

1 Introduction
Application of vinyl sulfones or α,β-unsaturated sulfones in organic synthesis has increased dramatically, and to a limited extent, in biology. Vinyl sulfones are now widely used as useful intermediates in organic synthesis because vinyl sulfones serve efficiently as Michael acceptors as well as 2π-partners in cycloaddition reactions [1–16]. Carbohydrates, on the other hand, are used extensively as chiral building blocks for the synthesis of various complex molecules [17–24]. The preliminary requirement of such a synthesis is the functionalization of the sugar molecules at the monosaccharide level. It is therefore logical to argue that vinyl sulfone-modified (VSM) carbohydrates have the potential for utilization in organic synthesis because after using the vinyl sulfone moiety as a tool for functionalization, desulfonylation under strategically selected conditions [25] would easily generate an array of modified carbohydrates. It is expected that the “in-built” chiralities in sugar molecules would contribute significantly in determining the stereochemical outcome of reactions of VSM carbohydrates.
As part of a research program dealing with the modification of carbohydrates, we intended to investigate a vinyl sulfone-based strategy for the functionalization of carbohydrates for primarily accessing amino- and branched-chain sugars. Limited contributions of other groups in this area have been discussed in detail elsewhere [26–28]. This review attempts to highlight the strategies used by us in combining the vinyl sulfone functional group and carbohydrates as extensively as possible and studying their properties as reactive intermediates.
2 Background
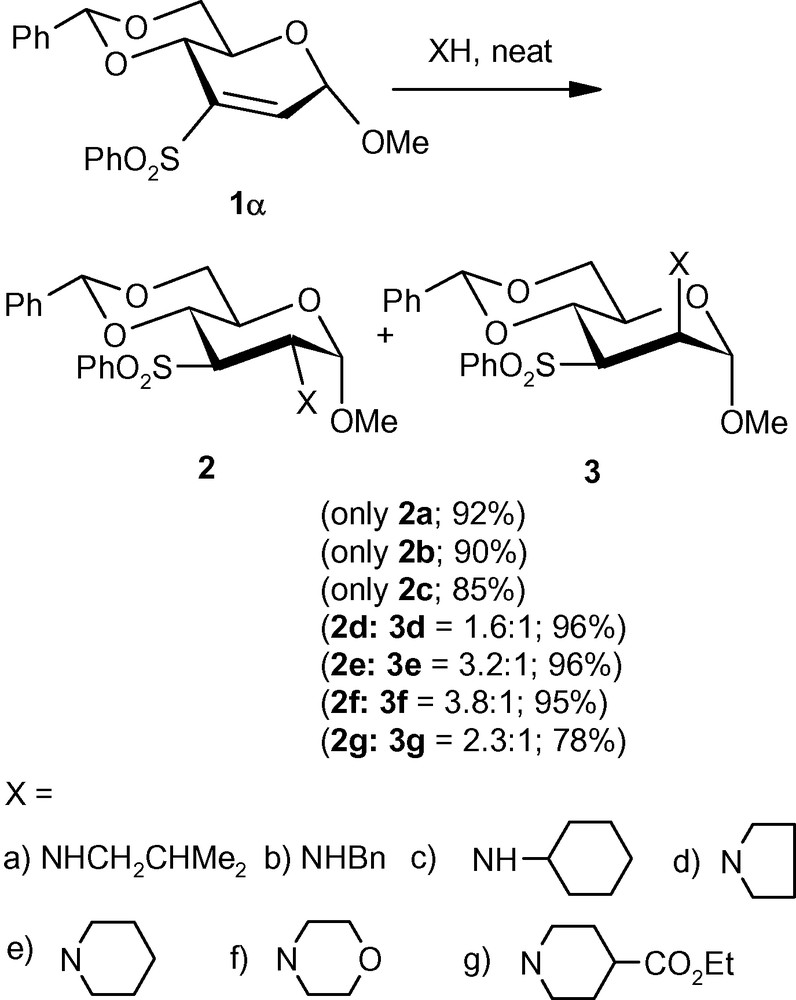
To develop methodologies for the synthesis of new aminosugars, a VSM hex-2-enopyranoside 1α was treated with various primary and secondary amines [29]. Primary amines, such as isobutylamine, benzylamine and cyclohexylamine were found to add diastereoselectively to produce single isomers 2a–c with the d-gluco- configuration (Scheme 1). The secondary amines, pyrrolidine, piperidine, morpholine and ethyl isonipecotate, on the other hand, generated a mixture (isomeric at C-2), also having the d-gluco- 2d–g as the major isomers, respectively. The major isomers 2d–g were separated by crystallization. One of the minor manno-isomers 3d was isolated and unambiguously characterized (Scheme 1) [29].
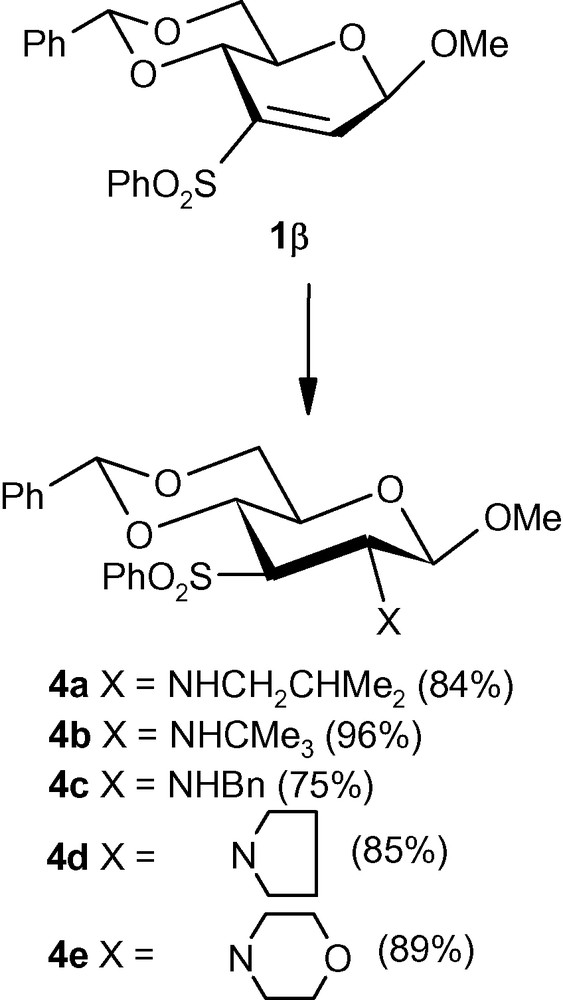
To examine whether the anomeric configuration influenced the diastereoselectivity of the addition of various nucleophiles to 3- sulfonyl-2-enopyranoside systems, the β-anomeric vinyl sulfone 1β was treated with isobutylamine, benzylamine, tert-butylamine, pyrrolidine and morpholine. The primary as well as secondary amines were found to add diastereoselectively to produce single β-D-gluco- isomers 4a–e, respectively (Scheme 2) [29]. It should be noted that 1α did not react with sterically bulky tert-butylamine and unreacted starting material was recovered from the reaction mixture. Attempted reactions under forcing conditions or prolonged reaction time caused extensive degradation of the starting material. The β-anomer 1β, on the other hand, reacted smoothly with the same amine at elevated temperature to produce a single isomer 4b in excellent yield (Scheme 2) [30].
From the above observations, we concluded that the anomeric configuration was playing a crucial role in determining the stereochemical outcome of this type of addition reaction. We therefore decided to expand the scope of this study to other pyranosides as well as furanosides. However, a thorough study on the diastereoselectivity of the addition of a range of nucleophiles to vinyl-sulfone modified carbohydrates required an easy access to a relatively large amount of a wide variety of VSM carbohydrates including anomerically pure analogues 1α/1β, 5α/5β, 6α/ 6β, 7α/7β, and 8–13 (Fig. 1).
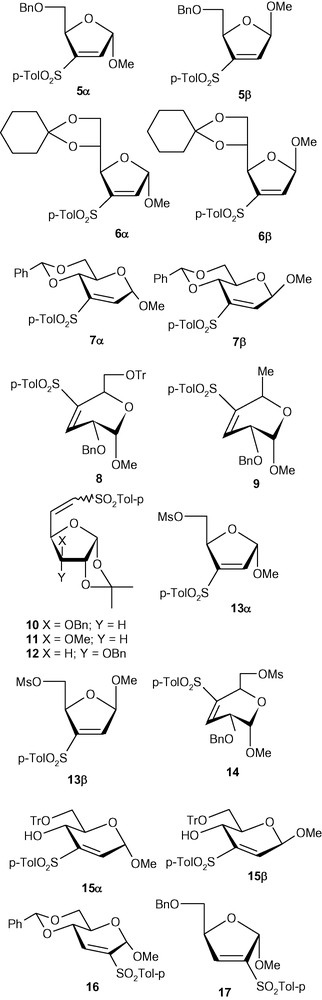
VSM carbohydrates.
3 Vinyl sulfone-modified pyranosides and furanosides
3.1 Synthesis
The requirement of anomeric purity of compounds imposed greater restrictions on the choice of methodologies for the synthesis of a particular pair of anomers starting from a single and easily accessible starting material. A retrosynthetic analysis of the route to 1α/1β, 5α/5β, 6α/ 6β and 7α/7β necessitated the introduction of an arylthio group at the C-3 position of pentose or hexose sugars. One of the easiest ways of forming a C–S bond is the regioselective opening of epoxides derived from carbohydrates. Alternatively, the arylthio group at the C-3 position of a hexose or a pentose sugar could be introduced by displacing leaving groups at C-3 position of easily accessible starting materials [31,32]. Similar strategies would also lead to C–S bond formation at C-4 and C-5 leading to the synthesis of 8 [33], 9 [34] and 10–12 [35] respectively. Vinyl sulfones 13–17 constitute a special class of Michael acceptors used for specific transformations (Sections 3.5–3.7).
VSM pent-2-enofuranosides 5α and 5β were first synthesized from easily accessible precursors 18 and 21. The known lyxo-epoxide 18, synthesized from d-xylose, was converted to the vinyl sulfone 5α following thiation (18 → 19), oxidation by magnesium monoperoxyphthalate hexahydrate (MMPP) (19 → 20), and mesylation followed by elimination (Scheme 3). The known ribo-epoxide 21 was converted into the corresponding vinyl sulfone 5β following similar sequence of reactions (Scheme 4).
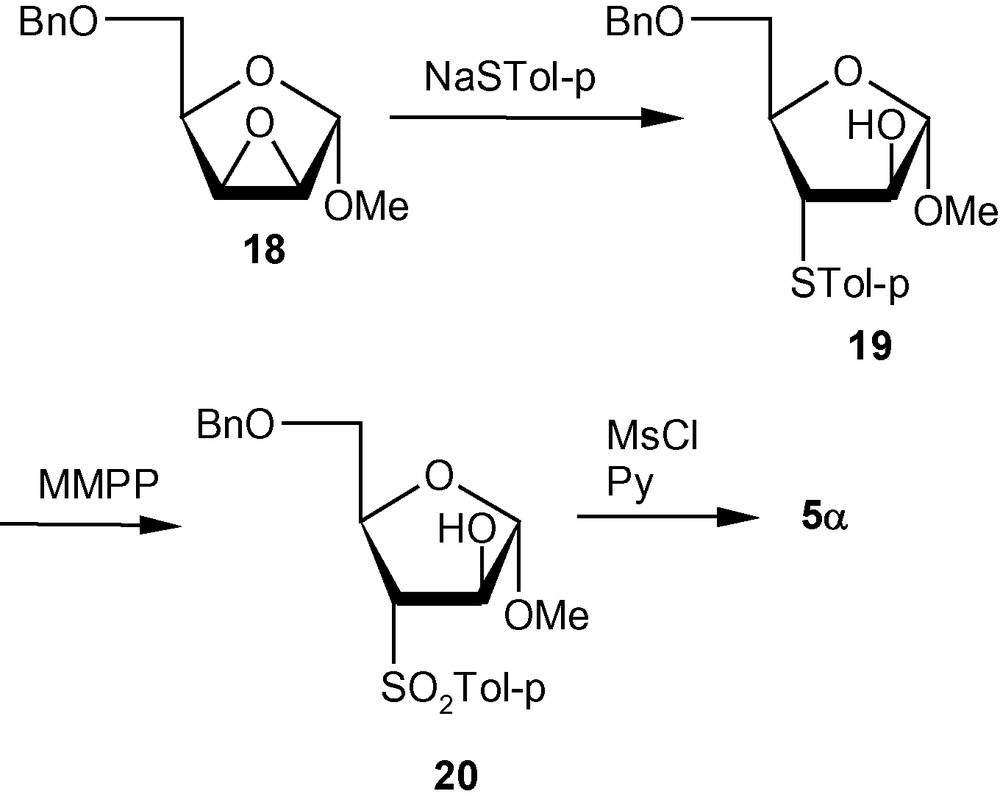

The separate synthetic routes for the starting epoxides 18 and 21, however, increased the number of steps and reduced the overall yield of the final products. Therefore, it was necessary to devise different approaches towards the synthesis of VSM pent-2-enofuranosides [31,32].
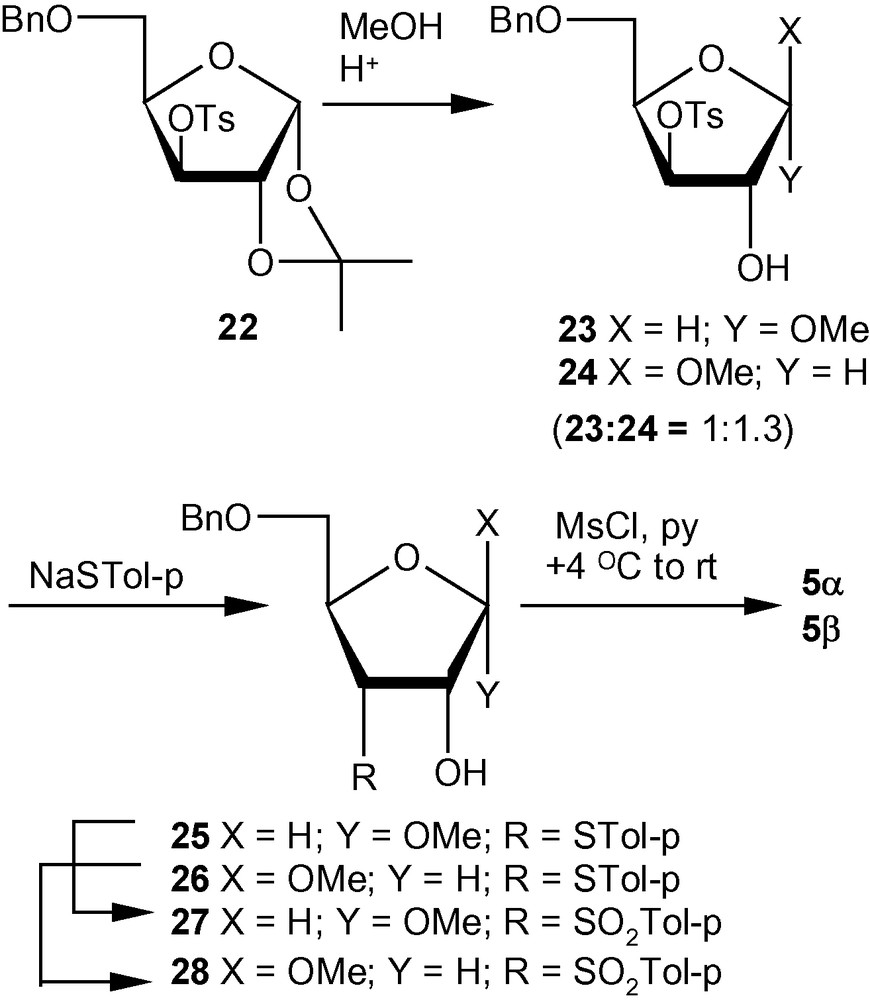
An examination of the percentage compositions of methyl furanosides of d-ribose, d-arabinose, d-xylose and d-lyxose revealed that the ratios of α- and β-furanosides present in equilibrium were 1:3.4, 3.1:1, 1:1.5 and only α-isomer, respectively [31,32]. Thus, the pattern of glycosylation of various pentose sugars led to the selection of a d-xylo derivative-based strategy for the synthesis of an anomeric mixture close to the ideal ratio of 1:1. Therefore, methanolysis of 22 produced an anomeric mixture of 23 and 24 in 1:1.3 (α/β) ratio. Nucleophilic displacement of the tosyl group in the mixture of 23 and 24 by p-thiocresolate proceeded smoothly at an elevated temperature to afford a mixture of ribofuranosides 25 and 26. Compounds 25 and 26 were separated and converted via 27 and 28 into the desired VSM carbohydrates 5α and 5β (Scheme 5) [31,32].
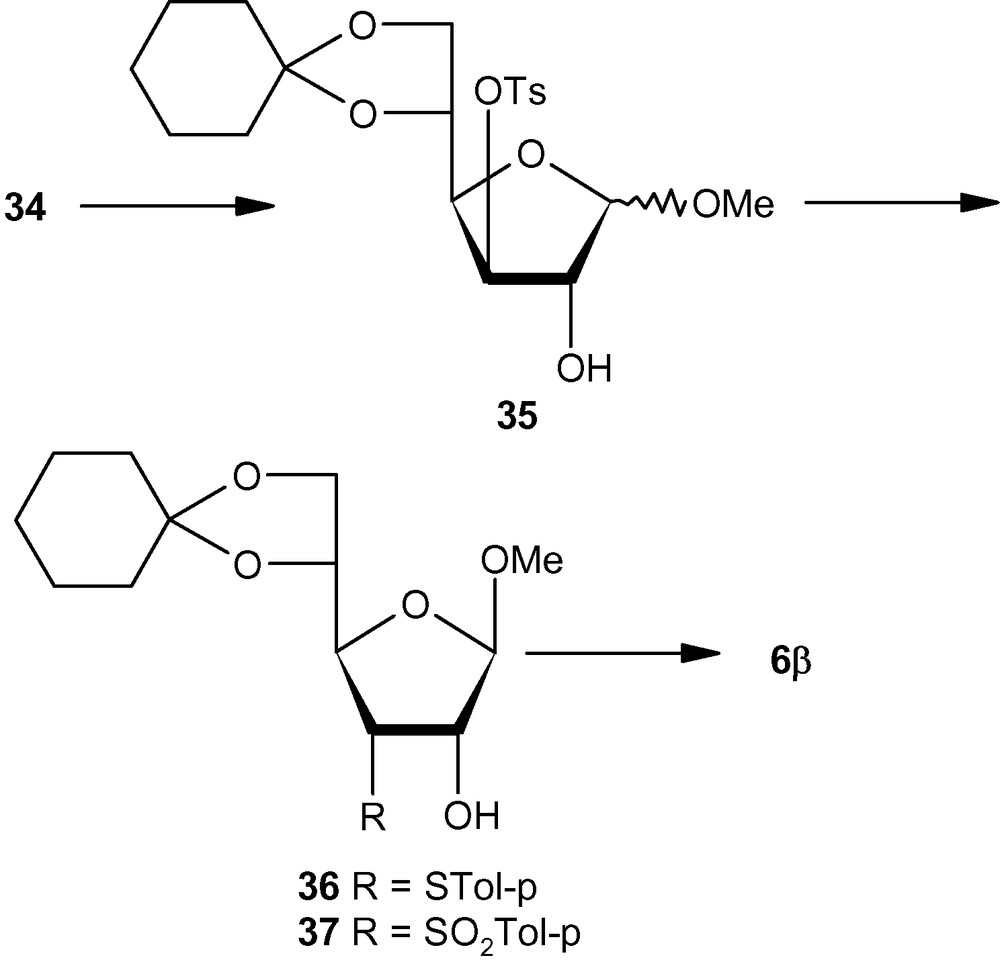
Compared to pent-2-enofuranosides 5α and 5β, the synthesis of hex-2-enofuranosides 6α and 6β was far more complicated because the anomeric ratio of methyl glycosides obtained from 29 (Scheme 6) or 34 (Scheme 7) was far from the ideal value of 1:1 [36]. It was therefore necessary to have two different approaches from the most easily accessible glucofuranose intermediates like 29 or 34. Thus, benzoate 29 on one-pot methanolysis and cyclohexylidenation produced a mixture, which contained the α-anomer in a major amount. Tosylation of the mixture produced 30, which on treatment with base produced an anomeric mixture of epoxides. The α-anomeric epoxide 31 was separated from the anomeric mixture and converted into the desired α-anomeric vinyl sulfone 6α via sulfide 32 and sulfone 33 in the usual way (Scheme 6) [36]. For accessing the β-anomer 6β, the tosylate 34 was converted into a mixture of methyl glycosides 35 in which the β-anomer was predominant. Displacement of the tosyl group by thiolate produced 36, which on oxidation produced 37. Mesylation of 37 led to the formation of β-anomeric vinyl sulfone 6β (Scheme 7) [36].

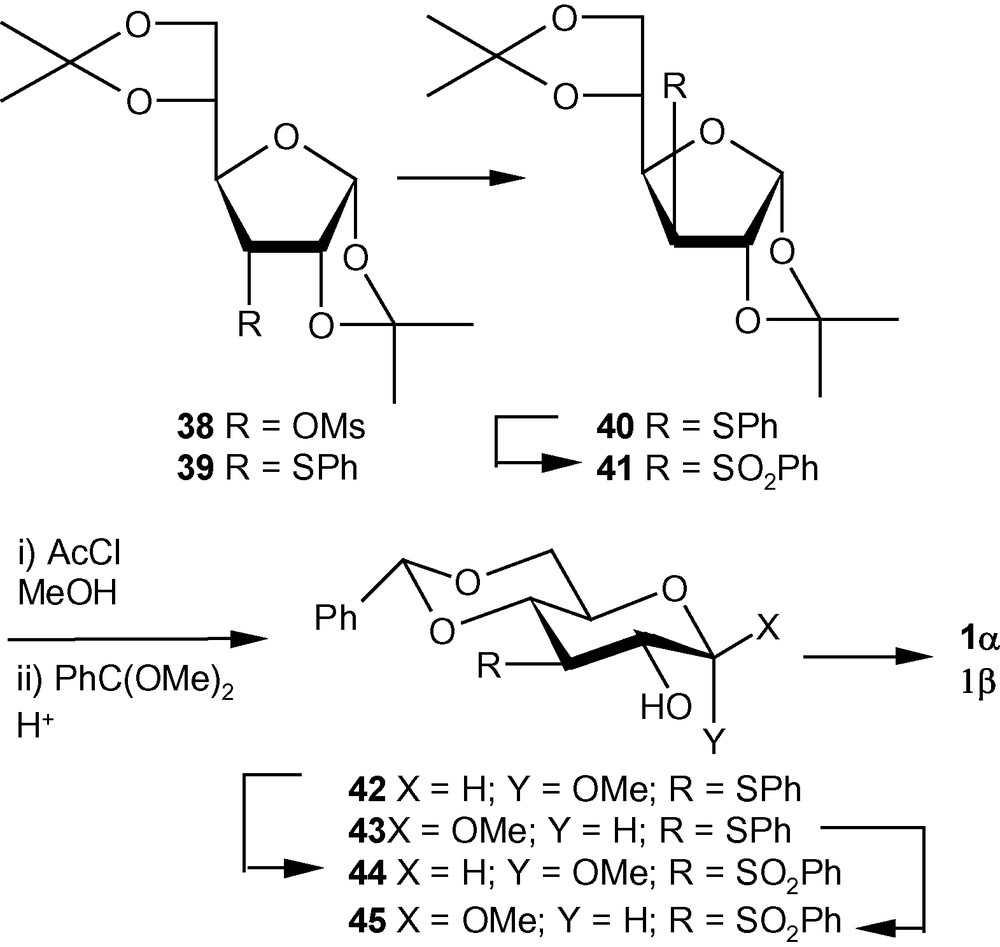
VSM hex-2-enopyranoside, 1α/1β and 7α/7β were initially synthesized from sugar epoxides by modifying the method reported by Sakakibara and coworkers [26–28]. However, this strategy turned out to be too long to be useful for accessing relatively large amounts of the anomerically pure compounds. Because of the fact that the arylthio group at the C-3 position of a hexose sugar could be introduced by displacing the leaving groups at the C-3 position as well, materials like 39 and 40 (Scheme 8) [32] were considered as starting sulfide derivatives for providing an easy access to both 1α and 1β from a single synthetic intermediate. It was known that the equilibrium mixture of methyl d-allosides in MeOH contained > 30% of furanosides, whereas d-glucose produced methyl d-pyranosides almost exclusively. Although the reported ratio of α- and β-anomers was not close to the ideal value of 1:1 needed for the synthetic strategy, in this case it was more important to obtain the methyl pyranosides without any contamination of the corresponding furanosides. In fact, methanolysis of the allo derivative 39 generated more than six products. Therefore, the gluco derivative 40, obtained from the mesylated allo derivative 38, was deprotected and glycosylated in a single operation by using acetyl chloride and MeOH to afford a mixture of 3-deoxy-3-phenylsulfide hexopyranosides; the diols, thus formed were collected as the benzylidene derivatives 42 and 43 in 2.2:1 ratio and in good yields [31,32]. The anomers were separated by chromatography and were converted separately into the corresponding sulfones 44 and 45 in excellent yields using MMPP in MeOH. In an alternative approach, compound 40 was oxidized to the corresponding sulfone 41. Compound 41 was deprotected and glycosylated to generate a mixture of anomeric sulfones, which were collected as the benzylidene gluco derivatives 44 and 45, respectively, in good yields in 1:1.8 ratio. After separation, the sulfones were converted into the desired VSM hex-2-enopyranosides 1α and 1β in the usual way (Scheme 8) [31,32].
In line with the synthetic routes discussed above, one of the easiest ways of forming a C–S bond at C-4 is the regioselective displacement of suitably oriented and protected sulfonates or regioselective ring opening of epoxides derived from the corresponding pyranosides. However, it is reported that ring-opening reactions at C3–C4 of epoxy sugars is not strictly regioselective. Moreover, the retrosynthetic analysis revealed that 4-thiopyranosides would be more easily accessible from 4-O-mesylates. Therefore, 4-O-mesylate 48 (Scheme 9) [33] was used as a common starting material for the synthesis of both 8 and 9. Thus, the known diol 46 was tritylated at room temperature by using trityl chloride and pyridine to afford 47 within 48 h. Mesylation of 47 afforded the required mesylate derivative 48 in 81% overall yield. Fully protected α-glucomesylate 48 was treated with p-thiocresol in DMF in the presence of 1,1,3,3-tetramethylguanidine (TMG) at 150–160 °C to afford galacto-derivative 49, as expected in good yield within 5 h. Treatment of compound 49 with methanolic NaOMe at reflux temperature for 7 h afforded compound 50 in 97% yield. The corresponding sulfone derivative 51 was generated in quantitative yield within 6 h at room temperature by oxidizing 50 with (MMPP) in MeOH. Compound 51 was subjected to an elimination reaction by using MsCl in pyridine at 0 to +4 °C to afford 8 in 88% yield (Scheme 9) [33].

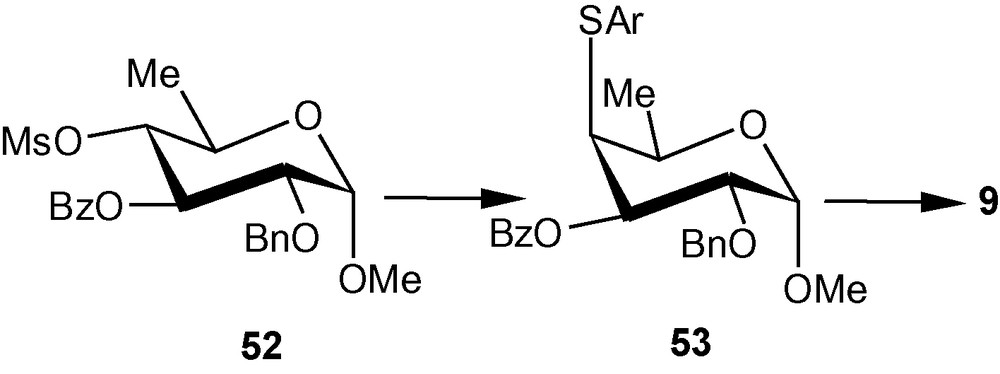
For the synthesis of 6-deoxy analogue of 8, the sugar derived mesylate 52 was reacted with p-tolylthiol in N,N-dimethyl formamide (DMF) in the presence of TMG at about 150–160 °C to afford 53 in 89% yield within 5 h. Compound 53 was finally converted to the required vinyl sulfone 9 (Scheme 10) [34].
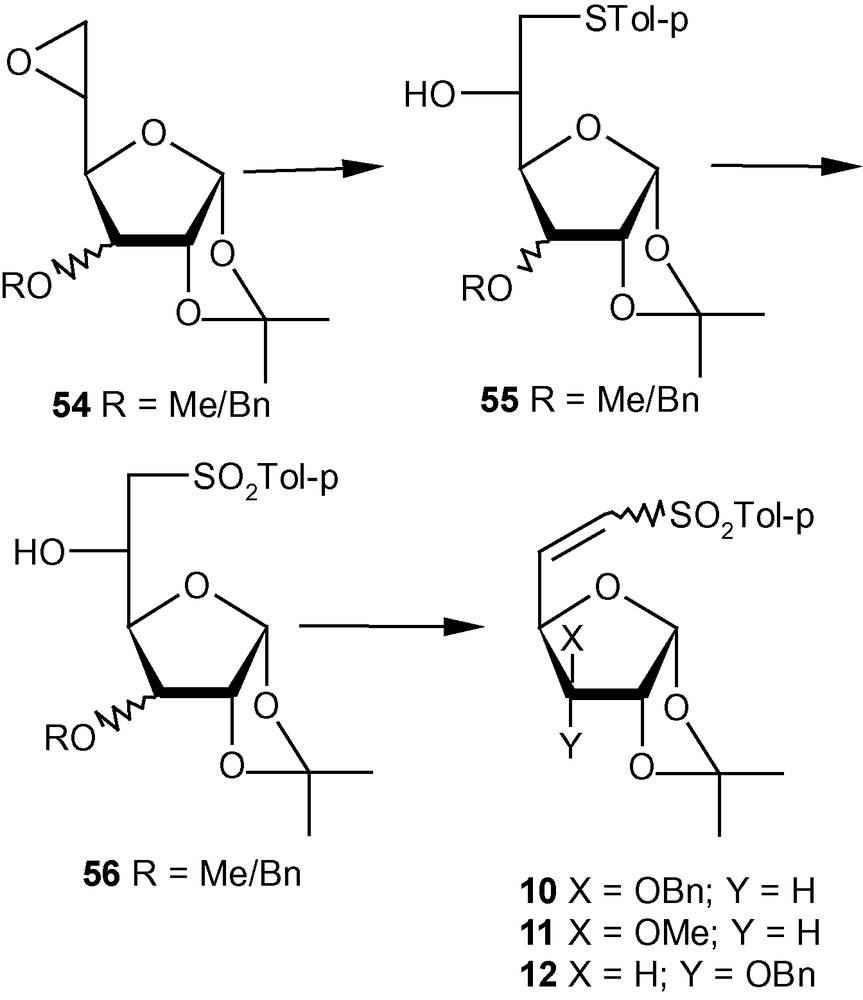
VSM hex-5-enofuranoside 10 and its analogues 11–12 were prepared from the easily accessible epoxides, represented by the general structure 54. Thus, epoxides 54 were reacted with sodium tolylthiolate to obtain 55 in high yields. Oxidation to 56 followed by mesylation and elimination afforded the desired vinyl sulfones 10–12 (Scheme 11) [35]. Vinyl sulfone 10 was obtained as a mixture of E and Z isomers in 3:1. Interestingly, the variation of the group at C-3 affected the E/Z ratios to some extent. Thus, by changing from OBn in 10 to OMe in 11, the E/Z ratio changed from 3:1 to 3:2; in the case of 12, however, only E isomer could be detected. Since the presence of OBn in 10 or OMe in 11 at C-3 caused the formation of Z isomer (alongside E isomer) as opposed to the exclusive formation of E isomer in the case of 12, it may be argued that the stereoselectivity of abstraction of protons from C-6 of 56 during the elimination reactions of the mesylated products was dictated by the stereoelectronic properties of the group present at C-3 on the β-face of furanosides [35].
3.2 Functionalization at C-2
As mentioned earlier, the addition of primary amines to C-2 of both 1α and 1β exclusively produced C-2 equatorial (gluco) products. Secondary amines on reactions with 1β produced only gluco derivatives but with 1α produced mixtures containing the gluco derivative as the major component [29]. Reactions of tert-butylamine with 1α and 1β indeed exemplified an extreme case of anomeric configuration influencing the addition of nucleophiles to enopyranoside systems 1α/1β. The X-ray analysis of the single crystal of 4b revealed that the pyranose ring assumed a boat conformation to facilitate the positioning of the methoxy group of C-1 away from the group at C-2. It was observed from the X-ray analysis of 4a, 4b and 4c (Scheme 2) that the magnitude of the conformational angles O1-C1-C2-N1 increased in the order 4a → 4c → 4b confirming this argument [30,37]. However, in the case of α-methoxy series (compounds 2; Scheme 1), any increase in this particular conformational angle is prohibited because of the axial disposition of C-1 methoxy group. Thus a bulkier group, greater than a critical size, could not be accommodated at the C-2 position of the α-anomers [30]. However, all our efforts to deliver primary amines effectively and exclusively from the β-face of the pyranose ring failed.
Although the diastereoselectivity of formation of products 3d–g may be explained to some extent on the basis of the stereoelectronic repulsion of amines by the anomeric configuration of 1α, an in-depth analysis was necessary to explain the pattern of addition of amines. Quantum chemical ab initio and DFT calculations performed on these systems established that neutral nucleophiles like amines add onto the Michael acceptors via a concerted mechanism; the transfer of proton to the α-position of the sulfonyl group involves a proton relay process thereby lowering the activation energy for this type of reactions to produce diequatorial compound like 7α-B (7α → 7α-A → 7α-B; Scheme 12) [38]. The explanation is applicable to the reaction patterns of both 1α/1β and 7α/7β. The formation of minor product 3d from 1α, however, seems to be a stepwise process (Scheme 13) [38]. The transition state 1α-Ts1, formed via the abstraction of pyrrolidine proton by the second pyrrolidine ring transforms into an activated complex 1α-AC for the anti-face proton transfer to the olefinic double bond. The protonated pyrrolidine in 1α-AC interacts with sulfone oxygen near the newly formed carbanion, which eventually leads to the formation of the second transition state 1α-TS2 with the final transfer of proton from the amine to generate the 3d (Scheme 13). 1α-TS2 is slightly lower in energy compared to the complex, which suggests that the proton transfer process from the protonated pyrrolidine could take place instantaneously without involving these steps [38].


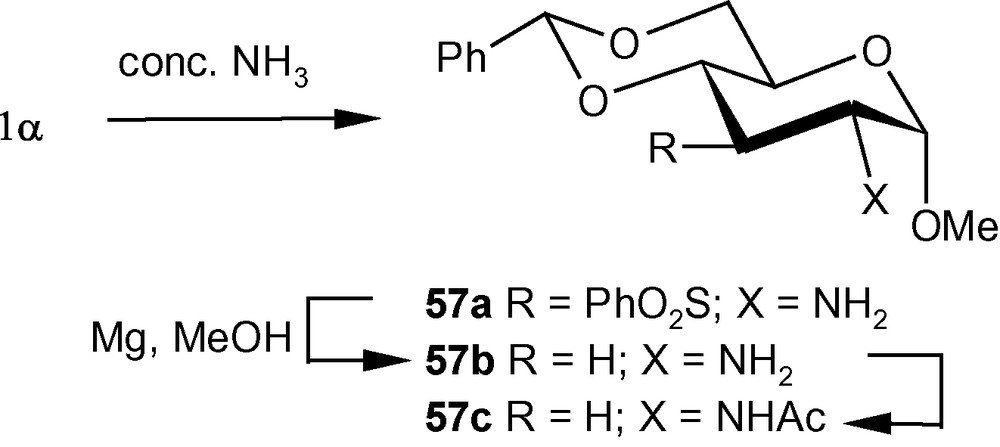
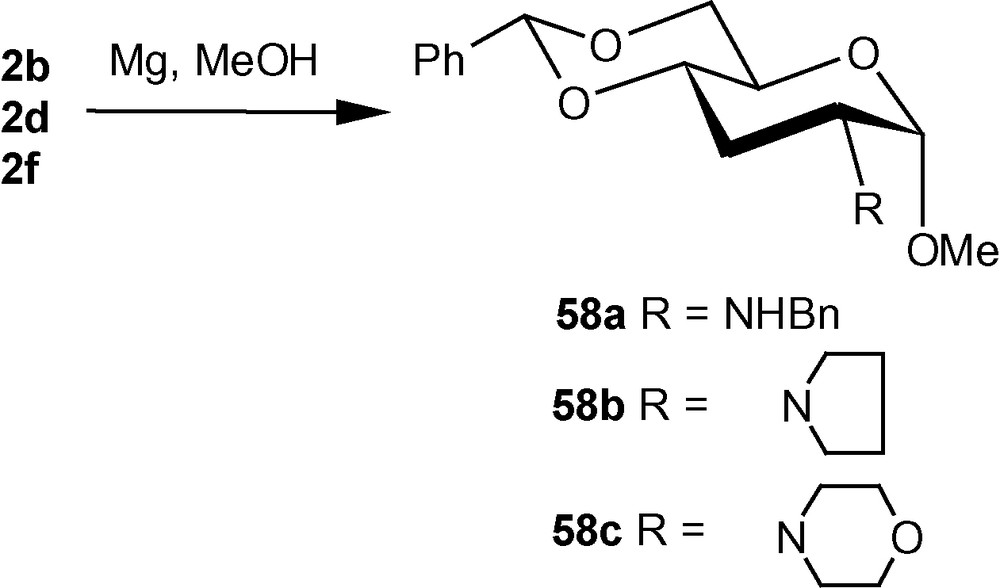
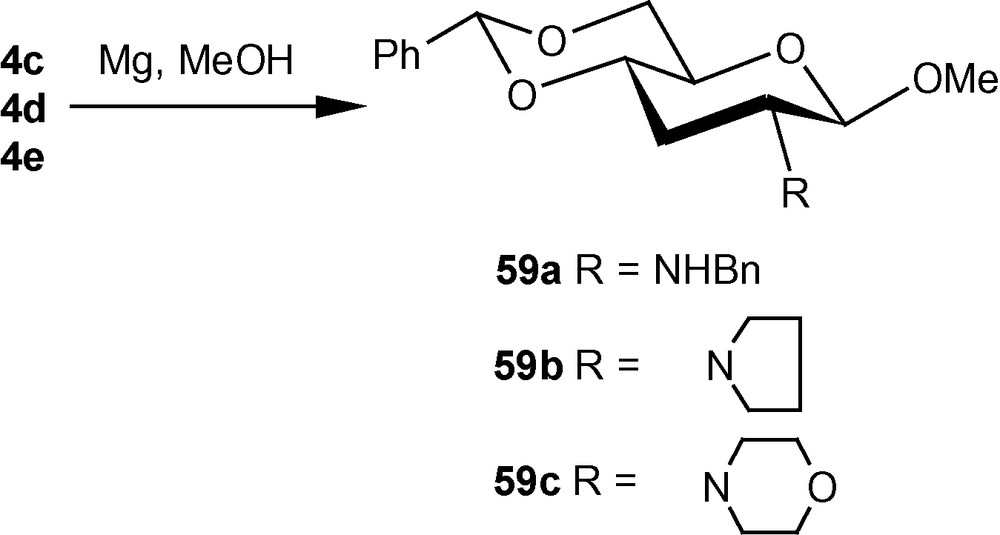
The diastereoselective addition of primary amines to 1α/1β has been applied to the synthesis of a naturally occurring aminosugar d-lividosamine and its analogues [39]. d-lividosamine (2-amino-2,3-dideoxy-d-glucose) was isolated from Streptomyces lividus, and it is present in aminoglycoside antibiotics such as lividomycin-A/lividomycin-B. Thus, compound 1α was treated directly with conc. aq. ammonia in dioxane to produce a mixture containing mainly compound 57a. The mixture was desulfonylated and the free amino product 57b was acylated. Pure aminodeoxysugar 57c, a known intermediate for the synthesis of d-livodosamine, was crystallized from benzene-pet. ether mixture in 65% overall yield (Scheme 14) [39]. Analogues of 57b were easily obtained by desulfonylating 2b, 2d and 2f to 58a–c in high yields, respectively (Scheme 15) [39]. In the β-series, 4c–e were also desulfonylated to 59a–c, respectively (Scheme 16) [39]. None of the known methods of amination of pyranosides could have been used as a general route for the synthesis of d-lividosamine and its analogues, either because of the undesired configuration or position of the C–N bond and/or the additional functionalization of the C-3 hydroxyl group required for the deoxygenation of the C-3 center [39,40].
The importance of branched chain sugars led us to study independently the reaction pattern of carbon nucleophiles to 1α/1β. Thus, the nucleophiles generated from nitromethane and dimethylmalonate, reacted with 1α to produce single isomers 60a and 60b respectively (Scheme 17) [41]. On the other hand, NaCH2NO2 and NaCH(CO2Me)2 reacted with 1β to produce the single isomers 61a (56%) and 61b (98%), respectively (Scheme 18) [41]. It should be noted that, in contrast to the addition pattern of amines to 1α, carbon nucleophiles exclusively added to C-2 from a direction opposite to that of the disposition of the anomeric methoxy group [41].

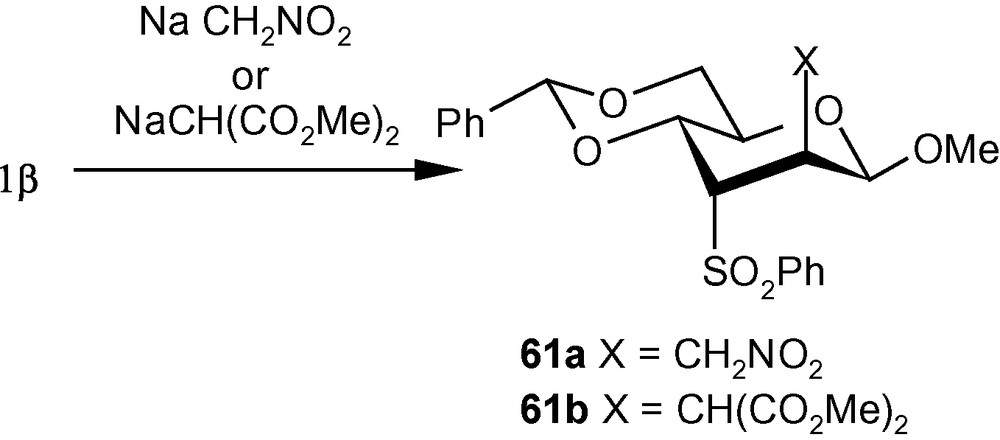
VSM pent-2-enofuranosides 5α/5β also reacted with a wide range of nitrogen [36,42] and carbon nucleophiles [41]. Thus, the reaction of 5α with 30% aqueous ammonia and a series of neat amines produced exclusively compounds 62a–e in high yields (Scheme 19) [42]. The reaction of 1,2,4-triazole with 5α in the presence of TMG in DMF at ambient temperature produced a single isomer 62f in high yield [31]. Compound 5α on reactions with carbanions generated from NaCH2NO2 and NaCH(CO2Me)2 produced branched-chain sugars 62 g and 62 h, respectively [41]. The β-anomer 5β, on the other hand reacted with imidazolyl ion to produce a separable mixture of a xylo- and a ribo- analogue 63a and 63b respectively in 1:1 ratio (Scheme 20) [31]. Although in this case a mixture was generated, both 63a and 63b resulted from the attack of the imidazolyl ion from a direction opposite to the disposition of the anomeric methoxy group. However, 5β on reactions with NaCH2NO2 and NaCH(CO2Me)2 produced branched-chain sugars 63c and 63d respectively [41]. Interestingly, vinyl sulfone 5β on reactions with 30% aqueous ammonia, neat benzylamine, cyclohexylamine, and pyrrolidine at ambient temperature, produced single isomers 63e–i in high yields (Scheme 20) [36]. In all these cases, the arabino derivatives were the sole products. Only morpholine produced an inseparable mixture of 63i/63i’ in 1:1 ratio (Scheme 20) [36]. It should be noted that 5α produced the expected d-arabino derivatives 62a–h (Scheme 19) where the nucleophiles were delivered from a direction opposite to the disposition of the anomeric configuration; [42] 5β with three nucleophiles also produced the expected d-xylo- and d-ribo-products 63a–d (Scheme 20) [31,41]. However, in contrary to our expectations, 5β on reactions with another set of nucleophiles (amines) produced aminosugars 63e–i with the d-arabino configuration [36].
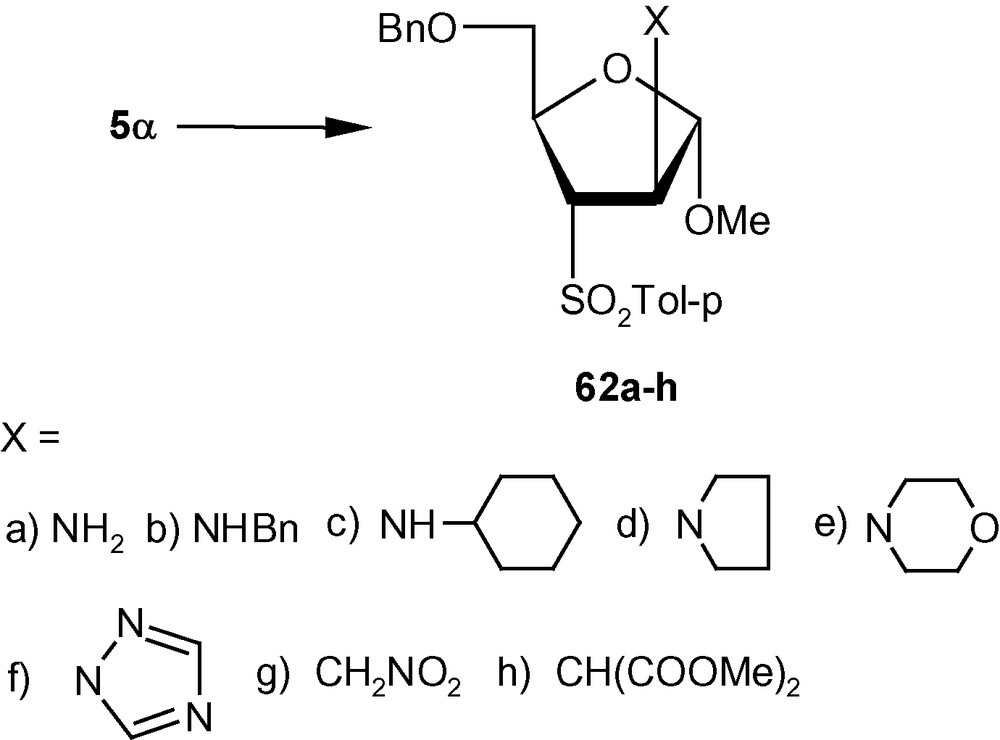
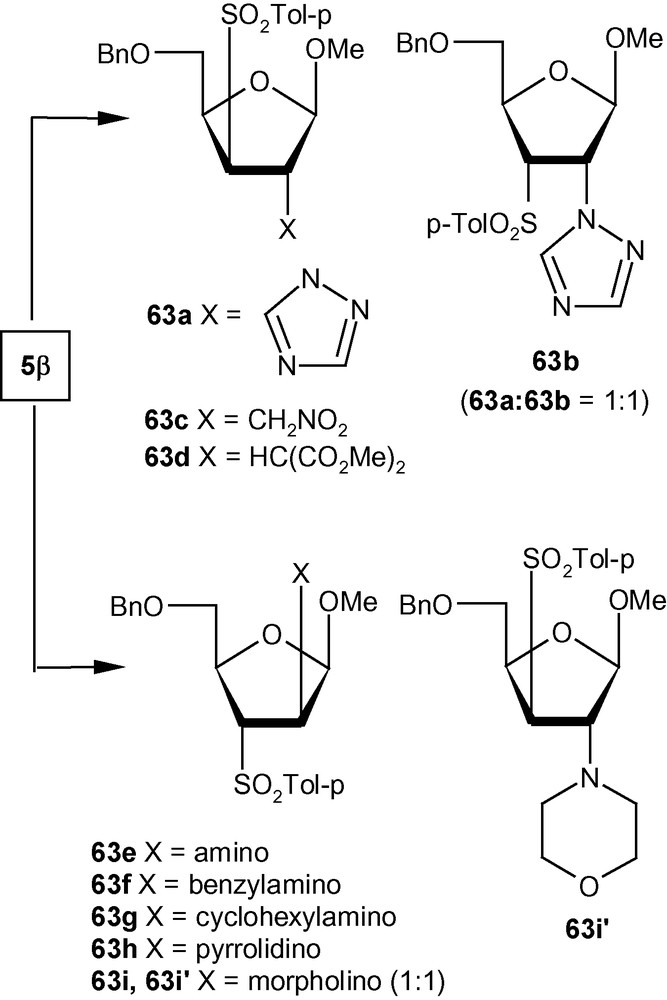
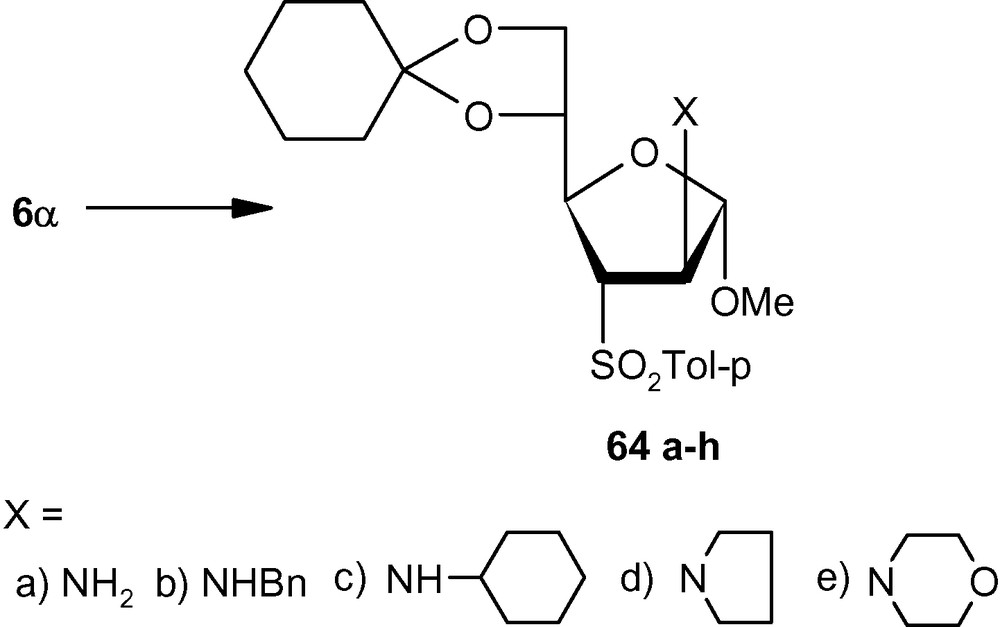
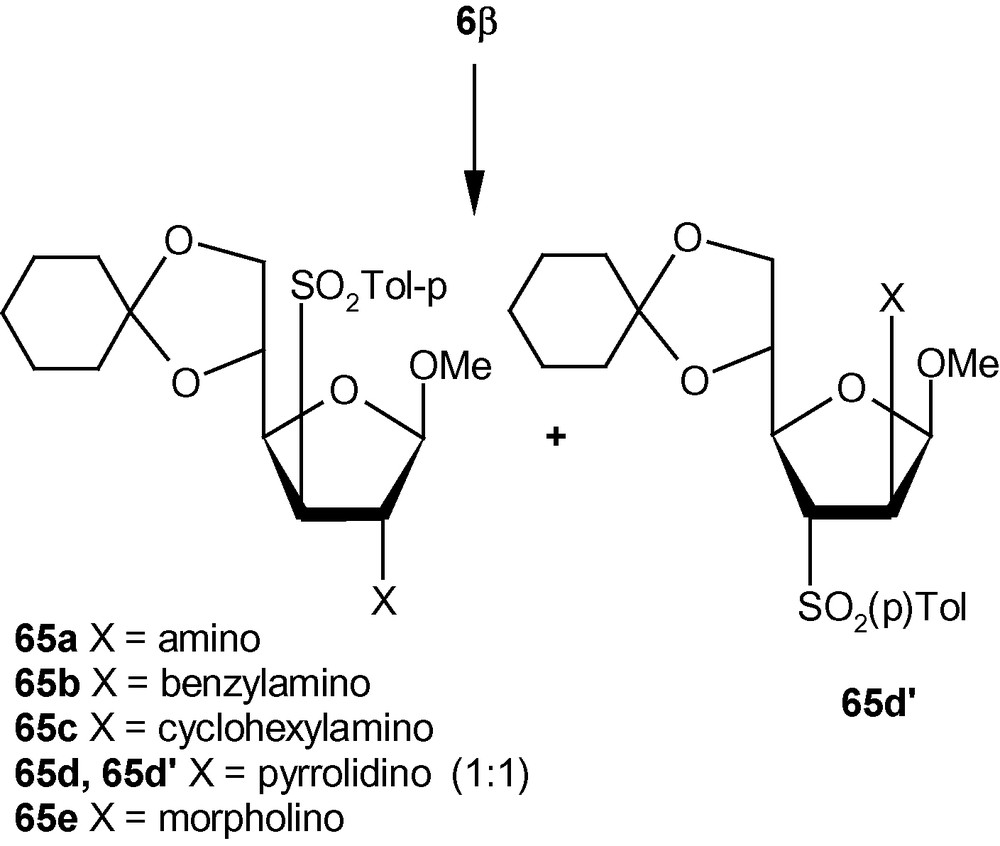
To establish the fact that the formation of 63e–i was indeed the result of an “unusual” addition reaction, we intended to examine the similar addition reaction patterns of related VSM hex-2-enofuranosides 6α and 6β. Hexofuranoside 6α on reactions with 30% aqueous ammonia, neat benzylamine, cyclohexylamine, pyrrolidine, and morpholine at ambient temperature, generated single isomers 64a–e in high yields (Scheme 21) [36]. Similarly, vinyl sulfone 6β on reaction with the same group of amines generated single isomers 65a–c and 65e in high yields. Only pyrrolidine afforded an inseparable mixture of compounds 65d/65d′ in a ratio 1:1 (Scheme 22) [36]. It was therefore clear from these experiments that, except for the formation of 65d′, amino nucleophiles were always delivered to C2 of 5α, 6α, and 6β from a direction opposite to that of the disposition of the anomeric methoxy group. It may therefore be emphasized that the addition pattern of amines to 5β was unusual in nature (Scheme 20) [36]. Interestingly, in the case of another β-anomeric VSM carbohydrate 5β, at least one nucleophile (pyrrolidine) showed the tendency of attacking C2 in the unusual way [36].
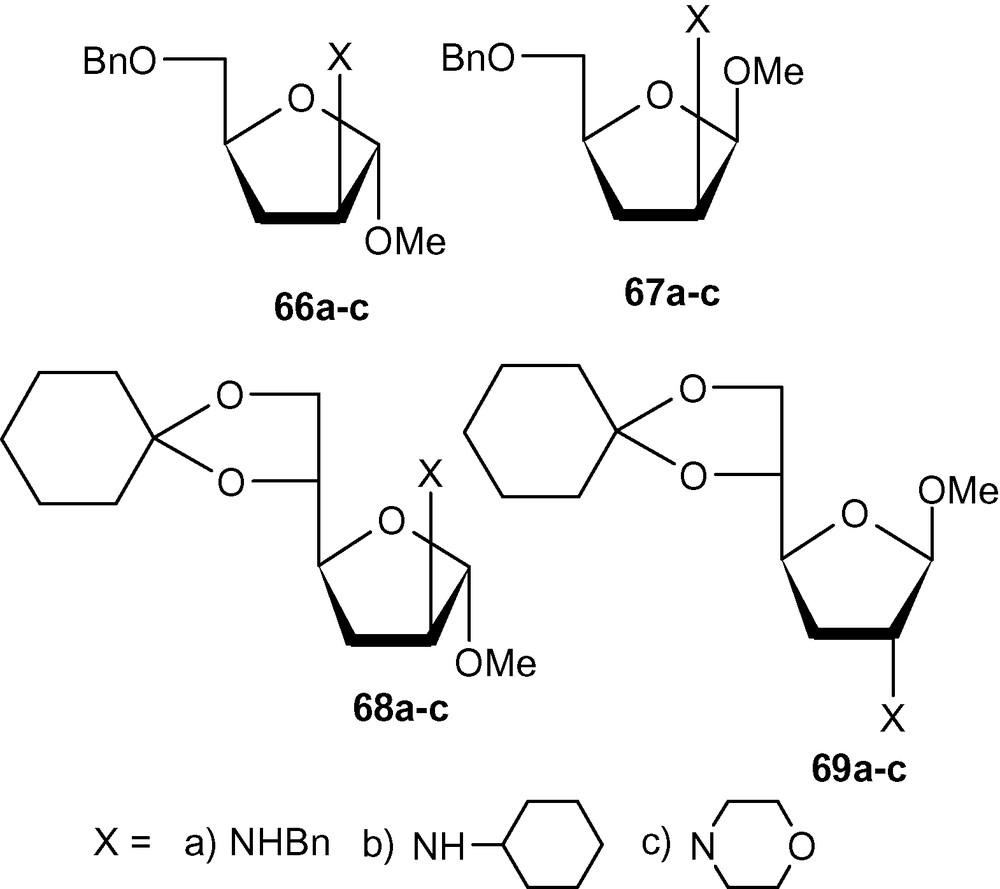
Successful synthesis of deoxyaminosugars using carbohydrate vinyl-sulfones would depend on the critical desulfonylation step. Although sulfonyl groups were successfully removed from pyranosyl derivatives (Schemes 15 and 16), all attempts to desulfonylate furanosyl derivatives with conventional reagents failed. After several experiments, we could desulfonylate a wide range of furanosides with Mg-MeOH-NiBr2 system to produce dideoxyaminosugars 66a–c, 67a–c, 68a–c and 69a–c (Fig. 2) [42].
The desulfonylation of a wide range of furanosides with the Mg-MeOH-NiBr2 system.
3.3 Functionalization at C-3
Barring only two reports on the use of systems like 2-sulfonyl-hex-2-enopyranoside as a Michael acceptor and 4-sulfonyl-hex-3-enopyranoside as a partner in a cycloaddition reaction [28], the strategy for the functionalization of the C-3 carbon of a VSM carbohydrate largely remains unexplored. We therefore, intended to synthesize and study the reaction patterns of a 4-sulfonyl-hex-3-enopyranoside 8 derived from d-glucose. Although, compound 8 on reactions with primary amines produced inseparable mixture of adducts, carbon nucleophiles generated from nitromethane and dimethylmalonate afforded single d-gluco adducts 70a and 70b, respectively (Scheme 23) [33].

The 6-deoxy analogue 9 on reactions with neat benzylamine at 80–90 °C generated a mixture from which the major product 71a was isolated in 79% yield (Scheme 24) [34]. Similarly, reactions of 9 with neat n-butylamine at elevated temperature afforded 71b in 83% yield. Aq. ammonia (30%) at room temperature produced a mixture from which the major product 71c was isolated in 81% yield; the product was however characterized as the N-benzoyl derivative 71d. Compound 9 on the other hand, on reactions with carbon nucleophiles generated from nitromethane or dimethylmalonate in the presence of t-BuOK in THF at reflux temperature, generated single compounds 72a and 72b in 73 and 78% yields respectively (Scheme 24) [34].

Although it is difficult to pinpoint the exact reasons for the diastereoselectivity of addition of nucleophiles to 8–9, it may be argued that for the formation of branched chain derivatives 70a–b (Scheme 23) and 72a–b (Scheme 24), the incoming nucleophile added to C-3 from a direction opposite to the disposition of C-2 OBn group; additionally the bulky substituent at C-3 preferred to orient itself at the equatorial position and thus among all other possibilities, the most stable products were formed. It should be noted that compounds 70a–b and 72a–b represent a special class of branched-chain sugars reported for the first time [33,34].
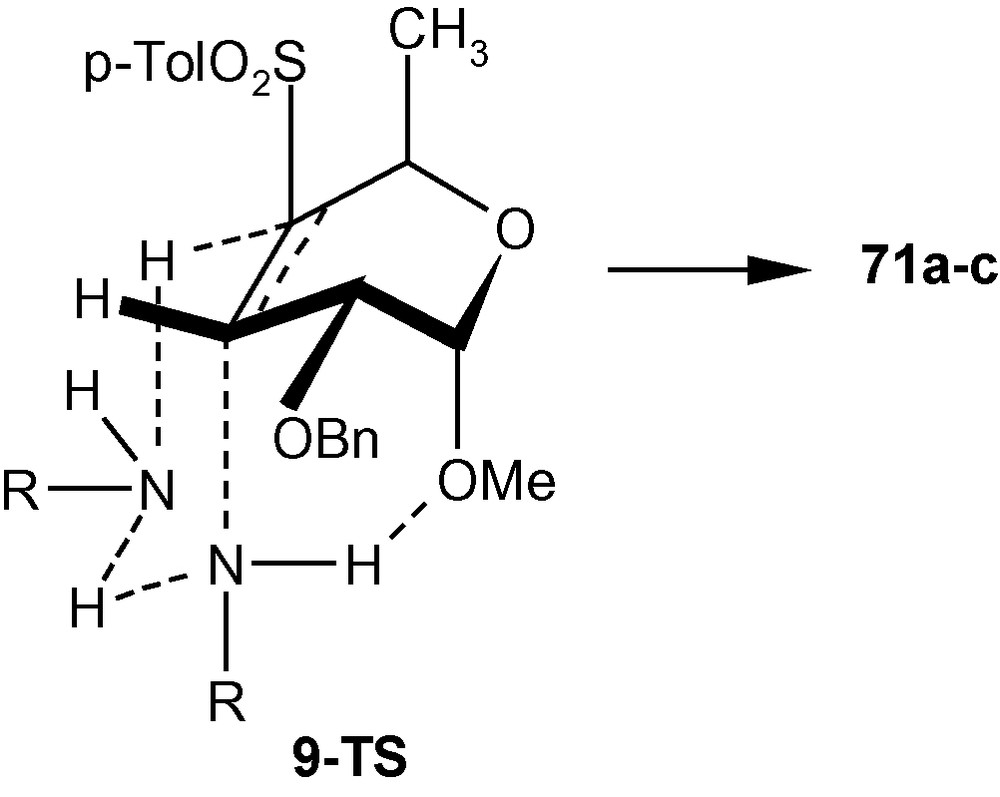
Since amines were expected to add to the C-3 position of 9 from a direction opposite to that of the disposition of the C-2 benzyloxy group to afford a d-gluco derivative having four (C2, C3, C4 and C5) equatorial bonds, the formation of products 71a–c with C–N axial bond at C-3 and C–S axial bond at C-4 was surprising. However, we have mentioned earlier that the addition of neutral species like amines to a Michael acceptor may take place through concerted mechanism involving a proton relay process (Scheme 12) [34]. Considering the same mechanistic pathway, we argued that the cis- addition of amines to compound 9 generated the trans- product 71a–c. It is highly probable that the preferential formation of trans-diaxial geometry in the transition state over the trans-diequatorial geometry resulted from the positioning of amines via six-membered transition states as depicted in Scheme 25. It is also probable that an additional H-bonding such as R–N---H---OMe stabilized the transition state 9-TS further to such an extent that the formation of tetra-equatorial products (like 72a–b) was overruled in favor of the formation of C3-C4 diaxial products 71a–c (Scheme 25) [34].
3.4 Functionalization at C-5
Methods for the functionalization of the C-5 position of hexose sugars are limited in number because the 5-O-sulfonylated hexoses are reluctant partners in nucleophilic displacement reactions and 5,6-epoxides (e.g. 54; Scheme 11) are regioselectively opened at C-6 when reacted with nucleophiles [35,43]. Therefore, the 3-O-benzylated gluco derivative 10 was reacted with neat benzylamine and isopropylamine to produce 73a/74a (9:1) and 73b/74b (9:1), respectively. In both cases, the l-ido derivatives 73a and 73b were the major products (Scheme 26) [35]. It should be noted that all addition reactions were performed using the inseparable E/Z mixtures of 10 and 11. It is therefore not possible to comment on the influence of these geometrical isomers, if any, on the stereochemical outcome of these reactions.

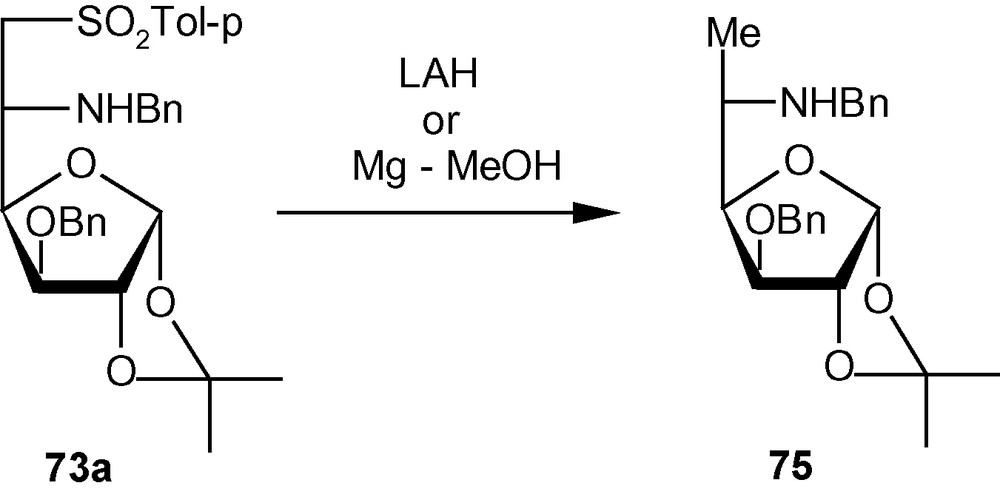
The 3-O-methylated gluco derivative 11 reacted in a similar fashion with benzylamine and isopropylamine to produce 73c/74c (9:1) and 73d/74d (9:1), respectively. In this case too, the l-ido isomer was the major product. It is noteworthy that the stereoelectronic effect of OMe at C-3 of compound 11 is sufficient to impose diastereoselectivity in favor of the l-ido derivative. The influence of C-3 substitution on the diastereoselectivity of addition was established further when the allo derivative 12 showed a significant to complete lack of diastereoselectivity of addition when reacted with benzylamine and isopropylamine. Compound 12, with a significantly reduced steric bulk at C-3 because of the presence of a hydrogen atom instead of β-OBn/OMe at C-3, produced inseparable mixtures of benzylamino and isopropylamino adducts in ratios of 1:1 and 3:2, respectively. A secondary amine piperidine reacted with 10 and 11 to produce 73e/74e (1:1) and 73f/74f (6:1) respectively (Scheme 26) [35]. The allo isomer 12 produced the piperidino adduct in a 1:1 ratio. It was possible to desulfonylate 73a to 75 using both LAH and Mg in MeOH (Scheme 27) [35].
This strategy was exploited further for the diastereoselective C–C bond formation at C-5 of hexofuranosyl carbohydrates using carbon nucleophiles [43]. Thus, vinyl sulfones 10 and 11 were reacted with NaCH2NO2 to generate the addition products 76a/77a (5.5:1) and 76b/77b (> 9:< 1), respectively, in very good yields (Scheme 28) [43]. The other nucleophile NaCH(CO2Me)2, reacted at a much faster rate with 10 and 11 to generate mixtures of the addition products 76c/77c (5:1) and 76d/77d (9:1), respectively, in very good yields [43]. The branched-chain sugars 76 and 77 were converted to a wide range of products 76e-h and 77e-h (Scheme 28) [43] using a variety of synthetic manipulations. Some of the latter compounds were utilized further for the synthesis of various modified carbohydrates 78–82 as well as carbocycles 83 and 85 (Fig. 3) [43]. As discussed in the case of the amino compounds (Scheme 26), the diastereoselectivity of the addition of carbon nucleophiles to 10 and 11 was controlled to a great extent by the configuration and substituents present at C-3. Therefore, as expected, the allo derivative 12 showed a complete or significant lack of diastereoselectivity of addition when reacted with carbon nucleophiles [43].
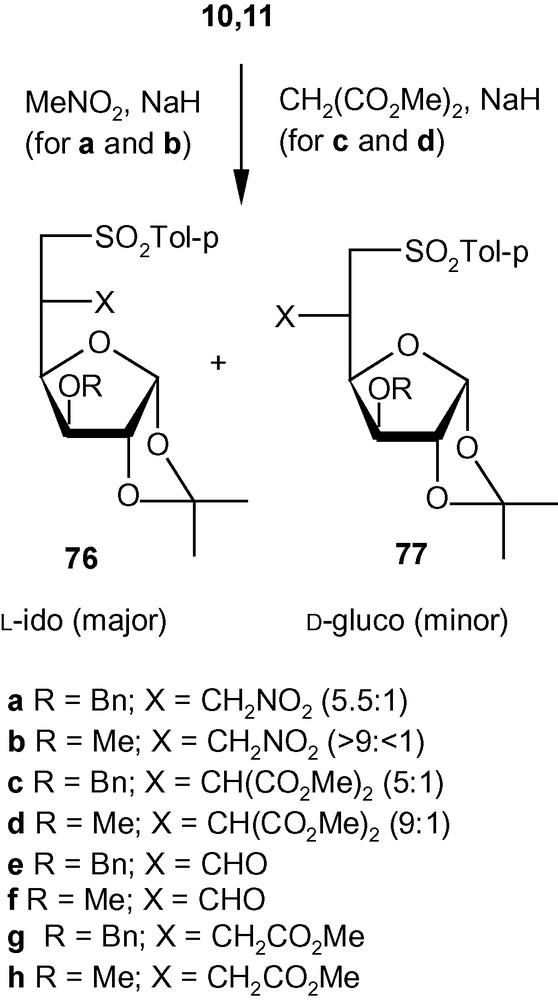
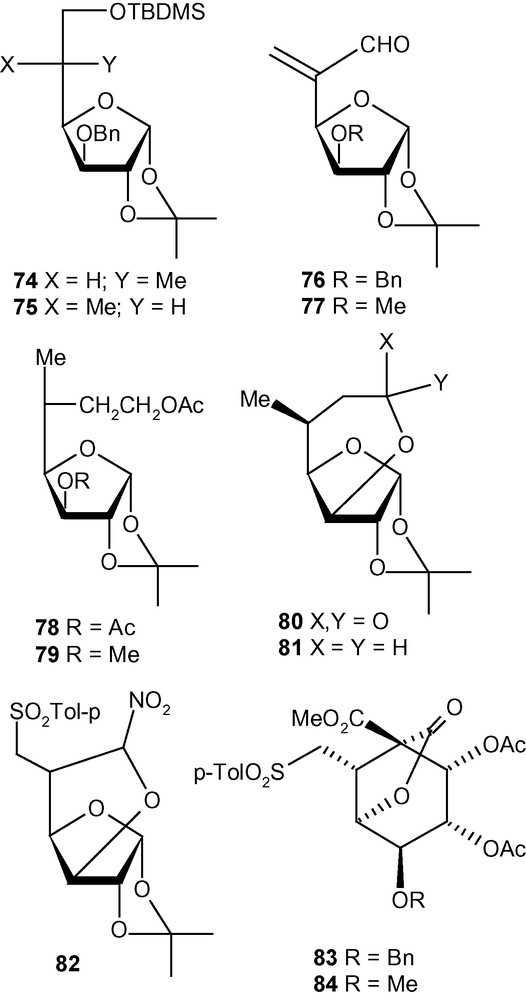
The synthesis of various modified carbohydrates and carbocycles.
To explain the diastereoselectivity of addition of amines to 10–12, we postulated the formation of an H-bonded transition state 10-TS or 11-TS having the geometry of a six membered ring (Fig. 4) [35]. This system fixed the transition state in the l-ido configuration. We argued that the stereoelectronic interactions between the R group (OBn/OMe) would allow the amine nucleophile to take up the position as shown in Fig. 4. Minimum interactions of primary amines with OBn (10-TS) or OMe (11-TS) allowed the amines to attack C-5 in a diastereoselective fashion via the H-bonded intermediate. A more severe interaction of OBn (10-TS) with a bulky secondary amine piperidine did not allow the formation of the hydrogen-bonded intermediate but highly reactive piperidine reacted with 10 anyway without any selectivity. A moderate interaction between OMe (11-TS) and piperidine allowed most of the reaction to proceed through a 6-membered intermediate. In the absence of any such intermediates in case of 12, both primary and secondary amines attacked C-5 from both sides without any diastereoselectivity [35]. However, the same concept is not applicable in the case of carbon nucleophiles because the negatively charged carbon nucleophiles are not capable of forming H- bonds with OMe or OBn groups present at C-3 positions of 10 and 11. An alternative explanation based on the model reported for the addition of primary amines to E (or Z)-6-bromo-5,6-dideoxy-1,2-O-isopropylidene-3-O-methyl-α-d-xylo-hept-5-eno-l,4-furanurononitriles was invoked to explain the product distribution in this case [43]. Thus, a carbon nucleophile was repelled by the OMe group of 11 and was forced to attack the double bond from the other side resulting in the formation of the l-ido product preferentially (Fig. 5A) [43]. In the case of 12, however, the carbon nucleophile was weakly repelled by the ring-oxygen and attacked the double bond from both sides resulting into a loss of diastereoselectivity of addition (Fig. 5B) [43].
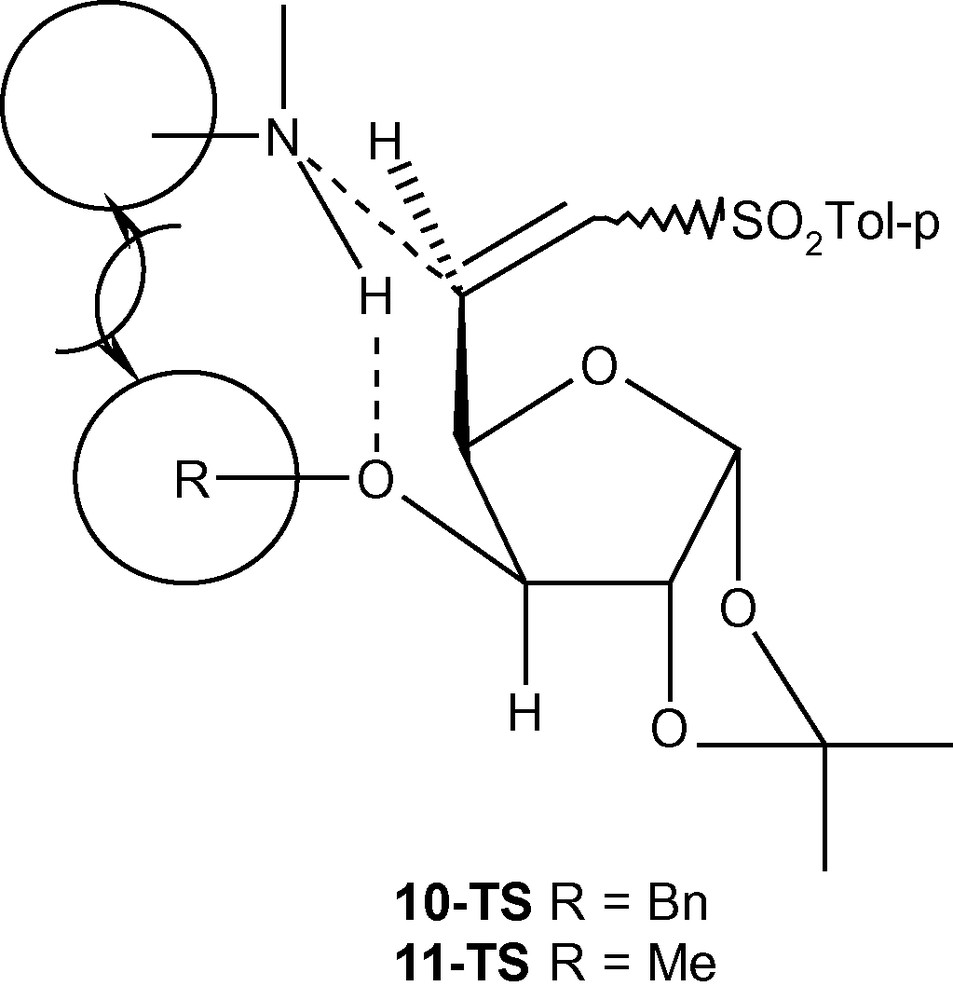
The formation of an H-bonded transition state 10-TS or 11-TS.
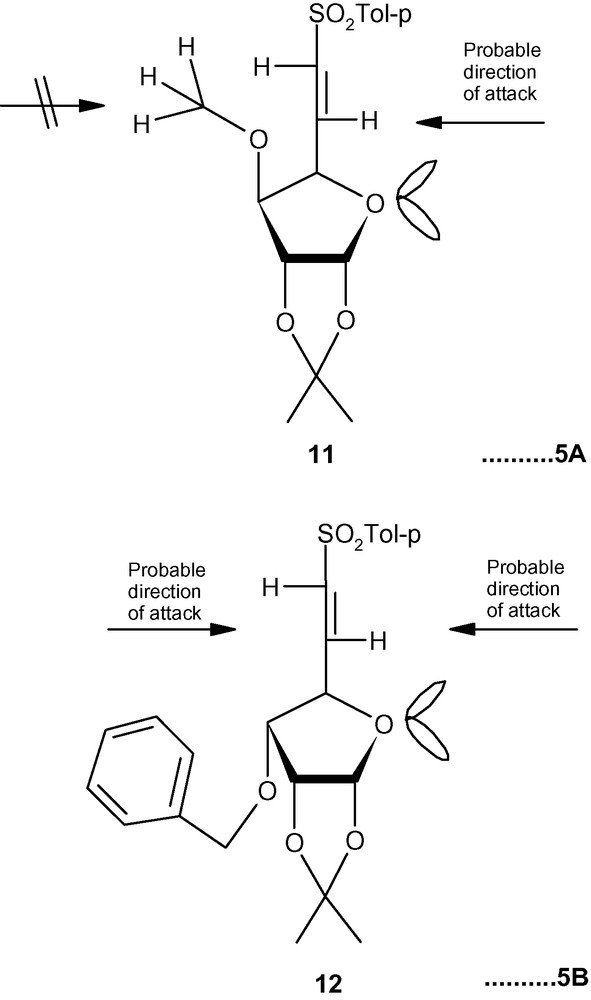
A: the preferential formation of l-ido product; B: the loss of diastereoselectivity of addition.
A critical situation arose in the case of 10. In theory, reaction of 10 should have resulted into the same diastereoselective product formation as was the case for 11 (Fig. 5). However, we presumed that an additional factor like the equilibrium between two rotamers 10A and 10B blocked both sides of C-5 (Fig. 6) resulting into a lower diastereoselectivity still favoring the formation of l-ido isomer 76a [43].

The equilibrium between two rotamers 10A and 10B block both sides of C-5.
3.5 Cyclopropanated carbohydrates
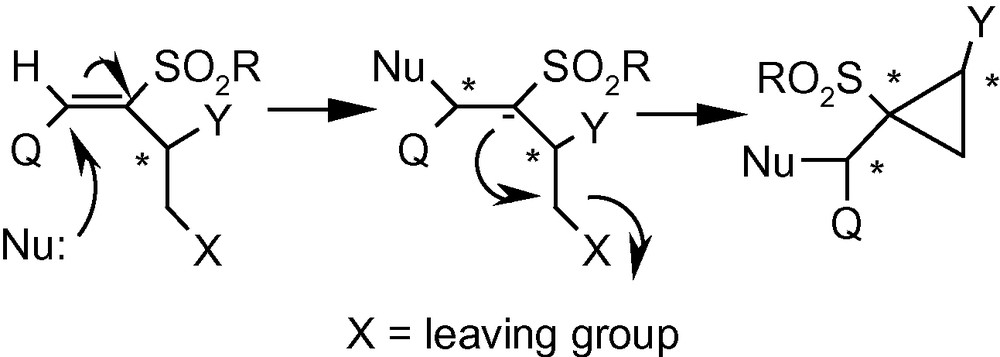
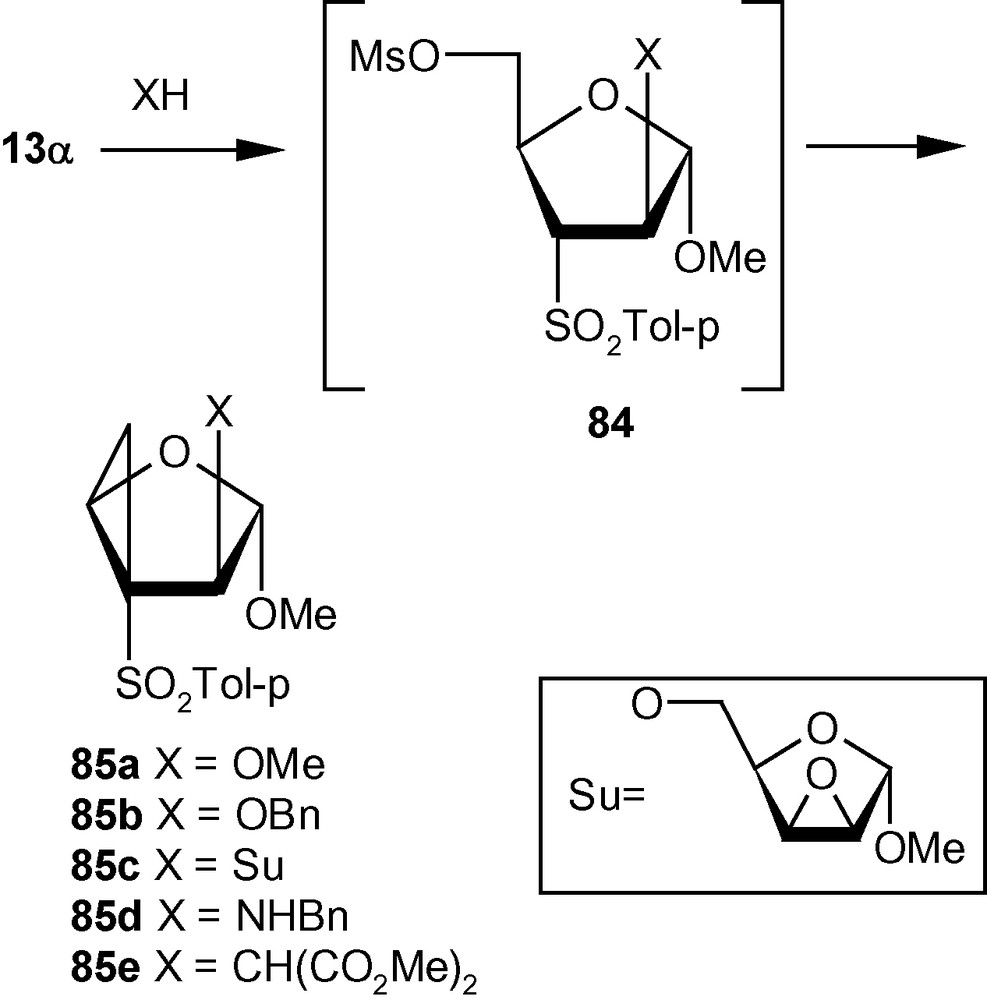
Integration of cyclopropanes and carbohydrates has been identified as an important area of research in cyclopropane related synthetic chemistry. This particular combination provides access to a class of strained and reactive cyclopropanes embedded in chiral appendages like carbohydrates. On the basis of the argument developed on Michael Initiated Ring Closure reactions (Scheme 29), we proposed that VSM carbohydrates with suitably positioned leaving groups, such as 13α, 13β and 14 should be capable of generating cyclopropanated carbohydrates [44]. Thus the VSM carbohydrate 13α was reacted with NaOMe or NaOBn, to get the cyclopropanated carbohydrates 85a and 85b, respectively in excellent yields (Scheme 30) [44]. A bulky, sugar-derived nucleophile, methyl-2,3-anhydro-lyxofuranoside in the presence of NaH easily produced a disaccharide 85c in high yield. Interestingly, reactions of benzylamine, and the sodium salt of dimethylmalonate with 13α did not produce any cyclopropane derivative but generated only addition products instead. However, these intermediates represented by the general structure 84, on treatment with K2CO3/MeOH or NaH yielded cyclopropanes 85d and 85e (Scheme 30) [44].
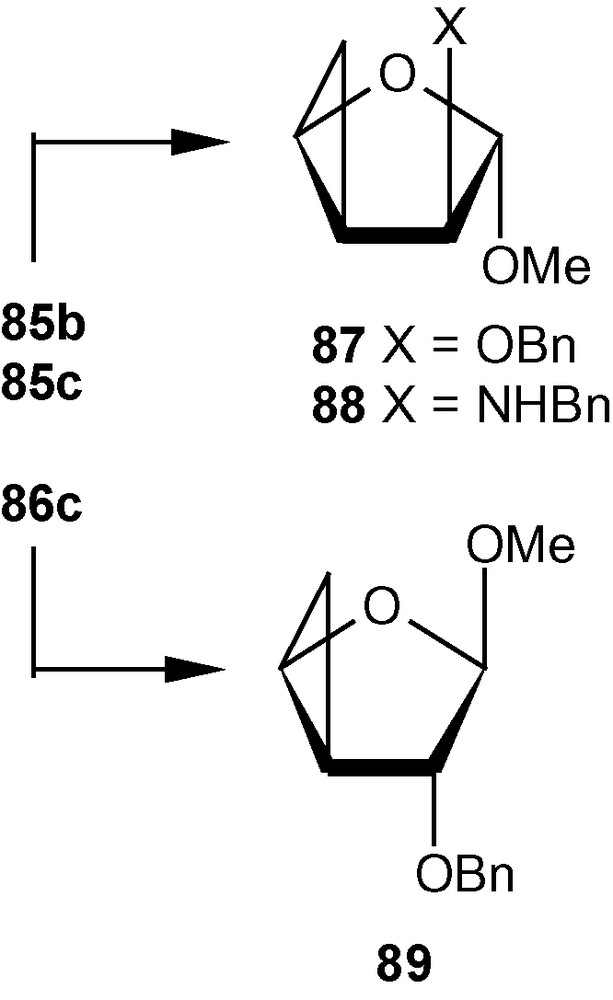
The other vinyl sulfone 13β, under similar conditions produced cyclopropanated carbohydrates 86a/86b (1:1), 86c, and 86d (Scheme 31). Mg in MeOH desulfonylated 85b and 86c to cyclopropane ethers 87 and 89 respectively in high yields. The aminocyclopropane 85c was desulfonylated to 89 with Mg-NiBr2-MeOH at 60 °C in good yield (Scheme 32) [44].

Further synthetic manipulation of 85e and 86d led to the formation of fully protected and enantiomerically pure cyclopropanols 90 and 91 (Fig. 7) [44].
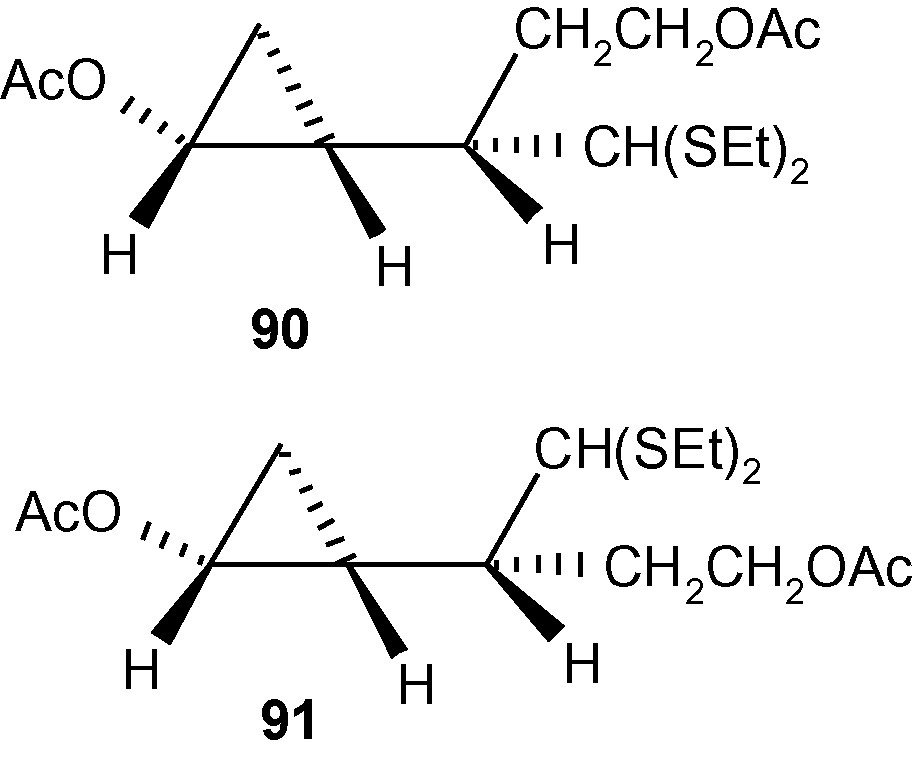
The formation of fully protected and enantiomerically pure cyclopropanols 90 and 91.
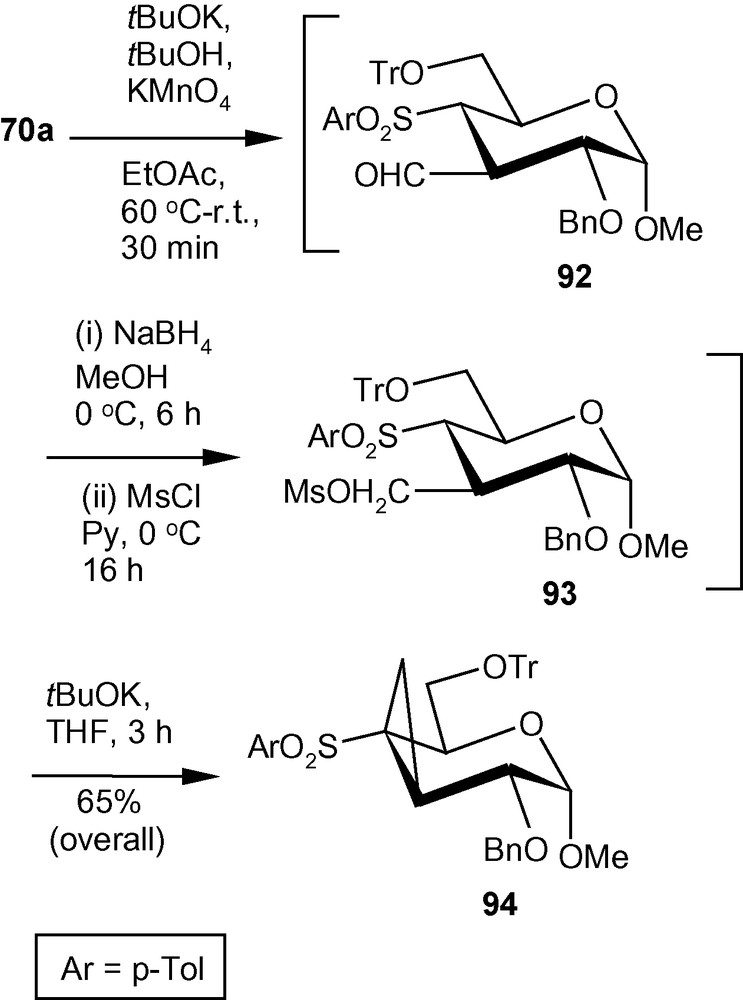
To synthesize C3-C4 cyclopropanated pyranosides from the branched-chain sugar 70a, it was subjected to a modified Nef- carbonyl synthesis to generate the corresponding aldehyde 92 (Scheme 33) [33]. The aldehyde was then reduced with NaBH4 to the corresponding alcohol and the alcohol was mesylated under standard conditions. The crude mesylate 93 was subjected to the ring closure involving intramolecular alkylation reaction in the presence of tBuOK dispersed in THF to afford compound 94 in 65% overall yield (Scheme 33) [33].
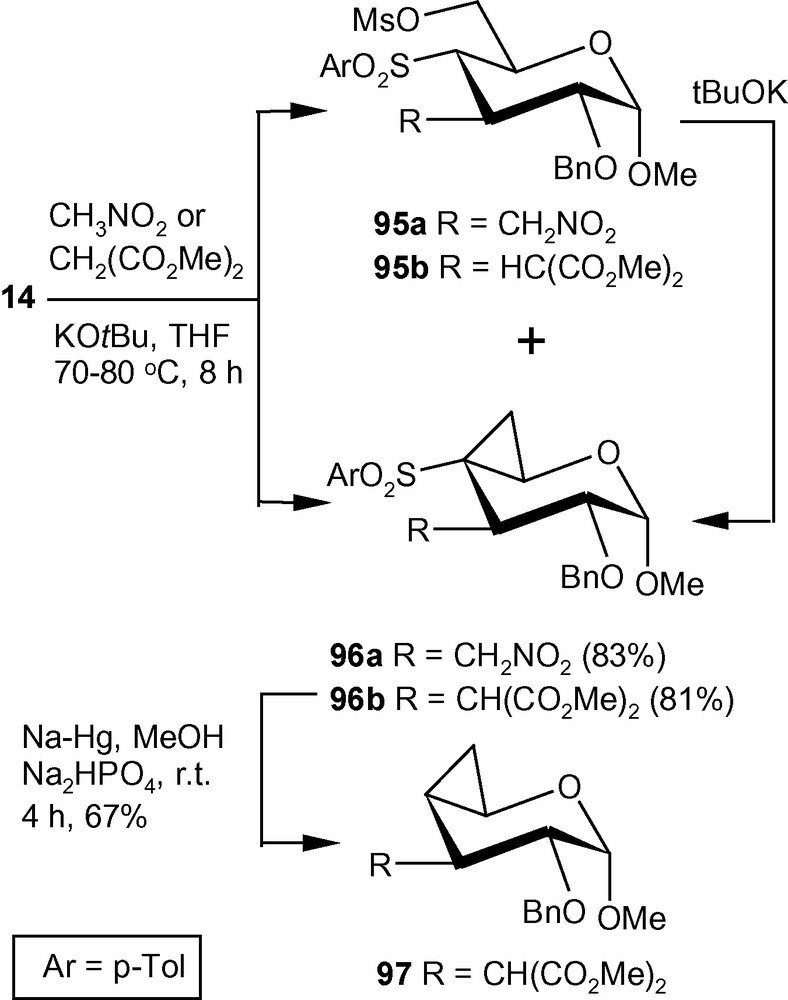
Compound 14 on reactions with nitromethane in the presence of tBuOK in THF at reflux temperature, generated a separable mixture of compounds 95a and 96a (Scheme 34) [33]. The mixture was separated over silica gel and compound 95a was treated with a suspension of tBuOK in THF for 2 h at room temperature to afford compound 96a in 83% combined yield. Similarly, dimethylmalonate in the presence of tBuOK in THF afforded compound 95b and 96b. Separation of the mixture over silica gel and treatment of compound 95b with tBuOK in THF afforded compound 96b in 81% combined yield. Compound 96b on treatment with Na-Hg in dry MeOH at room temperature underwent desulfonylation reaction affording compound 97 in 67% yield (Scheme 34). Schemes 33 and 34 represent new strategies for the synthesis of a hitherto unknown C3-C4 cyclopropanated pyranoside and a C3-branched-, C4-C5 cyclopropanated pyranoside [33].
3.6 Isonucleosides
To broaden the scope of nucleoside-based therapeutics, a novel class of modified nucleosides in which nucleobases are linked to the non-aromatic carbons of carbohydrates has been designed. These “isonucleosides” are promising therapeutic agents of apparently very low toxicity and some of them show strong and selective anti-cancer and antiviral activities. The bond connecting the nucleobase and carbohydrate has higher degree of stability towards acids and enzymatic deamination when compared to those of nucleosides [45].
Vinyl sulfone 1α on treatment with imidazole, 1,2,4-triazole and thymine in the presence of TMG in anhydrous DMF at ambient temperature, generated single isomers 98a–c, respectively in high yields. Adenine, on the other hand, under similar reaction conditions afforded an inseparable mixture of two compounds 98d in 2.2:1 ratio (Scheme 35) [46]. β-vinyl sulfone 1β was also treated with the same set of nucleophiles under similar conditions to afford single isomers 99a–d, respectively, in moderate to good yields (Scheme 36) [46].


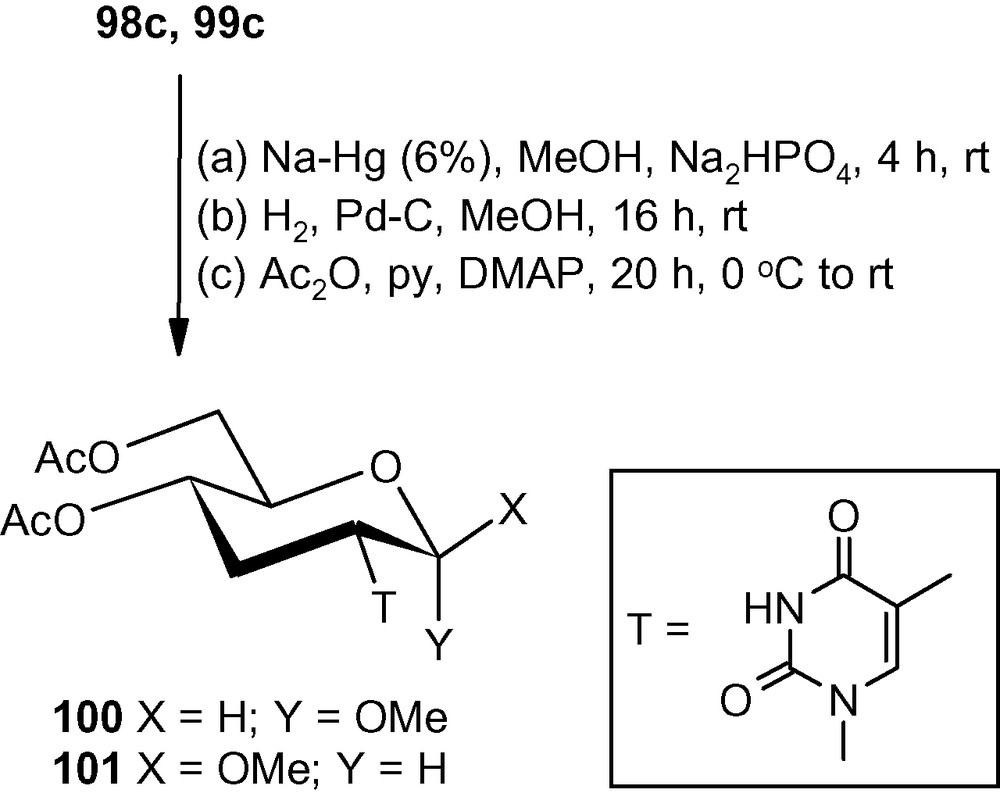
Vinyl sulfones 5α and 5β also reacted efficiently with these planar heterocycles [46]. However, attempted desulfonylation of furanosides using Na-Hg, MeOH-Mg or even with MeOH-Mg-NiBr2 used successfully in the synthesis of furanosyl deoxyaminosugars 66–69 (Fig. 2) led to extensive degradation of the starting materials [46]. Only the pyranosides were found to be stable towards Na-Hg (6%) mediated desulfonylation reactions. Thus, the thymine derivatives 98c and 99c on treatment with Na-Hg (6%) in a buffered system underwent efficient desulfonylation. Products of the desulfonylation reactions were directly deprotected under reductive conditions and finally acetylation produced the desired isonucleosides 100 and 101 in 72 and 64% overall yields, respectively (Scheme 37) [46].
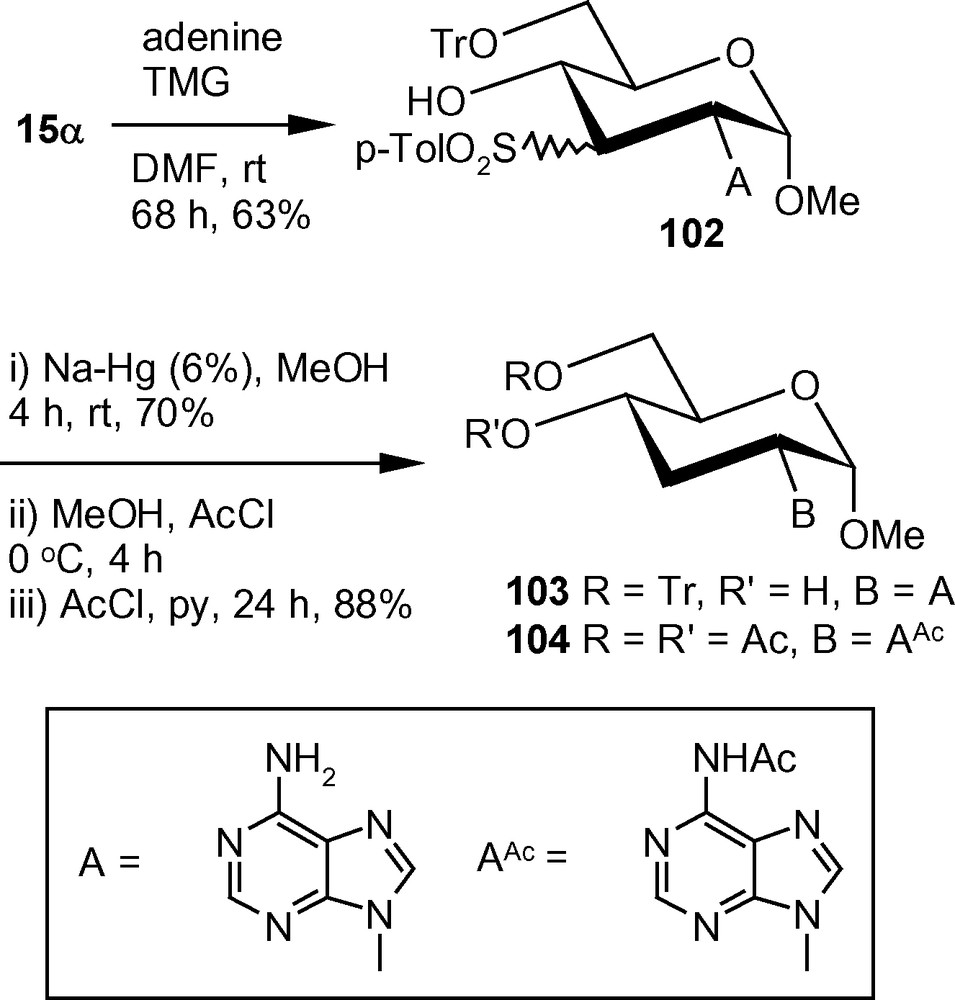
In an alternative approach, tritylated α-methoxy vinyl sulfone 15α on treatment with adenine in the presence of TMG in anhydrous DMF at ambient temperature, afforded an inseparable mixture of compounds 102 in 1:1 ratio. The mixture was subjected to Na-Hg mediated desulfonylation to afford a single compound 103 proving that the sulfonylated derivatives 102 was a mixture of C-3 epimers. The trityl group of 103 was removed easily under acidic condition and the deprotected product was isolated as the peracetylated derivative 104 in good yield in two steps (Scheme 38) [46].
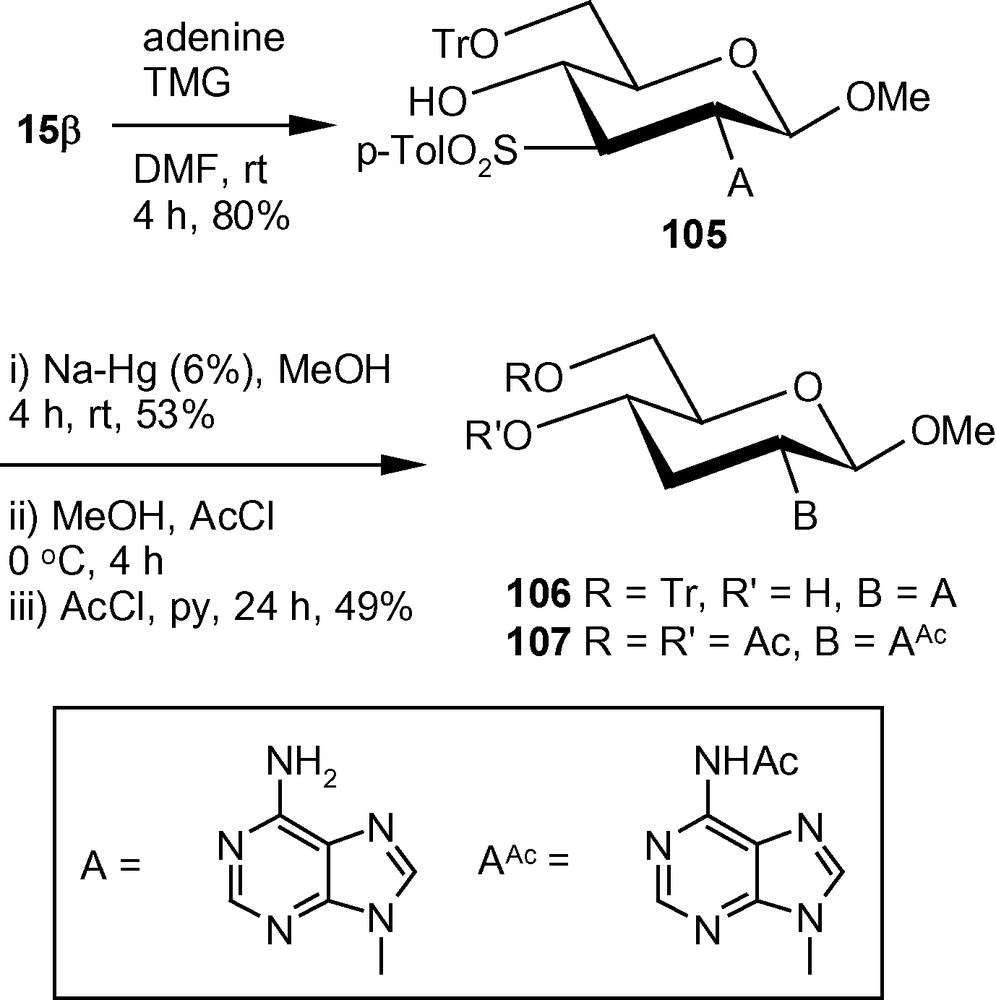
The β-methoxy vinyl sulfone 15β under similar reaction conditions reacted with adenine to afford a single isomer 105. Compound 105 was desulfonylated by Na-Hg to 106 in good yield. Compound 106 was detritylated under acidic condition and the product was isolated as the peracetylated derivative 107 in 49% yield in two steps (Scheme 39) [46].
3.7 Pyrroles
Pyrrole-containing compounds, in general, play crucial roles in nature and substituted pyrroles are important for research in pharmaceutical and material sciences. Although a variety of synthetic approaches for the preparation of pyrroles has been developed over the years, a perusal of the literature reveals that even now the synthesis of highly functionalized pyrroles remains a synthetic challenge in terms of regioselectivity and chemoselectivity [47]. Moreover, synthesis of β-substituted pyrroles was reported to be particularly difficult because the direct alkylation or acylation of pyrroles produced the desired products as minor components.
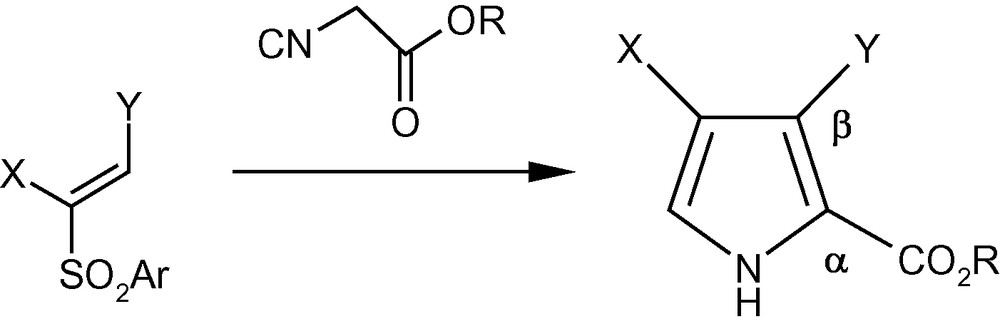
Conjugate addition of the anion generated from an isocyanoacetate to vinyl sulfones was put forward as a methodology for the synthesis of pyrrole-2-esters (Scheme 40) [47]. However, the strategy stagnated over the years for the non-availability of straightforward and general methodologies for the synthesis of polysubstituted vinyl sulfones. The serious shortcomings of currently available methods for the synthesis of polysubstituted pyrroles were compounded by the fact that strategies for the synthesis of pyrroles attached to chiral moieties are virtually nonexistent. Since we had access to different pyranosyl and furanosyl vinyl sulfones, we reacted vinyl sulfones 5α, 6α, 7α, 8–10, 16 and 17 with ethyl isocyanoacetate using standard protocols to afford a wide variety of densely functionalized pyrroles 108–115 (Table 1) [47]. Interestingly, the furanosyl rings of 5α, 6α, and 17 opened up during the reaction to directly afford polysubstituted pyrrole aldehydes 108, 109 and 115 respectively [47].
Chiral pyrroles from vinyl sulfone-modified carbohydrates.
4 Future prospects
Although carbohydrates are primarily modified via their sulfonates, epoxides or ketones [17–24], we have made an attempt to highlight the usefulness of the combination of vinyl sulfone groups and carbohydrates in the synthesis of wide-ranging modified carbohydrates as well as chirally pure non-carbohydrate molecules. It is clear from the examples cited in this review that this class of compounds, indeed, has the potential to act as an unlimited and versatile source of chiral building blocks. We are in the process of unearthing the vast potential of VSM carbohydrates and their downstream products as chirally pure target molecules as well as new chemical entities capable of eliciting responses in biological systems.
Conflict of interest
No conflict of interest is affecting either of the authors.
Acknowledgement
TP gratefully acknowledges the financial support from the DST, New Delhi, India and would like to thank all his past and present coworkers for their contributions towards the development of research on vinyl sulfone-modified carbohydrates in his laboratory. RB thanks CSIR, New Delhi, for a fellowship. DST is also thanked for the creation of 400 MHz facility under IRPHA program and DST-FIST for single crystal X-ray facility.