenation followed by mass spectrometry) runs with post-reaction catalyst samples were carried out in a flowing 20%H2/He mixture (25 cm3/min) at a 10 K/min ramp to 1100 K and followed by mass spectrometry (MA200, Dycor-Ametek, Pittsburgh, USA). Principal attention was paid to m/z 36 and 38, which are suggestive of HCl liberation from catalysts used.
2.2 Catalytic hydrodechlorination of tetrachloromethane
The reaction of CCl4 hydrodechlorination was carried out in the manner previously described [33]. Briefly, prior to the reaction, the catalyst charge (always ∼0.24 g) was dried at 373 K in an argon flow and reduced in flowing 20% H2/Ar (50 cm3/min), with a temperature ramp from RT to 433 K, and keeping the catalyst at 433 K for 2 h.
The reaction of HdCl of tetrachloromethane, provided from a saturator maintained at 273 K and bubbled in a flow of H2/Ar mixture (29 cm3/min), was carried out at 423 K and the H2:CCl4 ratio ∼14:1. The partial pressures of the reaction mixture were: CCl4 4.3 kPa, H2 60.5 kPa, Ar 36.5 kPa. Contact time (V/F) ∼0.5 s was applied. The HdCl reaction was followed by gas chromatography (HP 5890 series II with FID and a 5% Fluorcol/Carbopack B 10 ft from Supelco). A typical run lasted ∼20 h. The agreement in final conversion was fair, ±5%. Much better repeatability was found for product distribution, where final selectivities toward major products were in good agreement ±2%. Since all catalyst charges (∼0.24 g) contained nearly identical amounts of palladium, the conversion level of CCl4 was the first direct measure of the catalytic activity per gcat or per gPd. Very small catalyst grains (Table 1) allow us to neglect the possibility of internal diffusion limitations, which were found to play a role in the HdCl of 1,2-dichloroethane on carbon-supported Pd–Ag catalysts [42]. External diffusion limitations were also disregarded on the basis of additional kinetic experiments, as reported in [33]. Blank kinetic experiments with carbon supports showed very low conversions, ∼0.2%.
3 Results and discussion
3.1 Characterization of carbon-supported Pd–Au catalysts
The description of Pd–Au/Vulcan catalysts in Table 2 comes from [36]. It appeared that the catalysts contained quite small metal particles (crystallites), between 4 and 9 nm. As expected, the preparation method used in that work led to preparation of well-mixed uniform Pd–Au alloy crystallites (EDS and STEM results), characterized by a relatively narrow particle size distribution [36].
X-Ray diffraction, electron microscopy and temperature-programmed hydride decomposition (TPHD) methods were employed to characterize the state of Sibunit carbon-supported Pd–Au alloys. Again, the reverse “water-in-oil” microemulsion method led to preparation of homogenized Pd–Au alloy crystallites characterized by a relatively narrow particle size distribution (Table 1). Fig. 1 shows XRD profiles of reduced (and post-reaction, discussed further in text) samples of Pd–Au/Sibunit catalysts. Maxima of the profiles very well reflect the nominal compositions of Pd–Au alloys (based on the Vegard's law). Symmetrical reflections also indicate good alloy homogeneity and their broadness with application of the Scherrer formula led to calculation of crystallite size which appeared reasonably comparable (with one exception for Pd90Au10/Sibunit) with respective data obtained from electron microscopy (Fig. 2 a–d, and Table 2).
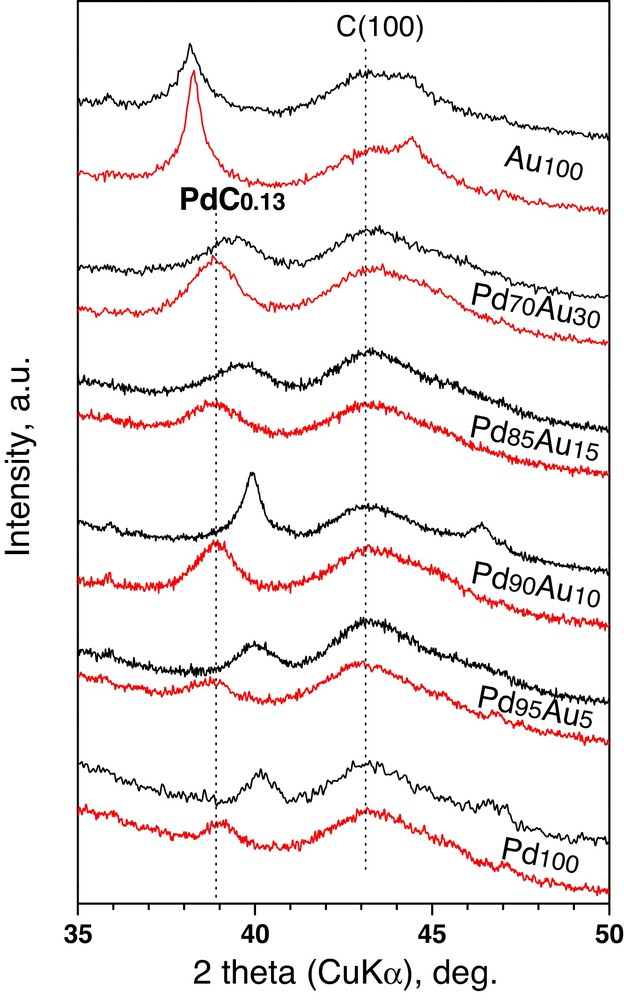
(Colour online.) XRD profiles of reduced and post-reaction samples of Sibunit carbon-supported Pd–Au catalysts. For catalyst notation see Table 2. Basic positions of carbon and PdC0.13 are marked.

(Colour online.) SEM images and metal particles size distribution diagrams (insets) of Pd–Au/Sibunit catalysts registered at magnification 200,000. (a) Pd100, (b) Pd90Au10, (c) Pd85Au15, (d) Pd70Au30.
Very good degree of Pd–Au alloying was also confirmed by the results of TPHD experiments, where decomposition of hydride phases was investigated. The well-known propensity of palladium to form β-hydride phases, characterized by respectable H/Pd bulk ratios (typically, up to ∼0.6 [43]) is expected to be gradually reduced by alloying with other elements, e.g. with gold. Luo et al. (Fig. 11 in [44]) measured the isotherms of hydrogen absorption and desorption for Pd–Au alloys at 303 K. Considering the plateau regions of different alloy compositions it is seen that the hydride phase no longer forms for Au contents higher than 15 at%. This is in agreement with the previously constructed phase diagram at 298 K, determined from the lattice parameters of the coexisting dilute (α) and hydride (β) phases [45]. Disappearance of the coexisting phases at 17 at% Au indicated the critical composition for the β-hydride formation at this temperature. Interestingly, the expansion of palladium lattice by alloying with gold results in the decrease of H2 equilibrium pressure for desorption. This tendency corresponds to a higher stability of PdAuH phases compared to PdH, so under conditions of TPHD runs, when the experiments were carried out at a constant flow of 10% H2/Ar mixture, PdAuH should be decomposed at higher temperature than PdH. In agreement with this expectation, Fig. 3 shows that indeed the decomposition of hydride phases is gradually reduced with alloying with gold. H/Pd ratio decreases from 0.29 for Pd100 to 0.18 and 0.12, for Pd95Au5 and Pd90Au10, respectively. Very low amounts of desorbed hydrogen uptake observed for Pd85Au15/Sibunit and Pd70Au30/Sibunit catalysts (∼0.03), are in line with the critical composition for PdAuH formation observed by Flanagan and co. [44,45]. In addition, the decomposition peaks are quite narrow, suggesting that the Pd–Au crystallites are uniform with respect to their bulk composition and particle size [41].
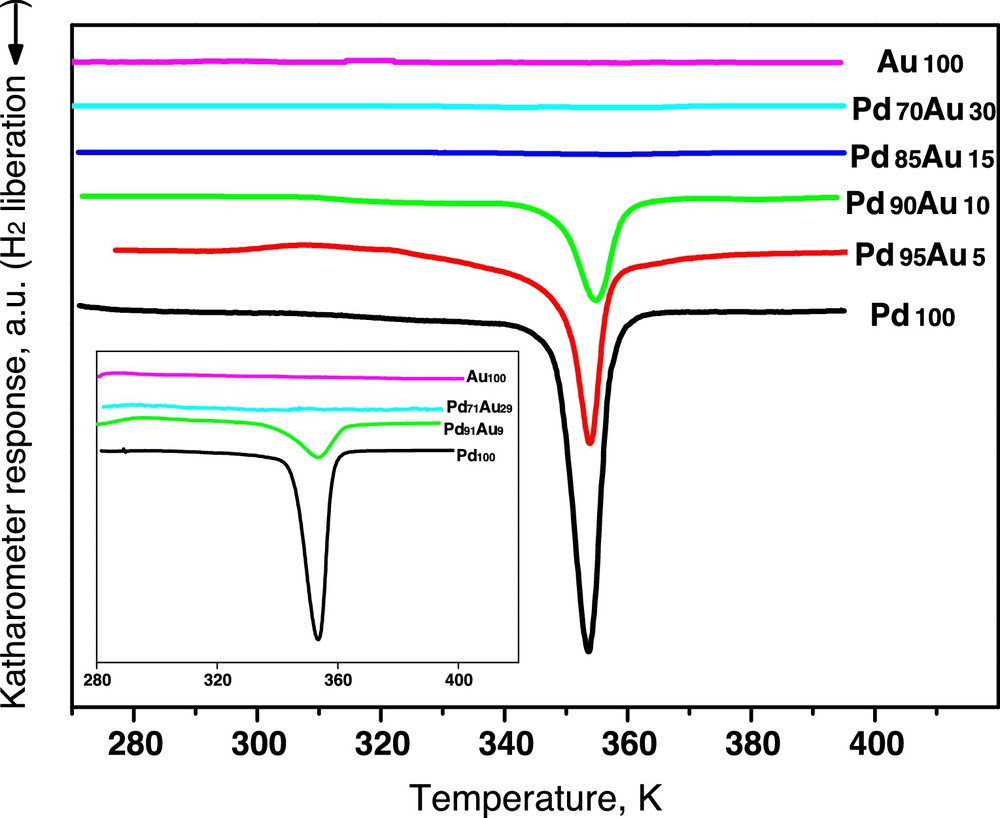
(Colour online.) Temperature-programmed hydride decomposition profiles of Pd–Au/Sibunit catalysts. Inset: results for Vulcan carbon-supported Pd–Au series.
3.2 Performance of carbon-supported Pd–Au catalysts in CCl4 hydrodechlorination
All catalysts were screened in the reaction of CCl4 hydrodechlorination at 423 K, i.e. at a temperature higher than that used in our previous studies [33] performed on Sibunit carbon-supported Pd–Au catalysts (363 K). The decision of increasing the reaction temperature arose from previous observation of Russian researchers that the time of stable catalyst performance generally increased as the reaction temperature was increased [7,8,10]. Our preliminary experiments confirm that observation. Kinetic runs performed at 363 K showed much less stable catalytic performance than those carried out at 423 K, so only the latter results shall be presented and discussed.
Fig. 4 shows the time on stream behaviour of all tested carbon-supported Pd–Au catalysts in CCl4 hydrodechlorination. The overall conversion of CCl4 on Au100/Sibunit was found quite significant, higher than that of Pd100/Sibunit. In the light of our previous experience [33] and the behaviour of Au/Vulcan (Fig. 3, inset), this unexpected (although repeatable) result acquires an unambiguous interpretation, supported by additional experimentation. Suspending this problem to future studies, we focus here on the effect of introducing gold to palladium.
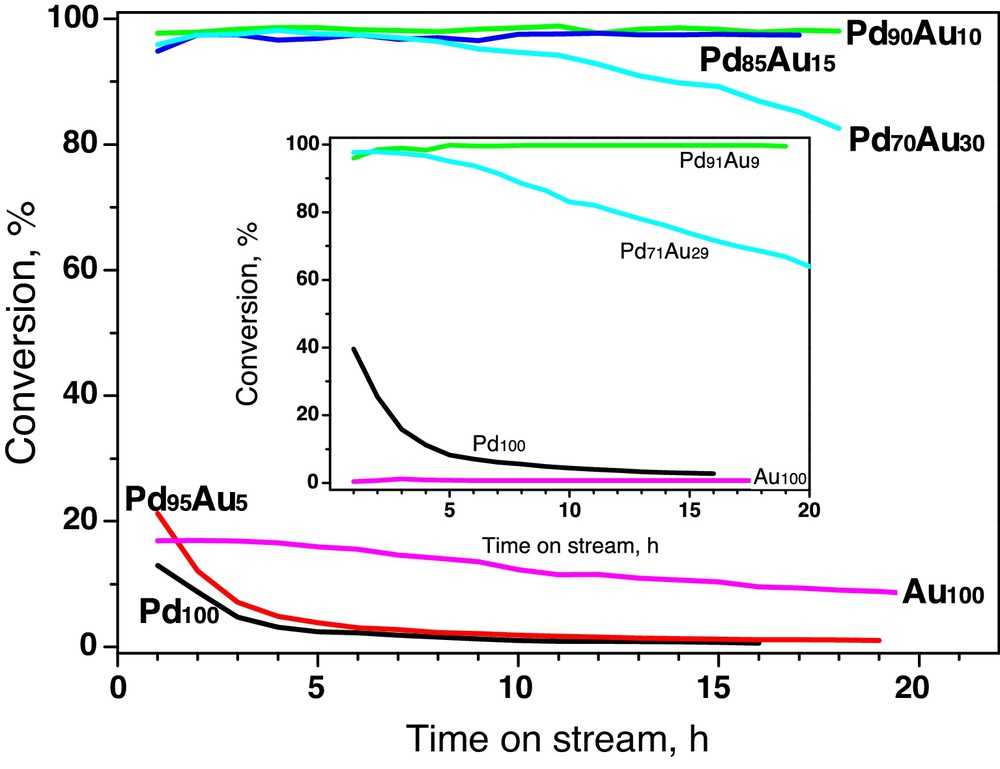
(Colour online.) Conversion changes during hydrodechlorination of CCl4 for Sibunit carbon-supported Pd–Au catalysts. Inset: time-on-stream behaviour of Vulcan carbon series. All samples ∼0.24 g.
Fig. 4 shows that irrespective of the kind of carbon support the performance of Pd100 suffers from rapid deactivation, in line with other reports [7–15,33]. Palladium deactivation is paralleled by variations in product distribution. One observes a systematic growth of methane (Fig. 5) and the role of dimeric C2HxCly products which start to play a dominant role when the catalyst is gradually more deactivated (not presented). A similar situation is observed for the Pd95Au5/Sibunit (Figs. 4 and 5). Apparently, insufficient amount of added gold leads only to minor changes in the catalytic behaviour of palladium.
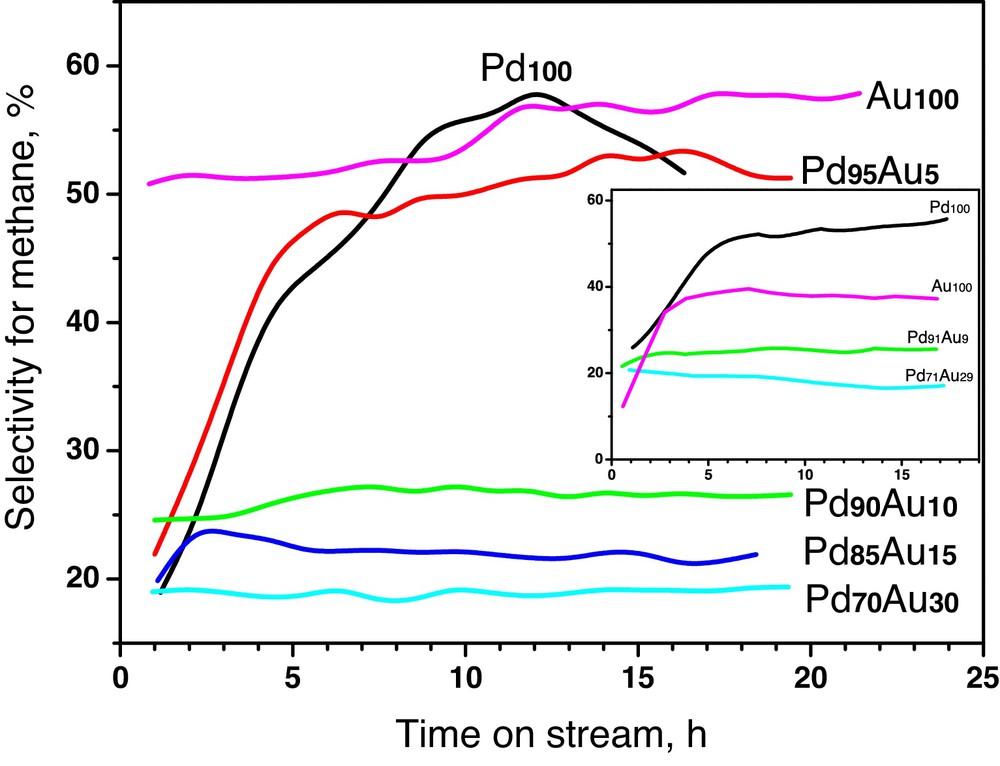
(Colour online.) Variations in the selectivity for methane during hydrodechlorination of CCl4 for Sibunit carbon-supported Pd–Au catalysts. Inset: time-on-stream behaviour of Vulcan carbon series.
On the other hand, the samples of Pd90Au10/Sibunit, Pd85Au15/Sibunit and Pd91Au9/Vulcan exhibited very high total conversion of CCl4, i.e. the result, which for nearly identical charges of catalyst (∼0.24 g) and comparable metal dispersions, directly indicates the superiority of these alloy compositions. Stabilized turnover frequencies given in Table 3 confirm a high value of these catalysts. Simultaneously, these catalysts showed the best selectivities towards C2+ hydrocarbons, which we regard as the most desired products [33]. Further addition of gold to palladium (to ∼30 at% Au) starts to depreciate the catalyst's performance, as it is seen for Pd70Au30/Sibunit and Pd71Au29/Vulcan. Both the overall conversion level (Fig. 4) as well as the product selectivities are inferior to the ones exhibited by Pd90Au10/Sibunit, Pd85Au15/Sibunit and Pd91Au9/Vulcan (Figs. 5 and 6).
CCl4 hydrodechlorination on carbon-supported Pd–Au catalysts. Final conversions, product selectivities and turnover frequencies at 423 K.
| Catalyst | Steady-state conversion, % | Product distribution, % | TOFa, s−1 | |||||
| Hydrocarbons | Chlorocarbons | |||||||
| CH4 | C2 | Cx>2 | ΣCn | CH3Cl + CH2Cl2 + CHCl3 | C2+HyClz | |||
| Sibunit series | ||||||||
| 2 wt%Pd100/Sibunit | 0.6 | 49.2 | 6.6 | 3.4 | 59.2 | 11.9 | 14.0 | 3.782E-4 |
| 2.19 wt%Pd95Au5/Sibunit | 1.0 | 49.1 | 5.9 | 0.5 | 55.5 | – | 44.5 | 7.477E-4 |
| 2.41 wt%Pd90Au10/Sibunit | 98.1 | 26.6 | 32.0 | 17.1 | 75.7 | 11.2 | 13.0 | 0.0793 |
| 2.65 wt%Pd85Au15/Sibunit | 97.4 | 21.9 | 24.0 | 28.0 | 73.9 | 8.5 | 17.6 | 0.0707 |
| 3.59 wt%Pd70Au30/Sibunit | 82.6 | 19.4 | 20.4 | 8.9 | 48.7 | 22.8 | 28.5 | 0.0474 |
| 2 wt%Au100/Sibunit | 8.4 | 57.9 | 1.1 | 8.0 | 67.0 | 16.9 | 16.1 | 0.0367 |
| Vulcan XC72 series | ||||||||
| 2 wt%Pd100/Vulcan | 2.7 | 55.7 | 3.6 | 3.1 | 62.4 | 3.4 | 34.2 | 0.0036 |
| 2.41 wt%Pd91Au9/Vulcan | 99.5 | 24.6 | 24.6 | 15.5 | 64.7 | 14.3 | 21.0 | 0.111 |
| 3.59 wt%Pd71Au29/Vulcan | 63.4 | 16.5 | 5.7 | 16.1 | 38.3 | 36.5 | 25.2 | 0.0518 |
| 2 wt%Au100/Vulcan | 0.7 | 36.1 | – | – | 36.1 | 12.0 | 51.9 | 0.0016 |
a Based on metal dispersion calculated from the relation FE = (1.12 × X/100 + 1.17 × Y/100)/dnm (SEM) for PdXAuY (for X and Y, Table 2).
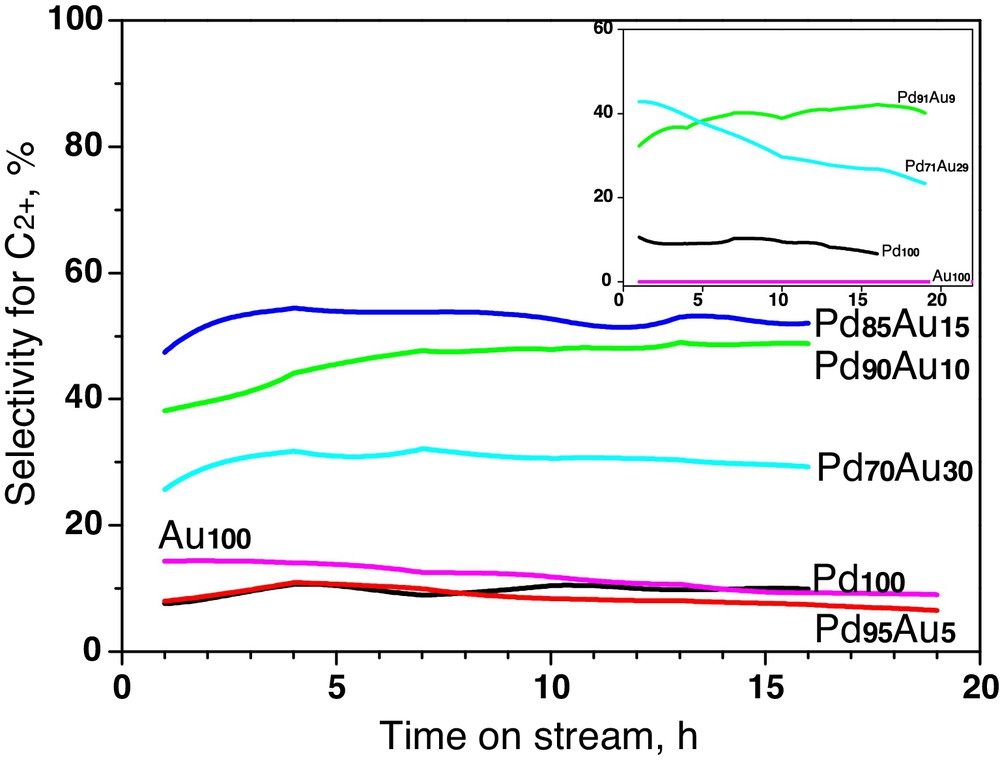
(Colour online.) Variations in the selectivity for ethane and longer hydrocarbons during hydrodechlorination of CCl4 for Sibunit carbon-supported Pd–Au catalysts. Inset: time-on-stream behaviour of Vulcan carbon series.
Thus, we consider that these studies showed that only the introduction of small definite gold amounts (10–15%) to palladium results in a considerable improvement of the catalytic behaviour. Our previous work only marked that the overall activity, catalyst stability and product selectivity of palladium are positively influenced by gold addition [33]. Reflections about the origin of synergistic effects for Pd–Au alloys in CCl4 HdCl are in the next subsection.
XRD studies of post-reaction catalysts showed a significant phase transformation for nearly all catalysts (excluding Au100). A considerable shift of XRD profiles toward lower diffraction angles (Fig. 1) suggests carbon incorporation into Pd/PdAu bulk. This effect was observed (and widely discussed) for Pd100/Sibunit catalysts subjected to CCl4 hydrodechlorination at 363 K [33], but not noticed for Pd–Au/Sibunit catalysts. Evidently, bare C1 species (from the CCl4 molecule after stripping off all chlorine atoms) enter Pd bulk before being hydrogenated to methane. The increase of methane selectivity (Fig. 5) during the reaction suggests that palladium sink becomes gradually more “saturated”.
Essentially identical positions of (111) fcc reflections for post-reaction samples of all Pd and Pd–Au catalysts at 2∼38.9° suggest the presence of a PdC0.13(0.15) phase [46,47]. This, in turn, suggests considerable phase changes. If palladium escapes from the alloy to form a carbide phase, then the rest of bimetallic material should become richer in gold. Since the PdC0.13 phase does not seem to be active in this reaction [33], then the resulting, Au-enriched, Pd–Au alloy must constitute a catalytically active phase. This hypothesis needs substantiation by additional HRTEM/EDS studies.
Finally, Fig. 7 shows the results of temperature-programmed hydrogenation (TPH-MS) of all catalysts subjected to a prolonged CCl4 reaction (ca. 20 h on stream). Large amounts of chlorine desorbed from Pd100 and Pd95Au5 catalysts indicate that, apart from carbonaceous coke, retained chloride species is also a possible cause of the catalyst's deactivation. It is in line with other results [48–51].
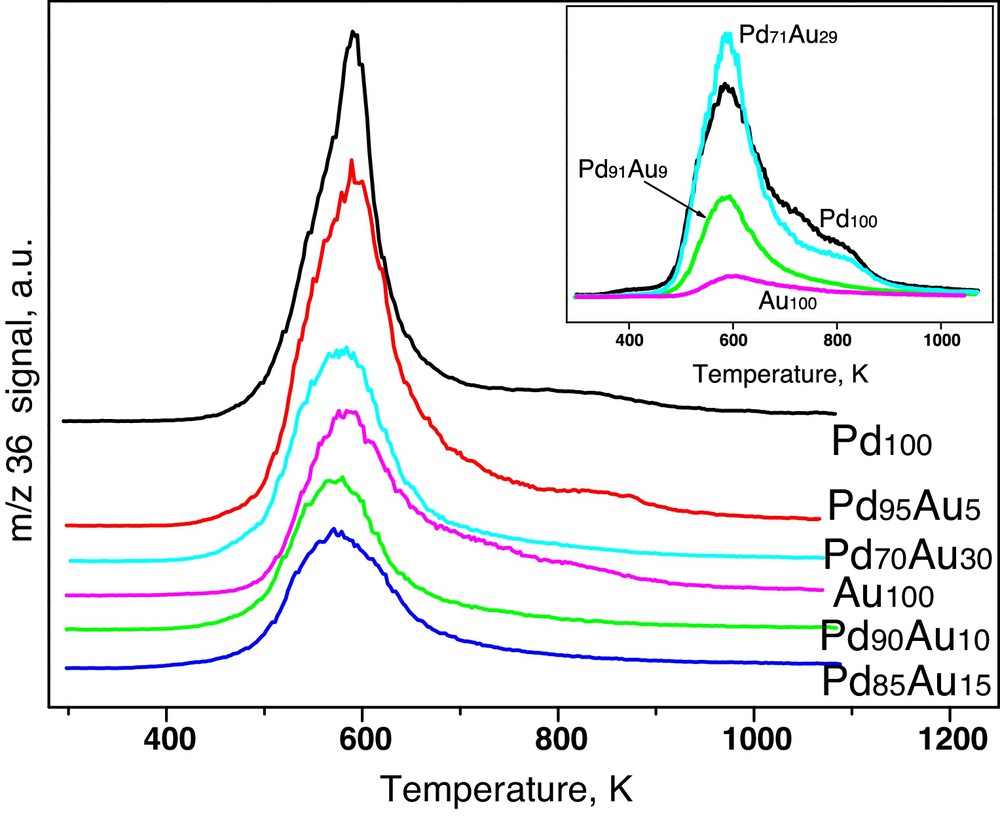
(Colour online.) Temperature-programmed hydrogenation (TPH) profiles of chloride species from post-reaction deposits from Sibunit carbon-supported Pd–Au catalysts. Inset: TPH from the Vulcan series.
3.3 Synergistic effects in CC4 hydrodechlorination on Pd–Au catalysts
Fig. 8 shows the relation of the turnover frequency with Pd–Au alloy composition, calculated on the basis of stabilized conversion, i.e. at the end of catalytic runs (all runs with 0.24 g samples). The overall activity pattern obtained with two series of carbon-supported Pd–Au catalysts shows a maximum at ∼10 at% Au. The fact that the results of both series of Pd–Au/carbon catalysts, characterized by so different porous structures, form a kind of an universal relationship is in line with our conclusion (in §Experimental) about the absence of internal diffusion limitations. Present results also confirm our previous results obtained on Pd–Au/Sibunit catalysts prepared by reductive deposition [33], where some bimetallic catalysts were more active, stable and selective (towards hydrocarbon products) than the monometallic Pd/Sibunit catalyst. It should be admitted that a beneficial HdCl behaviour of gold introduction to palladium was earlier reported [32].
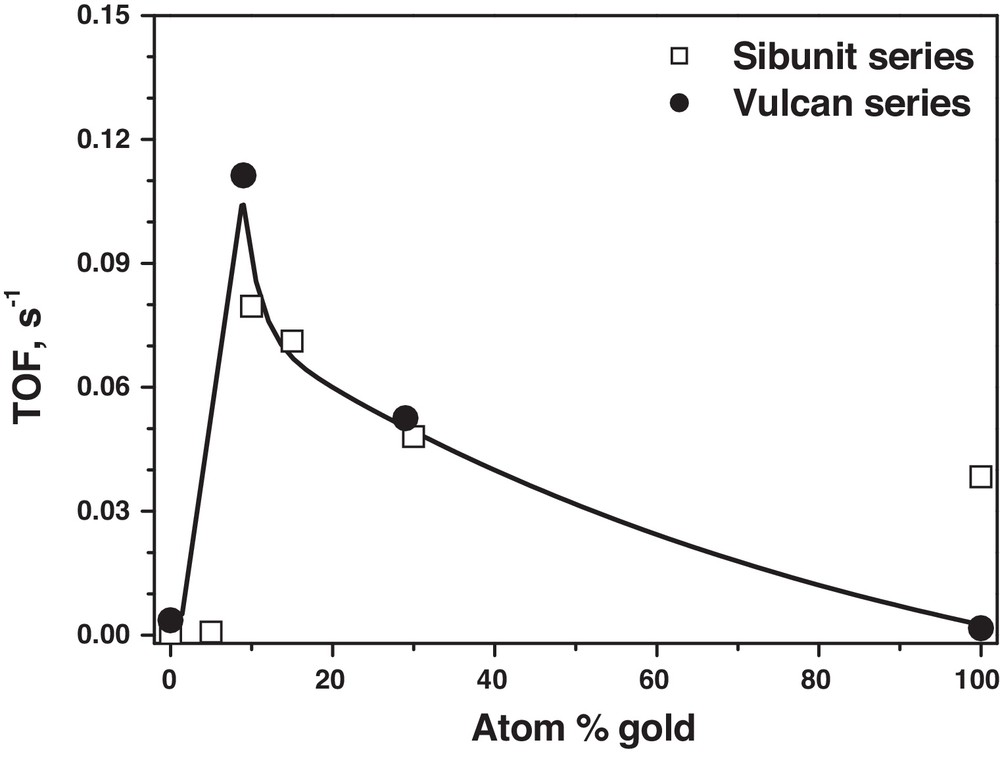
Activity of Pd–Au/C catalysts in hydrodechlorination of tetrachloromethane at 423 K as a function of alloy composition.
Synergistic effects observed for Pd–Au alloys are interpreted by the occurrence of two effects: ensemble (geometric) and ligand (electronic) ones [52]. The ensemble effect is a dilution of surface Pd by Au, leading to the disappearance of contiguous Pd site ensembles and the creation of isolated Pd sites. The maximum in activity at about 10 at% Au suggests that the geometric effect does not operate here, unless we are dealing with a significant gold segregation to the alloy surface. XPS studies were not undertaken in this work, however XPS studies of Pd–Au/Vulcan catalysts showed no special surface enrichment with gold [36]. The ligand effects occurring via direct charge transfer or by affecting bond lengths cause the Pd d band to be more filled, moving the d-band centre away from the Fermi level. This makes Pd more “atomic like” which binds reactants and products more weakly. It would relieve catalyst deactivation caused by self-poisoning and enhances the activity/selectivity. This effect would explain the better resistance to deactivation of Pd–Au alloys. Improvement in deactivation resistance to chloride species in HdCl (of mainly trichloroethene) reactions by for gold-supported palladium nanolayers was observed in numerous papers from the group of Wong [30,53–59], where both ensemble and ligand effects were invoked to describe the superiority of Pd–Au combinations.
Very small amounts of gold introduced to palladium make a pronounced and beneficial difference in the catalytic behaviour. Synergistic effects observed for 10–15 at% Au in the overall activity (Fig. 8), stability (Fig. 4) and selectivity to C2+ hydrocarbons (Fig. 6) suggest that a larger group of Pd surface atoms must be electronically modified by single Au atoms. However, such modification should not be too deep as a gold-rich surface would be inactive in providing active (dissociated) hydrogen needed for hydrodechlorination and removal of chloro-carbonic residues.
Our recent XPS data for Pd–Au/Vulcan catalysts showed the binding energy of Pd 3d5/2 within the range of 335–337 eV [table 2 in [36]). The monometallic Pd/Vulcan showed the Pd state of binding energy 335.3 eV corresponding to the Pd-metal. In the spectra of all Au-containing catalysts (starting from Pd91Au9/Vulcan), the binding energy of Pd-metal peak component was slightly shifted but also a less intense peak component at slightly higher energy of ca. 336–337 eV appeared. Since the Pd binding energy values slightly above 336 eV are commonly ascribed to the partially oxidized Pdσ+ species [60], therefore these XPS data could indicate a Pd–Au electronic interaction. A simplified picture of the Pd6Au1 ensemble is proposed in Fig. 9. The large size of the molecule of CCl4 (kinetic diameter ∼0.6 nm) implies its interaction with such a large surface ensemble.
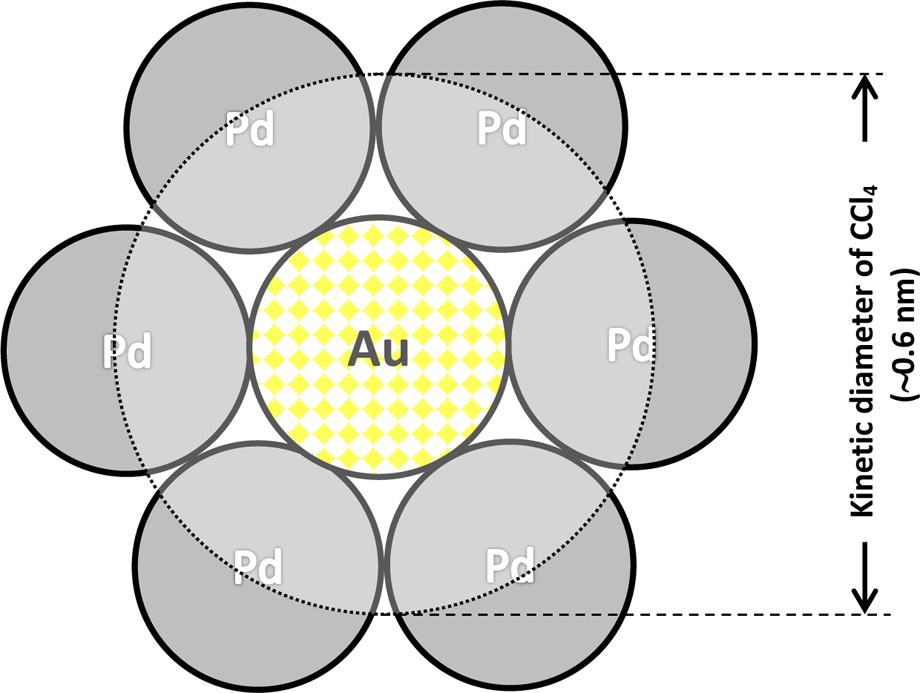
(Colour online.) CCl4 molecule at the mixed Pd–Au ensemble on (111) surface, composed of six Pd atoms and one central Au atom (scheme in accordance with the scale).
4 Conclusions
Two series of well-characterized carbon-supported Pd–Au catalysts showed interesting features in the reaction of tetrachloromethane with dihydrogen at 423 K. In contrast to monometallic palladium catalysts which quickly deactivated in time-on-stream, palladium–gold catalysts containing 10 and 15 at% gold appeared very stable for a long time, much more active and selective towards C2+ hydrocarbon products than the monometallic palladium and Au-richer Pd–Au catalysts. It appears that large surface ensembles composed of several contiguous Pd atoms and electronically modified by single Au atoms are beneficial for such exceptional catalytic performance.
Acknowledgements
This work was supported by the Polish National Science Centre within Research Project DEC-2011/01/B/ST5/03888.