

1 Introduction
Sulfones represent an important class of organic compounds with pharmacological activity and technological applications. This functional group has been studied as a synthetic intermediate in organic synthesis since it can be readily removed from the substrate [1–3]. As can be seen in the literature, sulfonyl carbanions can be alkylated or acylated, and hence are a relevant reagent for C–C bond formation [4–6]. Carbanions that are stabilized by carbonyl, cyano, nitro, and sulfonyl groups have a wide range of uses. Generally, carbanions with localized charge should be tetrahedral, and carbanions whose charge is delocalized should be planar. Elements such as sulfur, phosphorus, and halogens (except fluorine) contain d orbitals, and then are capable of delocalizing and stabilizing the negative charge. It is believed that carbanions containing cyano or sulfonyl groups are closer to “true carbanions” in comparison with carbanions activated by the nitro group, where the negative charge is delocalized on the oxygen atoms of the nitro group. Thus, the anion formed after the deprotonation of nitroalkane is stabilized by the electronic shift from the carbon atom to the nitro group. It is well known as nitroalkane anomaly [7–11]. Nitroalkanes show low intrinsic reactivity in protic solvent and normal or high in DMSO. On the other hand, in cyano carbanions the negative charge is localized on the acidic carbon acid [12]. It is known that the sulfonyl group is a strong electron-withdrawing group and then has the ability to stabilize the α-sulfonyl carbanion, but it is less clear how this stabilization takes place [13–15,7]. The delocalization of the negative charge away from carbon acid in an sp2 hybridized carbanion with a dπ–pπ bonding would involve a significant decrease in intrinsic rate constants compared with “normal acids”, i.e. a behavior typical of carbon acids activated by carbonyl or nitro groups. The role of the resonance effect due to a dπ–pπ bonding between the carbanion lone pair and 3d orbital of sulfur (A) has been questioned by theoretical studies. The results of the computational study [16,17] suggested a rather simple charge polarization effect (B). Another factor influencing the stabilization of the negative charge is negative hyperconjugation (C). Resonance structures A, B and C of sulfonyl carbanion are shown in Scheme 1.
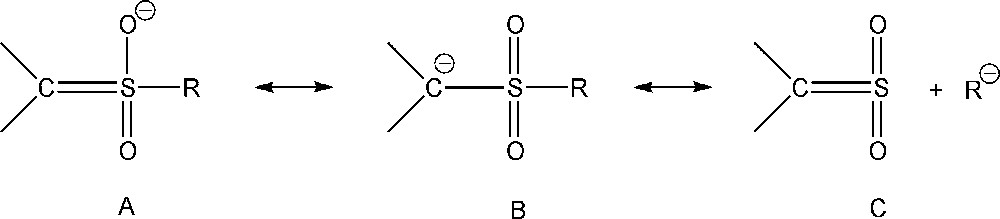
Resonance structures of sulfonyl carbanion.
The electron-withdrawing capability of the SO2CF3 group to stabilize the negative charge of the carbanion is the result of the polarizability effect. Goumont et al. [18] provided convincing experimental evidence of the unusually strong electron transmission ability of the highly acidifying SO2CF3 group, which was considered to be a consequence of its high polarizability. The SO2CF3 group seems to stabilize the excess negative charge via the polarizability effect while the resonance effect is very modest [18–20]. The sulfonyl group is more activating than the nitro one. The activation by the nitro group is favored in aqueous solution when solvation by hydrogen bonding stabilizes the nitronate anion. The situation is in contrast with the case of DMSO, when the stabilization of the polarizable negative charge occurs and favors the activation by a SO2CF3 group. This is consistent with totally different solvent dependences on the acidity of nitro and triflic carbon acids in H2O–DMSO mixtures [13,19]. A support for these observations is the comparison of the Hammett substituent constants (σ) reported by Sheppard and Yagupolskii et al. [21] for the SO2CF3 and the nitro one. The σ parameter for the SO2CF3 group shows that it is the strong electron-withdrawing group. The σ values obtained from studies of the ionization of various acids, for instance benzoic acids or phenols, show that the SO2CF3 group is more activating than the nitro one. The σp values obtained for substituted benzoic acids are 0.96 and 0.81 for SO2CF3 and nitro groups, respectively [22]. The structural analogs of 2,4,6-tris (trifluoromethylsulfonyl) benzene derivatives have been studied, but it is worth mentioning that the electronic properties of the SO2CF3 group are similar to those of fluorosulfonyl, the SO2F group, used in potentially biologically active sulfamides or netlike polymers [23]. The electron-withdrawing ability of the fluorosulfonyl group is nearly the same as that determined for the trifluoromethylsulfonyl one, but it is substantially higher than that of the nitro group, so aromatic sulfonyl fluorides can be involved in various processes of applied chemistry [24].
The investigations about the rates of deprotonation of disulfonyl carbon acids report that the rates of deprotonation of these species approach the rates of “normal” proton transfer from oxygen acids [25,26]. This observation is consistent with the negative charge in the carbanionic conjugate base being effectively localized upon carbon, with little resonance delocalization into sulfonyl groups. Aiken studied rate and equilibrium data for the deprotonation of a series of phenyl ring substituted phenyl[bis(ethylsulfony)]methanes in an aqueous solution. The kinetics show that the proton-transfer reaction indicates intrinsic rate constants that are three to four orders of magnitude lower than for normal acids, suggesting a considerable degree of charge delocalization. The obtained results suggested that after the ionization of disulfonyl carbon acids, partial delocalization of the negative charge on the carbanion occurs. This effect is somewhat larger than for cyano carbon acids, but it is considerably smaller than for nitro or carbonyl carbon acids. The acidifying power of sulfonyl groups is attributable not simply to electrostatic stabilization, but partly also to a mesomeric effect. The degree of mesomerism is, however, uncertain, because part of the intrinsic barrier may derive from steric effects, as is also observed for bromination [26]. As regards the structures of carbanions activated by sulfonyl groups, only some of them have been described by NMR [13,18,19,27,28], IR [29–32], X-ray [33–40], computational [35–37,40–43], kinetic and thermodynamic [19,20,26,44–46] studies.
This paper aims at discussing the spectroscopic and structural approaches to the reactivity of carbon acids activated by two sulfonyl groups. This review is also an attempt to compile the literature on the subject of disulfonyl carbon acids and indicate the prospects for further research. The examples presented in this paper have shown that these carbon acids can be utilized in organic synthesis and are an important class of compounds. Despite the continuous interest in the problems of stabilization of carbanions and their applications in organic synthesis, there is a need for a review and summary of the achievements.
2 Synthesis of disulfonyl carbon acids
The synthesis of sulfones is of interest due to their valuable applications as intermediates during the preparation of various chemically and biologically important molecules [1]. Disulfonyl carbon acids can be prepared by several methods. The oxidation of sulfides is the most straightforward method for the synthesis of these compounds [1,47–50]. The synthesis of thioacetals using application Bunte salts (esters of sodium thiosulfuric acid) was first described by Wastlake and Dougherty [51]. In this process, sodium alkyl thiosulfates react with aldehydes in the presence of an acid to give thioacetals (Scheme 2). The obtained products can be oxidized resulting in the corresponding sulfone.
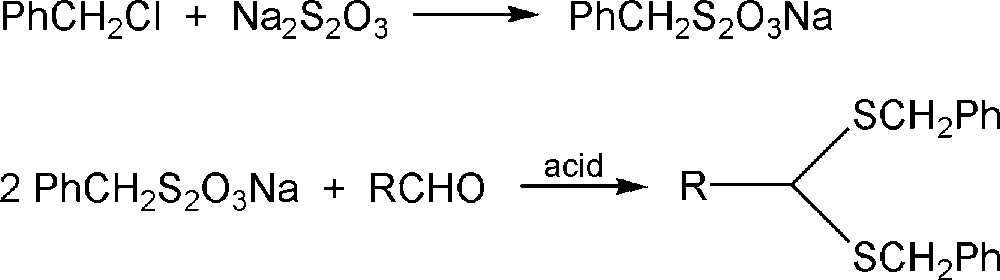
Synthesis of thioacetals using Bunte salts.
The oxidation of sulfides to sulfones was made using different oxidizing agents. Castro synthesized bis(methylsulfonyl)methane [6] using oxone (2 KHSO5·KHSO4·K2SO4) as an oxidant under anhydrous conditions, described previously by Trost and Curran [52]. Aiken et al. [26] synthesized a series of bis(ethylsulfonyl)methanes (Scheme 3) using a procedure reported by Cronyn [47] based on the reaction between ethyl mercaptan and an appropriate aldehyde and then the oxidation of disulfides by a hydrogen peroxide/acetic acid mixture [26].
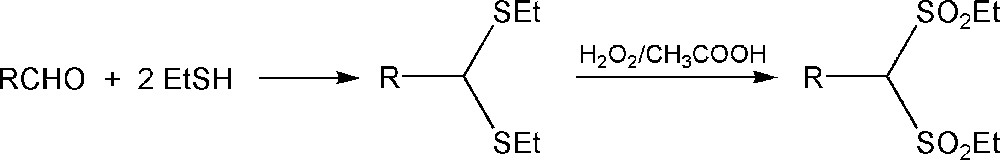
Synthesis of bis(ethylsulfonyl)methane derivatives.
The dynamic development of chemical catalysis led chemists to use a catalyst for the preparation of sulfones. A number of oxidants have been reported for the oxidation of sulfides to sulfones [53,54], such as direct oxidation by molecular dioxygen or hydrogen peroxide [55]. It was also reported that boric acid [56,57] can be used as a highly efficient and environmentally friendly catalyst for the selective oxidation of sulfides to sulfones using 30% hydrogen peroxide as an oxidant. There are a lot of procedures for oxidation with hydrogen peroxide and various transition metal catalysts such as Ti, Mo, Mn, V, W, Re, Cu [58–61]. An interesting and simple method for the oxidation of sulfides to sulfones was described by Zhang et al. [62]. They used a series of imidazolium perrhenate ionic liquids as green and efficient catalysts for the oxidation of sulfides to sulfones with aqueous hydrogen peroxide under mild conditions in a homogeneous phase [62].
3 Sulfones in organic synthesis
The use of sulfones in organic chemistry appears to be a highly developing field of chemical synthesis [1,2,5,63–69]. Sulfones seem to be a useful tool in organic synthesis due to their versatility, easy removal and easy chemical modification. The reactions of sulfones essentially depend on the generation of the α-sulfonyl carbanion. The formation of the α-sulfonyl carbanion is governed by the acidic behavior of the hydrogen atom. Carbon acids activated by two sulfonyl groups are also utilized in organic synthesis. Among these carbon acids, bis(phenylsulfonyl)methane (BSM) possesses acidity due to the presence of two sulfonyl groups and can form a stable α-carbanion. Bis(phenylsulfonyl)methane, a commercially available product, can be activated by mild bases forming a terminal “naked” alkyl carbanion. It should be noticed that the phenylsulfonyl group can be easily removed by using a more environmentally friendly Mg/HOAc/NaOAc reagent, which is a good replacement of a conventional toxic sodium/mercury amalgam [70–73]. Bis(phenylsulfonyl)methane was used in many organic syntheses. BSM was utilized in 1,4-addition reaction to afford hydrazone derivatives [74]. Byrne and co-workers used BSM for the preparation of carbocycles from the corresponding dibromide [75]. Ashgar et al. show that the carbanion obtained from bis(phenylsulfonyl)methane reacts with 4,6-dinitrobenzofuroxan to yield a σ-adduct that undergoes a base-catalyzed elimination to yield an alkene derivative [45]. In 2009, Garcia-Ruano et al. described the first organocatalytic addition of BSM to α,β-unsaturated aldehydes (Scheme 4) in the presence of a catalyst, lithium acetate, and THF at room temperature [64]. The optimized conditions (temperature, solvent and asymmetric iminium catalyst) allowed one to develop an asymmetric methyl or alkyl addition to α,β-unsaturated aldehydes with good yields and enantioselectivities. The reaction also reported to be applicable to the synthesis of derivatives of a series of aliphatic enals. Landa et al. found that the reaction worked well with aromatic enals [76]. Optimization of the methodology allowed one to perform reductive desulfonylation and to synthesize different derivatives. Additionally, Alba et al. described this reaction as a valuable method for the synthesis of isotope enantiomers. The replacement of BSM by 13C labelled compound can find a wide use in the synthesis of 13C-labeled biological products [77].
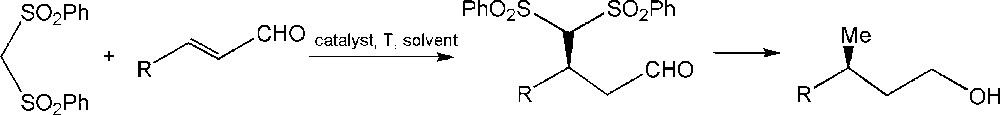
The Michael addition of bis(phenylsulfonyl)methane to α,β-unsaturated aldehydes.
The literature describes several methods concerning sulfones as C nucleophiles in the catalytic C–C bond-forming alkylation of enals [76,78,79] and oxazolones [80,81]. In these reactions, bis(phenylsulfonyl) derivatives are the synthetic equivalent of an alkyl group because the disulfonyl moieties can be easily transformed by reductive desulfonylation and allow the formation of a number of biologically active compounds [5,69].
The fluoro derivative fluorobis(phenylsulfonyl)methane (FBSM) was also used for a broad range of α,β-unsaturated aldehydes with a good yield [82,83]. Alba et al. reported that using aliphatic aldehydes one obtains high enantioselectivity and good yields. They reported that when aromatic aldehydes were used, the reaction became slower and the yields decreased. However, the substitution in the phenyl ring by an electron-withdrawing group, such as the cyano or the nitro group, gives high enantioselectivity and good yields. This method could be used for synthesizing derivatives and the facility of removal of the phenylsulfonyl moiety to give access to allylic methylation. Recently, Yang et al. [84], Furukawa et al. [85] and Companyó [84,83,86] proposed a highly enantioselective addition of bis(phenylsulfonyl)methane (Scheme 5) to Morita–Baylis–Hillman carbonates as electrophiles catalyzed by Cinchona alkaloid derivatives.
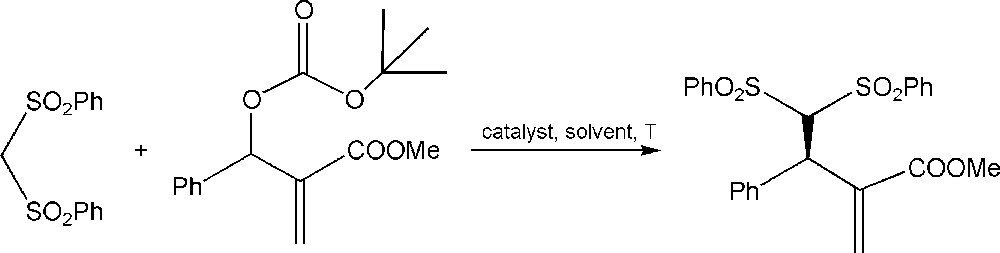
Alkylation of bis(phenylsulfonyl)methane with MBH carbonate.
An interesting method for the synthesis of ibuprofen using bis(phenylsulfonyl)methane has been described by Acemoglu et al. In this procedure, palladium-catalyzed asymmetric allylic substitution is used as the key step of the synthesis of α-aryl propanoic acids including ibuprofen (Scheme 6). The carbanion obtained by the deprotonation of bis(phenylsulfonyl)methane in the presence of sodium hydride in THF solvent can easily react with Select-Fluor® (Scheme 7), resulting in a more nucleophilic product, fluorobis(phenylsulfonyl)methane. The originally described procedure shows 50–60% yields [87,88].
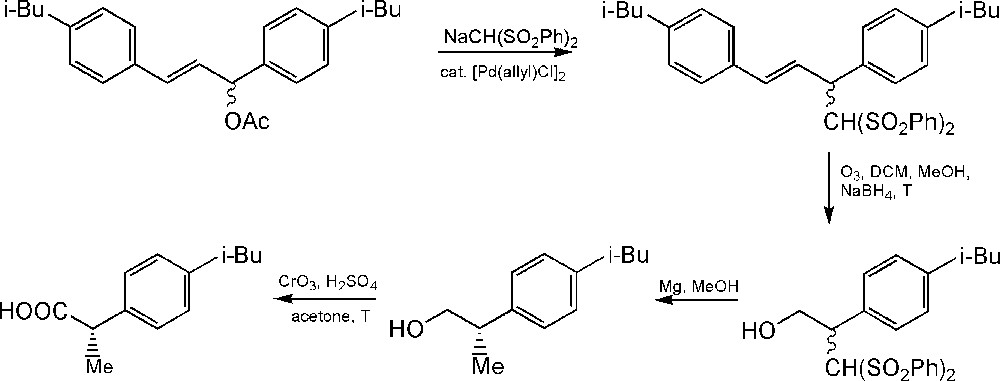
Synthesis of ibuprofen using bis(phenylsulfonyl)methane.
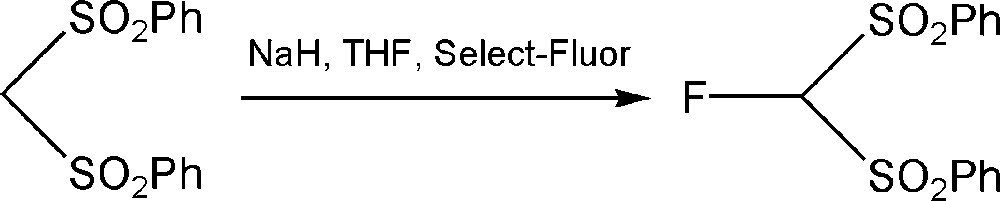
Synthesis of fluorobis(phenylsulfonyl)methane.
In 2006, FBSM was reported independently in the literature by Shibatas [87] and Hu's [88] research groups as a synthetic equivalent of the fluoromethide species in an enantioselective monofluoromethylation. FBSM can also be prepared by the oxidation of α-fluoro-α-phenylthiomethyl phenyl sulfide, previously obtained through the electrochemical fluorination of phenyl phenylsulfonylmethyl sulfide [89]. However, the fluorination reaction step gives moderate yields and difluorinated by-products. As can be seen above, fluorobis(phenylsulfonyl)methane FBSM has applications in organocatalytic synthesis. In 2013, the group of Prakash has achieved a method, presented in Scheme 8, between fluoromethyl phenyl sulfone and methyl benzenesulfinate, followed by the oxidation of the intermediate [90].
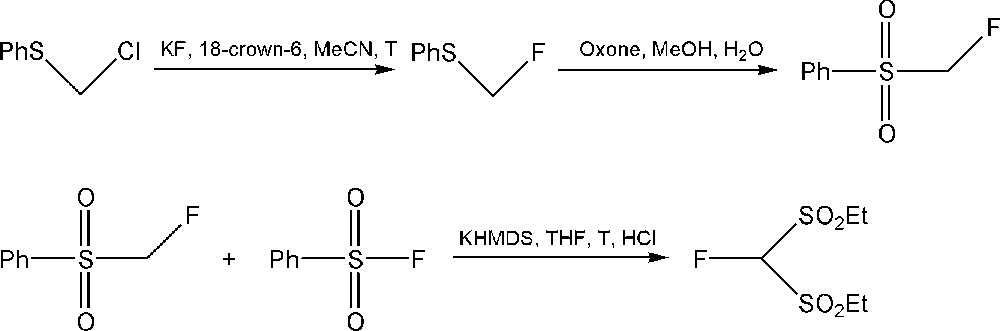
Synthesis of FBSM by the group of Prakash.
Now, BSM and FBSM are commercially available and their reactivity is comparable [84,85]. The anion derived from FBSM possesses good thermal stability and nucleophilicity towards many electrophiles. However, the fluorine substitution of the carbanion decreases nucleophilicity, showing a negative fluorine effect described by Ni et al. [88]. They compared the reactivity of epoxides with nonfluorinated carbanion PhSO2CH2– and fluorinated or chlorinated analogues. Fluorinated carbanions showed lower reactivity than chlorinated or nonfluorinated ones. The reported results indicate that substitution by the second phenylsulfonyl group attached to α-carbon atom delocalizes the electron density from the carbanionic center, resulting in a better stabilization of the fluorinated carbanion. Therefore, the substitution of the fluorinated carbanion by the electron-withdrawing group effectively increases the nucleophilicity of these carbanions, and the reactivity order is: (PhSO2)2CF– > PhSO2CFH– >> PhSO2CF2– [88].
Fluorobis(phenylsulfonyl)methane proved to be an efficient nucleophilic monofluoromethylating reagent, frequently applied in many reactions such as those with the above-mentioned epoxides or aziridine [88], stereospecific monofluoromethylation using Mitsunobu reaction conditions [65], the Mannich reaction, palladium-catalyzed enantioselective allylic monofluoromethylation [87], monofluoromethylation of alkyl halides [70] and many others. Some examples of the use of fluorobis(phenylsulfonyl)methane in organic synthesis are presented in Scheme 9.
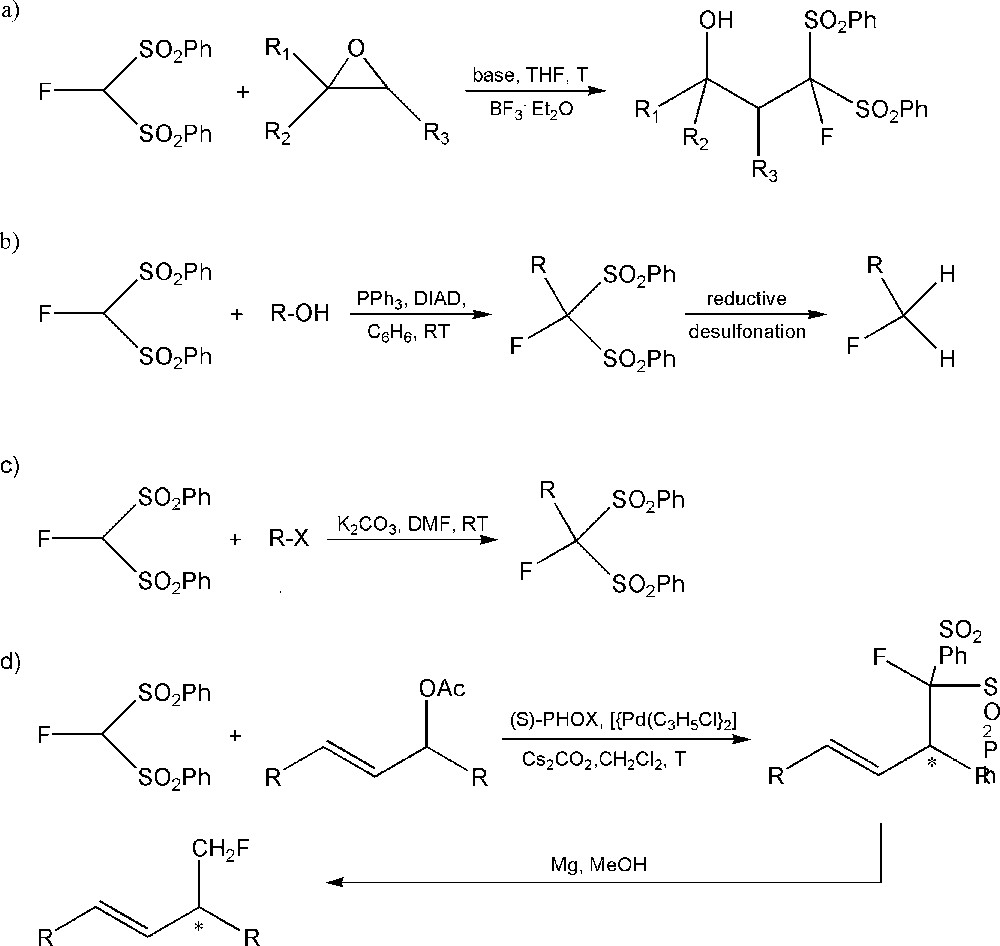
Examples of synthetic applications of fluorobis(phenylsulfonyl)methane: a: fluoroalkylation of simple epoxides; b: monofluoromethylation of alcohols by using Mitsunobu reaction; c: monofluoromethylation of alkyl halides; d: palladium-catalyzed enantioselective allylic monofluoromethylation.
4 Acidity of disulfonyl carbon acids
The electron-withdrawing sulfonyl groups activate the sp3-hybridized C–H bond. That effect has an influence on the neighboring α hydrogen atoms and increases the acidity of the proton. The reactivity of α-sulfonyl carbanions is controlled by the pKa of the sulfone α-protone. The successive substitution of protons with methyl sulfonyl groups in a methane molecule leads to an increase in the acidity of the derivatives and pKa values in water: 29, 12 and 0, respectively [91].
The presence of two sulfonyl groups has an influence on the stability of corresponding α-sulfonyl carbanions due the delocalization of the negative charge from the acidic carbon atom [28,94–96]. The sulfonyl group has a less acidifying effect than the carbonyl, nitro and triflone group. The substitution of one sulfonyl group in CH2(SO2Ph)2 (pKa = 12.25 in DMSO) by the carbonyl group increases the acidity by 0.85 pK unit (pKa of PhSO2CH2COPh = 11.4) [97]. As can be seen from Table 1, the marked difference is found for triflone carbon acids (15,16) and other disulfonyl carbon acids. The substitution at the α-carbon atom by the methyl group reduces the acidity, for instance CH2(SO2Ph)2 with pKa = 11.21 and CH3CH(SO2Ph)2 with pKa = 13.76. On the other hand, the phenylsulfonyl or benzylsulfonyl substituent increases the acidity by 1–2 pK units in comparison with the ethylsulfonyl or butylsulfonyl group due to the inductive effect of the phenyl group. This is consistent with previous observations by Bordwell evidencing that PhCH2X compounds are more acidic than the corresponding CH3X [97]. This difference shows that the carbanion-stabilizing effect of the benzyl or the phenyl substituent is large. As can be seen in Table 1, in aprotic solvent such as acetonitrile, p-NO2PhCH(SO2C2H5)2 is a stronger acid than p-CNPhCH(SO2C2H5)2, but in water the acidity is the same. This effect may be associated with different kinds of carbanion stabilization by the solvent. Therefore, the acidity of the compounds presented in Table 1 is larger in acetonitrile than in an aqueous solution. Bordwell has reported that the solvation of carbanion CH(CN)2– by DMSO and water is almost the same and both pKa values are also equal [97]. Berger et al. [7] studied trifluoromethylsulfonyl substituted benzyl carbanions and observed that these anions are more effectively solvated by ion–dipole interactions in DMSO than by the hydrogen bonding in methanol. The obtained pKa values in methanol are by five units larger than in DMSO. Terrier reported that 4-X-benzyl trifluoromethyl sulfones are weaker acids in water than in DMSO [13]. However, the trifluoromethylsulfonyl group appears less acidifying than the nitro group in an aqueous solution in comparison with DMSO. Thus, the acidity difference between the corresponding triflones and nitro carbon acid is lower in DMSO than in the H2O/DMSO mixture [13]. The replacement of nitro groups by more electronegative SO2CF3 groups leads to an increase in the activity of these compounds. For example, 2,4,6-tris(trifluoromethylsulfonyl)phenol (pKa = –1 or –2.5) is rather more acidic than 2,4,6-trinitrophenol (pKa = 0.38) [24]. On the other hand, 2,4,6-tris(fluorosulfonyl)phenol (pKa = 5.66) may be compared with the acidity of 2,4,6-tris(trifluoromethylsulfonyl)phenol (pKa = 4.93) in acetonitrile solvent [98]. The sulfonyl group is a weaker activating group than the cyano group and then cyano carbon acids are significantly more acidic than sulfonyl analogues. Thus, in DMSO, PhCH(CN)2 (pKa = 4.2) is by 10 and 7 pK units more acidic than PhCH(SO2CH3)2 and PhCH(SO2CH2Ph)2, respectively. Similar differences are observed in acetonitrile between p-NO2PhCH(CN)2 (pKa = 11.61) and p-NO2PhCH(SO2C2H5)2 (pKa = 21.7) or p-NO2PhCH(SO2CH2Ph)2 (pKa = 20.1) (Table 1). This phenomenon seems to indicate that the negative charge is more delocalized from the carbanionic carbon atom towards the sulfonyl groups than towards the cyano ones. It should be noted that the Hammett plots yield ρ values of +1.46 and +2.46 in acetonitrile for para-substituted disulfonyl carbon acids: p-X-Ar-CH-(SO2-C2H5)2 and p-X-Ar-CH-(SO2-CH2Ph)2, respectively [28]. It should be noted that the corresponding ρ value for 4-X-phenyl(4-nitrophenyl)cyanomethanes in acetonitrile is relatively low (ρ = +2.6) [99] and similar to the value obtained for carbon acids activated by two benzylsulfonyl groups. These values of ρ for disulfonyl carbon acids are lower in comparison with Hammett ρ values for other carbon acids, such as anilines, nitriles or phenols, which are usually > 4. The value of ρ obtained for carbon acids activated by two cyano groups: ArCH(CN)2 is 4.2 [97]. The higher values are also found for mono functionalized compounds in a polar aprotic solvent such as DMSO, with ρ values of 4.8 and 5.9 for ArCH2–SO2–Ph and Ar–CH–CN, respectively [97].
pKa values of some disulfonyl carbon acids.
| Compound | Formula | pKa | ||
| MeCN | 80% DMSO/H2O | H2O | ||
| 1 | CH2(SO2CH3)2 | – | 15.0 [97] | 12.7 [97] |
| 2 | CH2(SO2C2H5)2 | – | – | 11.95 [92] |
| 3 | CH2(SO2Ph)2 | – | 12.25 [97] | 11.21 [93] |
| 4 | CH3CH(SO2C2H5)2 | – | 16.7 [97] | 14.56 [93] |
| 5 | CH3CH(SO2Ph)2 | – | – | 13.76 [93] |
| 6 | PhCH(SO2CH3)2 | – | 14.3 [97] | – |
| 7 | PhCH(SO2C2H5)2 | 23.7 [28] | – | 12.01 [26] |
| 8 | PhCH(SO2C4H9)2 | – | 13.10 [50] | – |
| 9 | PhCH(SO2CH2Ph)2 | 23.0 [28] | 11.13 [48] | – |
| 10 | p-CNPhCH(SO2C2H5)2 | 22.6 [28] | – | 10.06 [26] |
| 11 | p-CNPhCH(SO2CH2Ph)2 | 20.8 [28] | 8.55 [48] | – |
| 12 | p-NO2PhCH(SO2C2H5)2 | 21.7 [28] | – | 10.08 [26] |
| 13 | p-NO2PhCH(SO2C4H9)2 | – | 9.80 [50] | – |
| 14 | p-NO2PhCH(SO2CH2Ph)2 | 20.1 [28] | 7.75 [48] | – |
| 15 | CH2(SO2CF3)2 | – | 2.1 [97] | – |
| 16 | PhCH(SO2CF3)2 | 7.83 [98] | – | – |
5 Structural studies
5.1 X-ray and theoretical models
The structural studies of sulfonyl carbanions have been the subject of many investigations [13,16,17,100,101]. The knowledge of the structure of carbanions provides information about their reactivity. The selected parameters obtained from X-ray data and calculation of the selected sulfonyl carbon acids and their carbanions are presented in Table 2. Theoretical models and crystal structure of α-sulfonyl carbanions show an elongation of the S = O bonds and a shortening of the S–Cα bond following ionization [16,100]. The shortening of the C–S bonds can be qualitatively explained by the effective overlap of the p orbital of the carbanionic carbon atom with the 3p orbital of sulfur resulting in a stronger C–S σ bond. A p-π–d-π overlap cannot be effective due to the fact that the 3d orbitals of sulfur atoms are too diffused and high in energy. The shortening of the Ph–Cα bond between the acidic carbon atom and the ipso carbon atom of the phenyl ring in a complex of 4-nitrophenyl[bis(ethylsulfonyl)]methane with 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) suggests the presence of the negative charge in this region [30]. In this complex, the carbanion and the TBD molecule are linked by a pair of relatively weak NH–O bonds. The structure described by X-ray results is consistent with FT–IR studies of this complex. The position of the band of the ν(NH+) vibrations with a maximum at 3278 cm−1 proves that hydrogen bonds are formed by the N–H protons of the protonated TBD molecule. Two ethylsulfonyl groups attached to the carbanionic center (Cα) are approximately coplanar with Cα⋅ The twist angle of the 4-nitrophenyl group is ∼35°. In these carbanions, the negative charge at the acidic carbon atom cannot be easily delocalized by conjugation over the phenyl ring attached to Cα, even though the twist angle is not particularly large. Two tetrahedral sulfonyl groups create little steric hindrance for coplanarity of trigonal Cα with the phenyl ring. It follows that conjugation in the disulfonyl carbon acids could be sterically hampered. The crystal structure of this complex shows also the planarity of the carbanionic center with the sum of the angles (359.2°) [30]. The sum of the analogous angles in 4-nitrophenyl[bis(ethylsulfonyl)]methane was 336.1° [102]. The sum of the angles is ∼359° for the four diethylsulfonyl stabilized carbanions, which indicates an sp2 hybridization of the carbanionic center in these carbanions [94]. Similar observations were made by Ochiai [41]. The solid-state structure of [CF3(SO2)2CBr]– shows that the sum of the Cα bond angles is 359.5°, indicating an sp2 hybridization of this disulfonyl carbanion [41]. These results are consistent with previous conclusions drawn by Bordwell, who noted that α-sulfonyl carbanions, as well as nitronate and enolate ions, are planar or adopt a nearly planar structure [103].
Selected structural parameters for sulfonyl carbon acids and their carbanions.
| Compounds | Bond length S–Cα | Bond length S–CR | Bond length S = O | Angle S–Cα–S | Dihedral angle C–S–Cα–S | Pyramidalization angle | Refs. |
| CH3SO2CH3 | – | 175.6cal 177.1exp | 143.8cal 143.5exp | – | – | – | [16] |
| CH3SO2CH2– | 161.8 | 179.7 | 146.2 | – | – | – | [16] |
| (SO2Ph)2CH2 | 1.786 | – | 1.432 | 115.5 | 177.5 | – | [33] |
| (SO2Ph)2CH– | 1.670 | – | 1.448 | – | – | 0.0 | [40] |
| p-NO2PhCH(SO2Et)2 | 1.823 1.831 | 1.756 1.762 | 1.428 1.431 | 112.85 | 164.86 170.76 | – | [102] |
| p-NO2PhC(SO2Et)2-TBD | 1.712 1.715 | 1.774 1.782 | 1.445 1.460 | 116.76 | 79.45 86.36 | 9.0cal 3.0exp | [30] |
| (SO2Ph)2CF– | 1.716 | – | 1.439 | – | – | 49.9 | [40] |
| [CF3SO2)2CBr]– | 1.670 | 1.844 1.845 | 1.435 | 125.4 | 88.2 89.2 | 0.0 | [41] |
| [(SO2Ph)2C-OCH3]– | 1.715 | 1.389 | 1.464 | – | – | 18.8 | [40] |
Interestingly, the structural parameters for methoxy[bis(phenylsulfonyl)]methane show pyramidalization angles of 18.8° [40]. Also, the fluorinated carbanions usually are not planar; for instance, fluoro[bis(phenylsulfonyl)]methane (FBSM) or fluoromethanes make a pyramidalization angle of 49.9° [40] or of 72–75° [104], respectively. Prakash et al. reported that the pyramidalization angles order is from 49.9° to 0°, with a decrease in electronegativity: F > OCH3 > Cl > Br, showing the gradual transition in hybridization of the carbanionic carbon atom from sp3 to typical sp2 [40]. On the other hand, the small energy difference between planar and pyramidal structures was reported for sulfonyl carbanions. The calculated energy difference for lithiosulfones is only 0.57 kcal·mol−1 [17]. This energy difference found for disulfonyl carbanions is also small. The calculations for phenyl[bis(ethylsulfonyl)]methane [94] and methoxy[bis(phenylsulfonyl)]methane [40] yield the energy difference only 0.1 and 0.2 kcal·mol−1, respectively.
5.2 NMR studies
Further confirmation of some of the above conclusions can be obtained from NMR analysis. NMR data could be helpful in the evaluation of charge distribution in the products of carbon acids deprotonation. An evaluation of the chemical shift changes that occur in forming a carbanion show that charge delocalization and hybridization provide more information about the structure of carbanion. The changes of electron density upon ionization result in a variation of the chemical shifts. It is known that the presence of a negative charge on the carbon atom α leads to a shielding of the nucleus and an upfield (ΔδC < 0) shift. The large downfield (ΔδC > 0) shift frequently observed for the α-carbons in carbanions is due to the rehybridization of the α-carbon from sp3 to sp2. Intermediate ΔδC values are associated with an hybridization between sp2 and sp3 of the carbanionic carbon atom, such as the unsubstituted benzyl anion, with ΔδC = 15.4 ppm [13,105]. The changes in chemical shifts and π electron densities after the ionization of disulfonyl carbon acids and the corresponding values for cyano and nitroalkanes are collected in Table 3.
Changes in the chemical shifts (ppm) and π electron densities (e) after ionization of disulfonyl carbon acids, cyano and nitroalkanes in DMSO.
| Compound | ΔδHα | ΔδCα | qCα | qC4 | q | Reference |
| PhCH(SO2C2H5)2 | – | –6.5 | 1.552 | 1.054 | 0.234 | [94] |
| PhCH(SO2CH2C6H5)2 | – | –6.0 | 1.527 | 1.055 | 0.223 | [94] |
| PhCHSO2C6H5 | –1.14 | 3.6 | 1.555 | 1.101 | 0.282 | [95] |
| PhCHSO2CH3 | –1.18 | 4.6 | 1.539 | 1.107 | 0.269 | [95] |
| PhCH2CN | –1.64 | 11.0 | 1.547 | 1.115 | 0.283 | [93] |
| PhCH3 | 15.4 | 1.52 | 1.19 | – | [13,105] | |
| PhSO2CF3 | –1.87 | 0.22 | – | – | – | [13] |
| PhCH2NO2 | 0.99 | 29.9 | 1.308 | 1.040 | 0.567 | [95] |
| p–NO2PhCH2NO2 | 32.2(a) | 1.296(b) | 1.050(b) | – | (a) [106] (b) [94] | |
| p–NO2PhCH2CN | 39.4 | 1.410 | 1.132 | – | [94] |
The results obtained for disulfonyl carbon acids agree with those previously reported by Terrier et al. [13] for benzyltriflones. The reported values show a small downfield shift of Cα and a large upfield shift of Hα for the studied triflones. The obtained value of ΔδCα = 0.22 for triflone PhSO2CF3 is similar to that observed for carbanion activated by the methylsulfonyl, phenylsulfonyl, and cyano groups. This similarity indicates that the negative charge density must be localized on the exocyclic carbon atom and charge delocalization through the phenyl plays a minor role in the stabilization of these carbanions [13]. This observation is supported by the fact that para-substitution in the phenyl ring by CN, NO2, SO2CF3 or CF3 group does not influence the chemical shift of Cα and Hα. The large downfield shifts of Cα (ΔδCα = 29.9) and Hα (ΔδHα = 0.99) associated with the ionization of phenylnitromethane are an evidence of the resonance stabilization of nitronate anion. The negative charge in the carbanion of disulfonyl carbon acids occurs on the Cα and is more similar to the case of cyanoalkanes than to that of nitroalkanes. Only part of the negative charge is delocalized to two sulfonyl group and the phenyl ring attached to the carbanionic center. These results are consistent with the fact that there is a small difference between π electron density in mono and disulfonyl carbanions, for instance: q = 1.539 and 1.555 for PhCHSO2CH3 and PhCHSO2C6H5, respectively, in comparison with 1.552 and 1.527 for PhCH(SO2C2H5)2 and PhCH(SO2CH2C6H5)2. The analysis of charge densities in disulfonyl carbanions shows that the delocalization of the negative charge is larger for the two benzylsulfonyl than for the ethylsulfonyl substituent attached to carbanionic center due to the stabilization of the negative charge on the sulfonyl groups by the benzyl substituent. It should be noted that the results obtained by the group of Prakash for the α-fluorocarbanion of fluoro[bis(phenylsulfonyl)]methane and its analogues support the fact that stabilization by sulfonyl groups occurs [40]. They calculated negative charges showing that each oxygen atom of the sulfonyl group bears more than 0.5 negative charge and the partial single bond character of S–O bonds [40].
Changes in chemical shifts occurring upon the deprotonation of phenyl[bis(ethylsulfonyl)]methanes are not large, but correlate well with the Hammett substituent constant: –6.5, –5.6, +1.4 and +6.5 ppm for H, Cl, CN and NO2, respectively. The same trend with values ranging between –7.9 and 4.2 ppm was observed for phenyl[bis(benzylsulfonyl)]methanes [94]. Interestingly, the calculated 13C NMR chemical shifts of carbanionic carbons and parent carbon acids reported by Prakash et al. show the gradual increase in ΔδCα values with the increasing electronegativity of the Cα substituent: ΔδCα = –24, –18, –12 and +14 ppm for Br, Cl, OCH3 and F, respectively, indicating that the electronegativity of the substituent plays a major role [40]. Changes in the chemical shift of the para carbon atom in the phenyl ring attached to Cα afford an evaluation of charge delocalization into the aromatic ring. They are less sensitive to changes in hybridization of Cα. For instance, benzyllithium shows an upfield shift for the para carbon of –21.6 ppm [107]. With an sp2 hybridized carbon atom and no steric restrictions of resonance, the maximum charge delocalization onto the para carbon results in a maximum Δδ of about –25 ppm [107]. In the case of phenyl[bis(ethylsulfonyl)]methanes, a ΔδCp value ranging between –8.7 and –10.6 is found, which is much lower than expected for, as reported by Browne et al., the maximum charge delocalization on the para carbon atom of the phenyl ring. Also values ΔδCp = –9.5 and –12.8 ppm, for PhCH2NO2 and PhCH2SO2CF3, respectively, were lower than for the maximum Δδ mentioned above. All these NMR-derived observations are consistent and lead to the conclusion that the dominant factor influencing the stabilization of the negative charge in α-sulfonyl carbanions is polarization rather than d–p resonance or negative hyperconjugation.
5.3 IR studies
The data from IR spectroscopic measurements are consistent with the above-presented ones obtained by X-ray and NMR studies. The results of IR spectroscopic studies of lithiosulfones carried out by Chassing et al. [29] show that in PhSO2CH2Li the force constant of the S–Cα bond increased and that a decrease in the S–O bond is reported, in comparison to the corresponding bonds in methylphenylsulfone. Additionally, shifts of the bands of νas(SO2) and νs(SO2) from 1285 cm−1 and 1148 cm−1 towards 1211 cm−1 and 1113 cm−1, respectively, were observed [29]. These results suggest that the negative charge in the α-sulfonyl carbanion is delocalized over carbon and oxygen atoms. The FT–IR spectra of complexes between disulfonyl carbon acids and strong organic bases such as TBD and MTBD shed more light on the problem of stabilization of α-sulfonyl carbanions. The data obtained from the FT–IR spectra of some disulfonyl carbon acids derivatives of methane indicate that there are two regions very sensitive for the charge delocalization within the complexes. The first charge-sensitive group is the para-substituted one in the phenyl ring attached to the carbanionic center. Delocalization of the negative charge in the cyanophenyl direction is confirmed by the shifts of the ν(CN) vibrations from about 2234 cm−1 to 2215 cm−1 after deprotonation. Such observations were made by Brzezinski et al., showing similar shifts in the spectra of the complex of 4-cyanophenyl with TBD and MTBD [108,109]. In the spectrum of complex of 4-nitrophenyl[bis(ethylsulfonyl)]methane with TBD in acetonitrile, the νas(NO2) and νs(NO2) vibrations are observed at 1532 cm−1 and 1352 cm−1, respectively. These bands vanished almost completely in the spectrum of this carbon acid and MTBD [30]. Thus, these results demonstrate that the nitro group is engaged in the conjugation process. Furthermore, the distribution of the negative charge in these complexes in the direction of the sulfonyl groups is revealed by the shifts of the νas(SO2) and νs(SO2) bands. For instance, in the spectra of 4-cyanophenyl[bis(ethylsulfonyl)]methane and 4-cyanophenyl[bis(benzylsulfonyl)]methane, the bands of the asymmetrical stretching vibrations νas(SO2) shift from 1330 cm−1 to 1290 cm−1 and two bands of symmetrical stretching vibrations νs(SO2) shift from 1140 cm−1 to 1095 cm−1 [32].
5.4 ESI-MS studies
The analysis of spectrometric data obtained by ESI-MS was the basis for proposing a general pathway for the fragmentation of complexes between carbon acids and a strong organic base such as TBD [32,110]. ESI-MS is a suitable method for the analysis of ionized species such as carbanions. The comparison of the ESI-MS spectra recorded at various cone voltages could provide information about the deprotonation reaction of carbon acids and the fragmentation pathways of the formed conjugate carbanions [32,110]. The fragmentation pathways of ethylsulfonyl and benzylsulfonyl carbanions are different, but final products are similar (Scheme 10). The results obtained by ESI-MS are consistent with other conclusions found for disulfonyl carbon acids, showing a different delocalization of negative charge in ethysulfonyl and benzylsulfonyl carbanions. The presence of a benzyl substituent has an influence on larger charge delocalization. The m/z signals recorded in acetonitrile as a solvent for the studied disulfonyl carbanions confirmed the proposed fragmentation pathways [32,110].
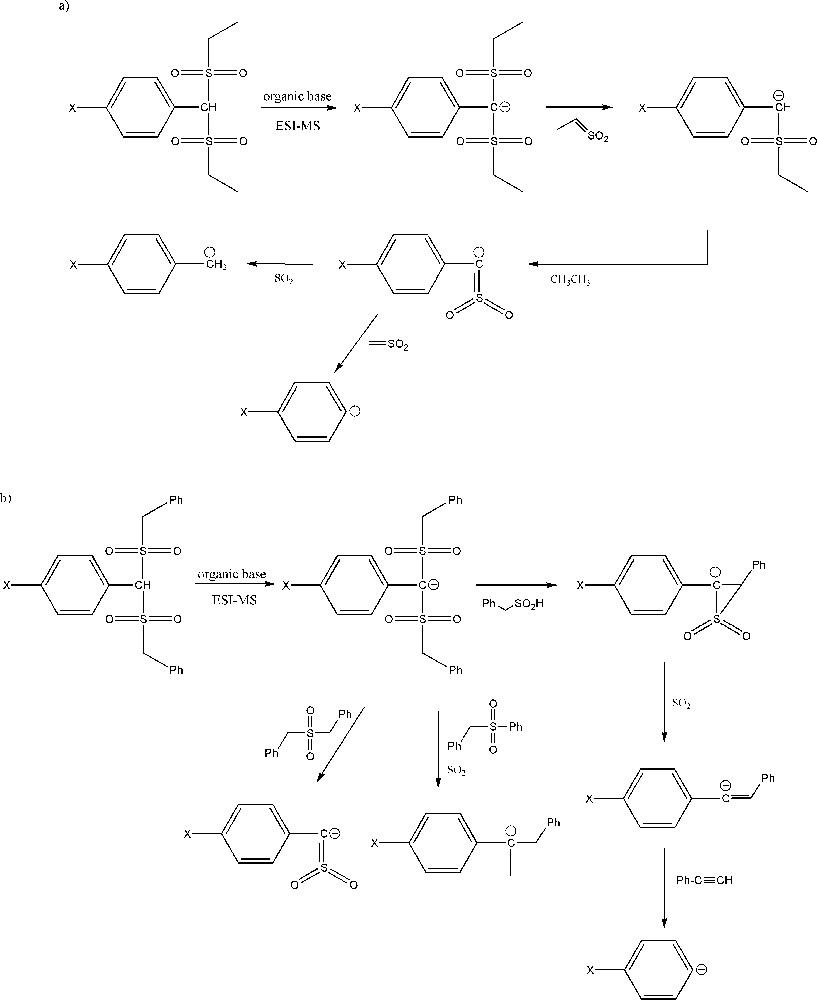
Fragmentation pathways of disulfonyl carbanions: a: ethylsulfonyl carbanions; b: benzylsulfonyl carbanions in acetonitrile as a solvent.
6 Conclusions
This review is a contribution to the understanding of the activating effect of sulfonyl groups attached to the carbanionic center. It should be noted that sulfonyl groups exert a significant effect in proton transfer processes and organic synthesis. Focusing on the structural changes induced by the ionization of α-sulfonyl and α-nitro carbon acids, the contrasting stabilization effects exerted by the substituent, i.e. polarizability for sulfonyl but resonance for nitro carbanions, is pointed out. The paper showed the structural characterization of complexes formed by title compounds derived from experimental and theoretical results. The crystal data agree very well with those obtained in solution and with computational studies. Despite the results obtained by experimental and theoretical investigations, there is still a need for a debate concerning the distribution of the negative charge in ionized sulfonyl carbon acid. The main findings show that sulfonyl carbanions play an increasing role in organic synthesis, especially in organometallics chemistry. Recently, intense research into developing synthetic methods for preparation of chiral carbanions has aroused much attention. The knowledge of the structure allows us to understand the reactivity as well as selectivity of the reaction.