

1 Introduction
The sun provides huge and inexhaustible energy to the Earth, and the major component is visible light. It is needless to say that plants utilize the energy for their growth as well as for oxygen evolution [1]. Many scientists in various fields have made challenges to analyze and understand the mechanisms of the complicated biological and chemical systems and to develop chemical systems that mimic functions and reactions occurring therein, occasionally called “artificial photosynthesis” [2]. In the field of inorganic chemistry, electron transfer and energy transfer have been the major concerns and many excellent chemical systems have been developed so far [3]. In contrast, little attention has been paid to applications to catalytic transformations and attempts have been limited. For example, the reduction of small inorganic molecules such as proton and carbon dioxide giving hydrogen and carbon monoxide, respectively, has been studied to a considerable extent [3]. Such studies are invaluable from the viewpoint of energy storage, but much less attention has been paid to catalytic organic transformations. Although attempts at them have been made sporadically and the obtained results have been reported [4], they have not been studied systematically until recently. Taking into account this situation, we have made challenges toward developing catalytic organic transformations with two different strategies. One is photoredox catalysis (For selected reviews on photoredox catalysis see [5]), which is the subject of the present account, and the other is bimetallic photocatalysis, which was reviewed previously [6].
On the other hand, introduction of fluorine functional groups to organic skeletons causes dramatic changes of their properties such as liposolubility, water repellency, stability, and heat resistance and, in particular, various medical and agricultural fluorinated chemicals have been developed and launched by making use of these favorable properties [7]. For these purposes, many kinds of techniques for fluorination of organic compounds have been developed and even now new fluorination methods are demanded.
We combined these two problems and set up development of novel trifluoromethylation methods of organic compounds involving carbon–carbon bond formation by the sunlight- or visible light-promoted photoredox catalysis as one of our research programs several years ago.
2 Photoredox catalysis
It has been recognized that photoreactions follow pathways different from those of thermal reactions so as to accelerate the reaction or even furnish different products [8]. Most of organic compounds, however, are colorless and, as a result, they cannot absorb sunlight (i.e. visible light). If one wants to utilize the sunlight energy (visible light), some photosensitizer should be added to absorb visible light. There are many kinds of photosensitizers ranging from organic dyes to metal complexes, and we have employed metal photosensitizers such as [Ru(bipy)3]2+ salts (1a in Fig. 1) and the isoelectronic cyclometalated pyridylphenyliridium complexes (e.g., Ir(ppy)3 (1b)) [9]. Because their photophysical properties have been studied for many years, their features, including those of the photo-excited states, are now understood in detail. They have intense absorption bands in the region of visible light and the lifetime of the photo-excited states is of the order of the microsecond, which is long enough to interact with and activate organic molecules.
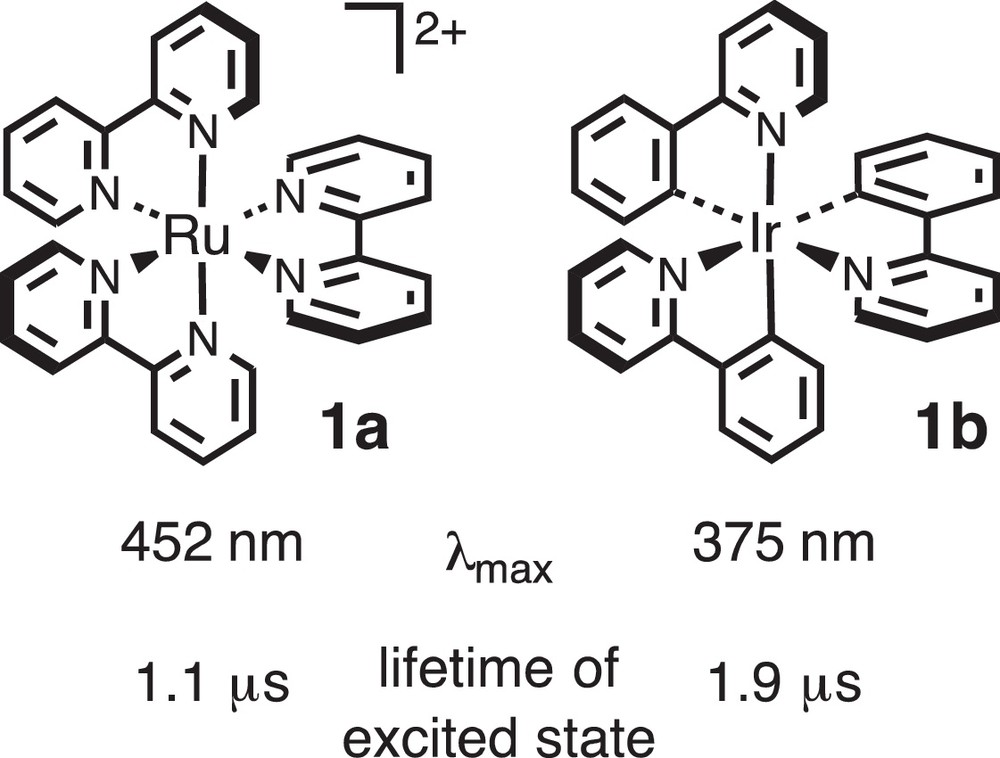
Structures and photophysical properties of representative photoredox catalysts.
The key concept of photoredox catalysis is summarized in Scheme 1 [5]. The first event occurring upon visible light irradiation is metal-to-ligand charge transfer (MLCT) to form the singlet photo-excited species, which is converted into the triplet excited species (M*) via intersystem crossing in an almost quantitative yield. The resulting triplet species with a μs-order lifetime has a hole in the lower SOMO (the solid oval line) and a higher energy electron in the higher SOMO (in the dotted oval line), which can serve as an oxidant and a reductant, respectively. If an electron donor is present nearby (the upper cycle in Scheme 1), electron transfer from the donor to the hole generates the anionic metal species (M−) together with the oxidized cationic organic species (D+). The resultant anionic metal species may be oxidized by an acceptor in the reaction system to generate the reduced acceptor (A−) in addition to the original ground-state neutral metal species M. Thus if an appropriate reaction system can be designed, a donor D and an acceptor A in the reaction system can undergo 1e-oxidation and 1e-reduction to form D+ and A−during the catalytic cycle, respectively. This cycle is called reductive quenching cycle (RQC), because the photoexcited species M* is reduced in the first step. But readers should note that the external substrate is oxidized in the first step of RQC. The catalytic cycle can also be initiated by transfer of the higher energy electron of the photoexcited species to an acceptor followed by oxidation of an external substrate, and this cycle is called oxidative quenching cycle (OQC; the lower cycle in Scheme 1).
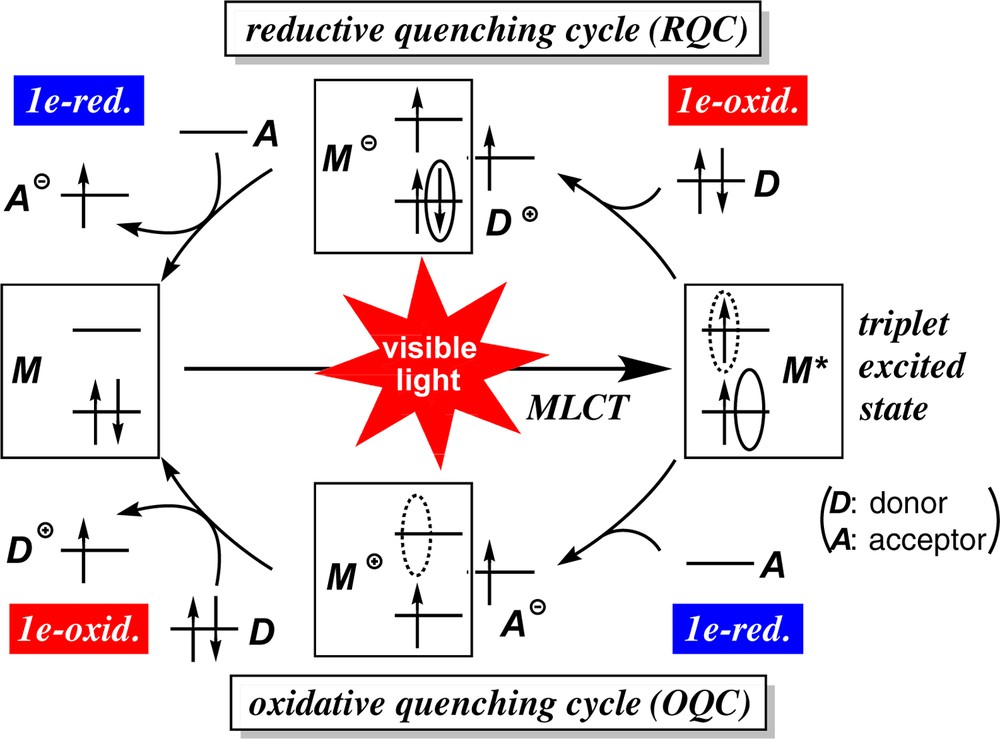
(Color online.) Photoredox cycles: oxidative and reductive quenching cycles.
This photoredox system has been utilized for many transformations, including the reduction of small inorganic molecules as mentioned above. In those systems, however, sacrificial reducing/oxidizing reagents (e.g., trialkylamine) were added to supply/remove electrons to/from the reaction substrate. In the present photoredox systems, however, if the unique redox system can be combined with chemical reactions demanding these two redox functions, no sacrificial redox reagents needs to be added, in other words, the system is redox-neutral, as it will be described in the following sections.
Catalytic photoredox reactions developed in our laboratory during the last several years are summarized in Scheme 2 [10] (For a review, see also [5l-o]) and are divided into two categories, where the reactions follow either RQC or OQC. All reactions involve C-centered organic radicals as key intermediates, and we have found that the OQC system is particularly useful for trifluoromethylation of olefinic substrates (reactions 2b–g), which is the main subject of the present account. Before discussion of trifluoromethylation, one typical example following RQC will be briefly described in the next section.
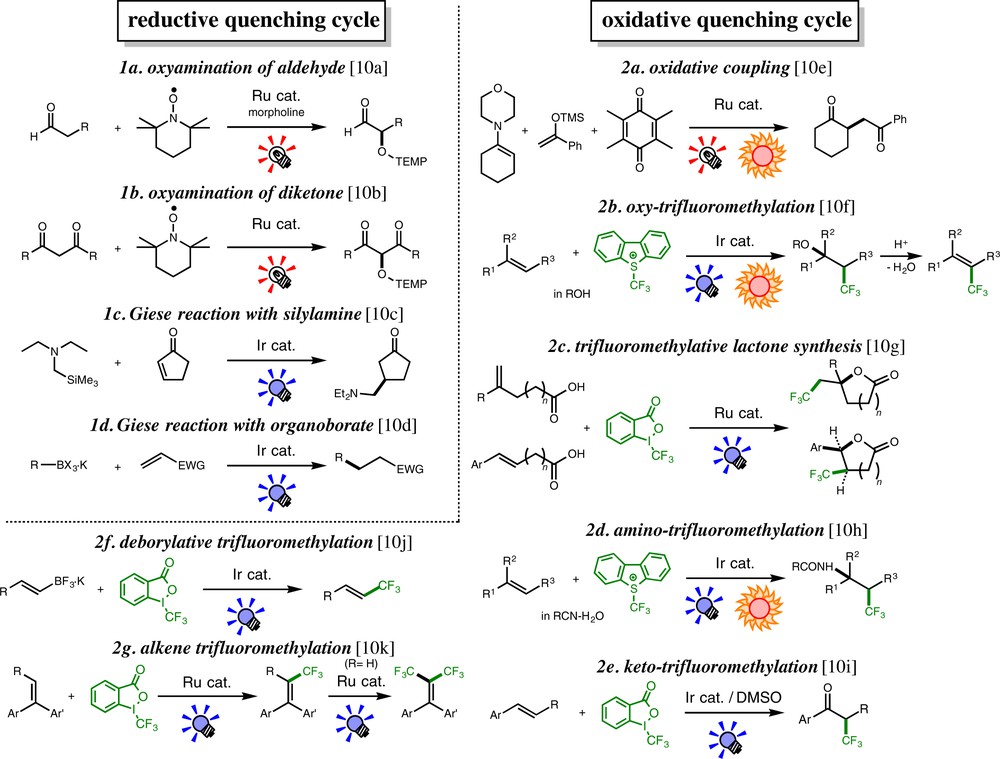
(Color online.) Photoredox-catalyzed reactions developed in our laboratory.
3 Reactions mediated by reductive quenching cycle (RQC) [10a–d]
Generation of organic radicals under mild reaction conditions has remained to be a problem to be solved even now. During the course of our study, we revealed that hydrocarbyl radicals can be readily generated from the corresponding organoborates by the action of photoredox catalysis [10d] When a mixture of potassium organotrifluoroborate 2 (K[R–BF3]) and enone derivatives 3 dissolved in an acetone–MeOH mixed solvent containing 1 mol % of the iridium catalyst 1c was irradiated by blue LED lamps, Giese-type reaction proceeded to give the C–C coupling products 4 in good yields (Scheme 3).

Giese-type reaction of organoborates.
The reaction cannot be driven by [Ru(bipy)3](PF6)2 1a·PF6, but they can be by the iridium catalyst 1c. The difference can be ascribed to the different reduction potentials of the photo-excited species (0.43 V (1a) vs. 0.91 V (1c); cf. oxidation potential for K[PhCH2–BF3]: 0.67 V); the reduction potential of the photo-excited iridium catalyst 1c is enough to oxidize the organoborate 2, but that of the ruthenium catalyst 1a·PF6 is not. As for the scope for the enone derivatives 3, various kinds of substrates including cyclic ones can be applied. On the other hand, as for the scope for the organoborates 2, tertiary alkyl and aryl derivatives gave the products in good yields, while secondary and primary alkyl derivatives turned out to be sluggish, presumably because of the instability of the corresponding alkyl radical intermediates. In these cases, however, the replacement of the fluoride substituents attached to the boron atom by less electronegative oxygen groups (B(OCH2)3CCH3; triolborate) improved the yields to a considerable extent. Heteroaryltriolborates and α-heterosubstituted methyl derivatives (K[X–CH2–BF3]; X= OR [10][10l], NR2 [10m]) also afforded the desired products in good yields.
The reaction mechanism can be interpreted as in Scheme 4. The photoexcited iridium species Ir* undergoes 1e-transfer from borate 2 to generate the organic radical R·and the reduced metal species Ir−. The radical adds to the β-carbon atom of enone 3 to generate the radical intermediate, which is reduced by Ir−to give the enolate intermediate and the original ground-state metal catalyst. Final protonation of the enolate furnishes the product 4. As is evident from the mechanism, no sacrificial redox reagent is needed and thus it turns out that the present catalytic photoredox system is redox-neutral.
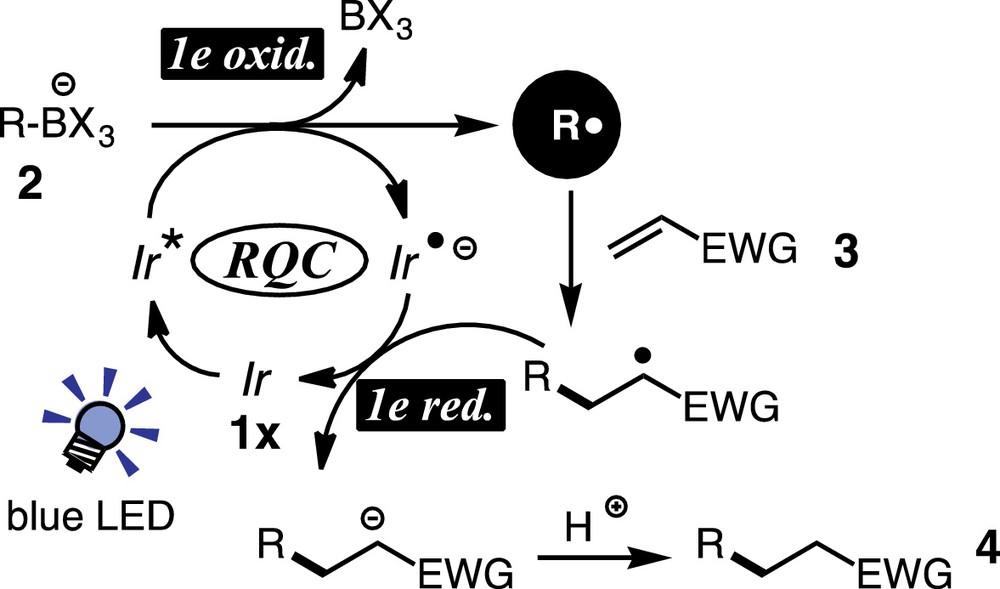
(Color online.) A plausible reaction mechanism for the Giese-type reaction.
4 Reactions mediated by oxidative quenching cycle (OQC) [10e–k]
The catalytic reaction following RQC indicates that organic radicals can be generated by 1e-oxidation of electron-rich organic substrates. Taking into consideration this result, it is readily expected that organic radicals may also be generated by 1e-reduction of electron-deficient substrates via OQC (Scheme 5). Then we examined the generation of the trifluoromethyl radical from electrophilic trifluoromethylating reagents such as Umemoto's reagent 5 (trifluoromethylsulfonium salt of dibenzothiophene) [11] and Togni's reagents 6 (hypervalent iodine species) [12].
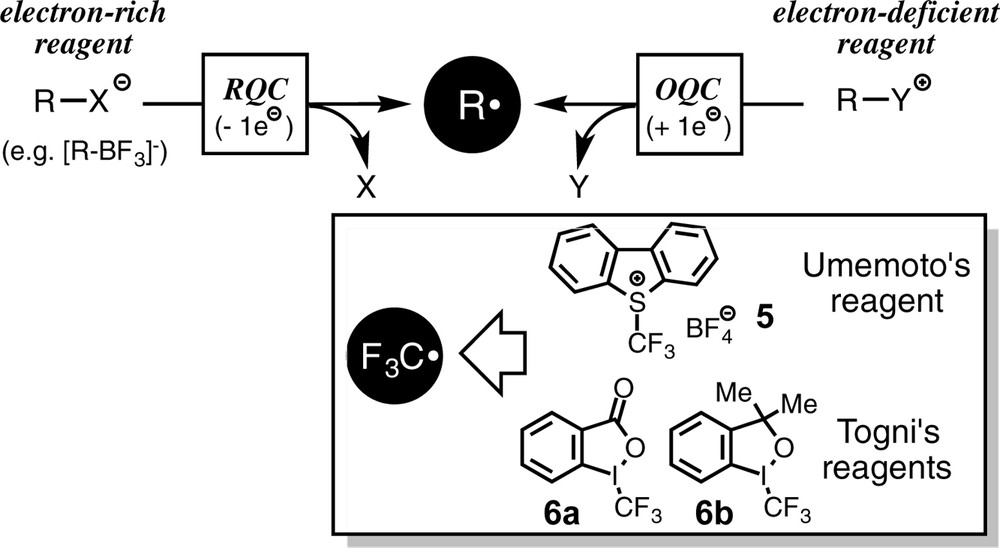
Generation of organic radicals via RQC and OQC.
4.1 Oxytrifluoromethylation of olefins giving 1-aryl-3,3,3-trifluoropropanols [10f]
When a mixture of styrene and Umemoto's reagent 5 dissolved in an acetone–H2O mixture (9 = 1) was irradiated by blue LED lamps in the presence of the iridium photosensitizer 1b for 2 h, 1-aryl-3,3,3-trifluoropropanol 7 was obtained in an almost quantitative yield as a result of regiospecific difunctionalization (oxytrifluoromethylation) of styrene, i.e. the CF3 and OH groups are introduced to the β- and α-carbon atoms of styrene, respectively, in a specific manner (Scheme 6). In this case, all examined Ru and Ir photosensitizers work well, but we prefer to use the neutral species Ir(ppy)3 1b because of its solubility in organic solvents. As will be described below, a significant solvent effect is observed for the present transformation, and such an effect can be studied only with such a soluble catalyst as 1b.
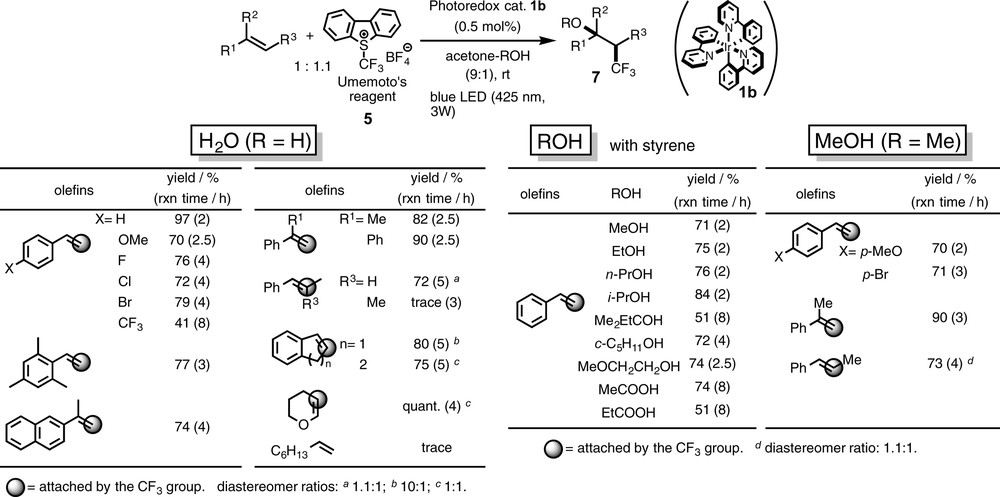
Oxytrifluoromethylation of olefins.
Scheme 6 also shows the scope of the present oxytrifluoromethylation, which can be applied to many kinds of styrene derivatives including naphthyl, α- and β-substituted styrenes, and even vinyl ether. Simple olefin such as 1-octene, however, turned out to be sluggish. In other words, olefin substituents, which can stabilize the radical at the α-position, such as aryl groups and hetero-functional groups are essential for the present transformation.
When the reaction was carried out in the presence of oxygen nucleophiles in place of H2O, various oxytrifluoromethylated products 7 such as ether (from alcohol) and ester (from carboxylic acid) were obtained in good yields (Scheme 6).
As described in the introductory part, we planned to develop catalytic organic transformations promoted by sunlight. Then we carried out the reaction under daylight and obtained the products in yields comparable to those obtained by irradiation with blue LED lamps (Scheme 7). Although we did not examine all reactions under daylight, all reactions we examined under daylight successfully afforded the desired products in good yields.
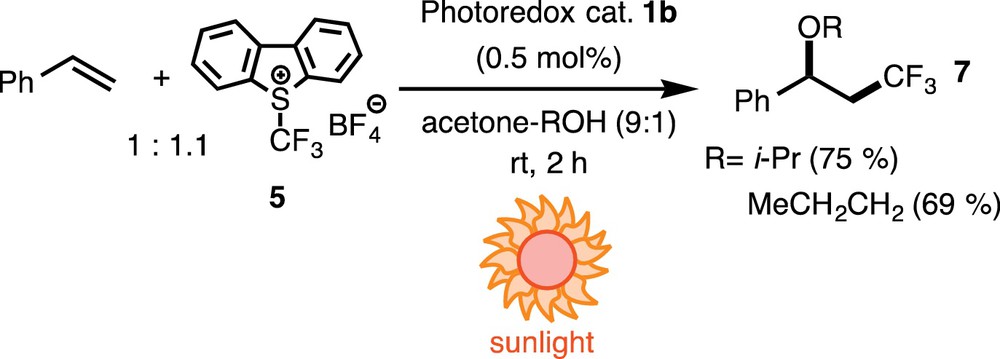
(Color online.) Sunlight-promoted oxytrifluoromethylation of styrene.
In order to obtain information on the reaction mechanisms, a couple of experiments were conducted. First, CV analysis revealed that Umemoto's reagent 5 (Eox = –0.75 V) could be readily oxidized by the photoexcited species of the metal catalyst (e.g., Ered(1b) = –2.14 V). Second, β-pinene with the bicyclic structure afforded a ring-opened product via a ring-opening process of the strained tertiary radical intermediate. Finally, Stern–Volmer plots revealed that luminescence from the photoexcited iridium species Ir* was not quenched by styrene but by Umemoto's reagent 5. These experimental results suggest the plausible mechanism based on OQC summarized in Scheme 8. The photoexcited metal species Ir* transfers one electron to 5 to form the oxidized metal species Ir+ together with trifluromethyl radical (after elimination of dibenzothiophene), which adds to the β-carbon atom of the styrene derivative to give the stable benzyl radical intermediate 8. Subsequent 1e-transfer from the radical to Ir+ regenerates the ground state Ir catalyst 1b and the benzyl cation intermediate 9, to which oxygen nucleophiles add to furnish the oxytrifluoromethylated product 7 after deprotonation. When compared with the mechanism shown in Scheme 4, the initial redox events are opposite but the sequential oxidation/reduction processes make both catalytic transformations redox-neutral.
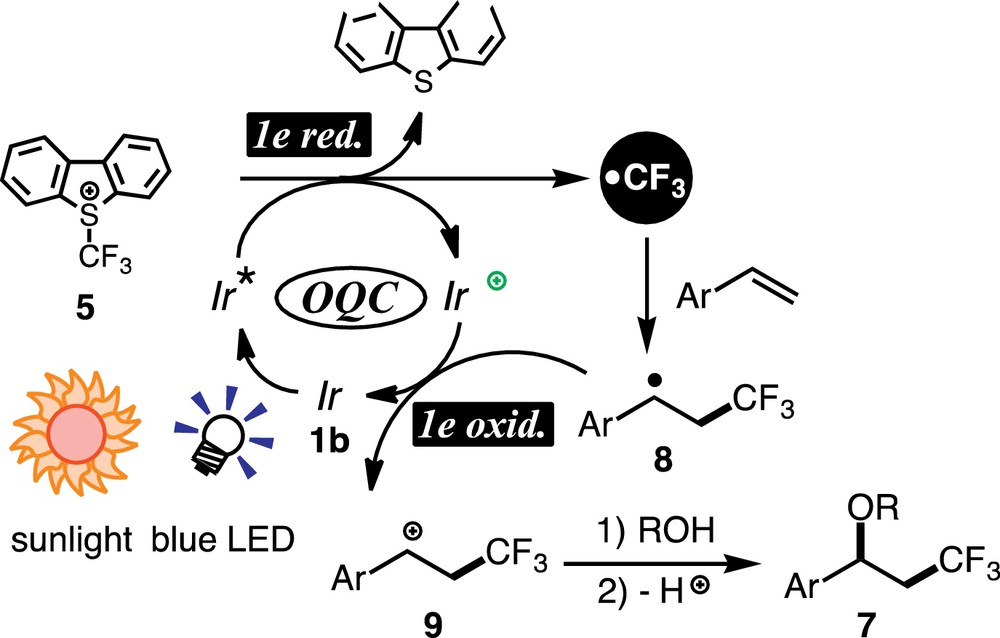
(Color online.) A plausible mechanism for oxytrifluoromethylation.
An intramolecular version of the present catalytic transformation provides synthetic methods for trifluoromethylated lactone derivatives from alkenoic acids (reaction 2c in Scheme 2) [10g]. In addition, combination of the oxytrifluoromethylation with acidic dehydration provides a new substitution method of a vinylic hydrogen atom by a CF3 group (10), and this process was applied to the synthesis of a precursor of panomifene, an anti-estrogen drug (Scheme 9) [10f].
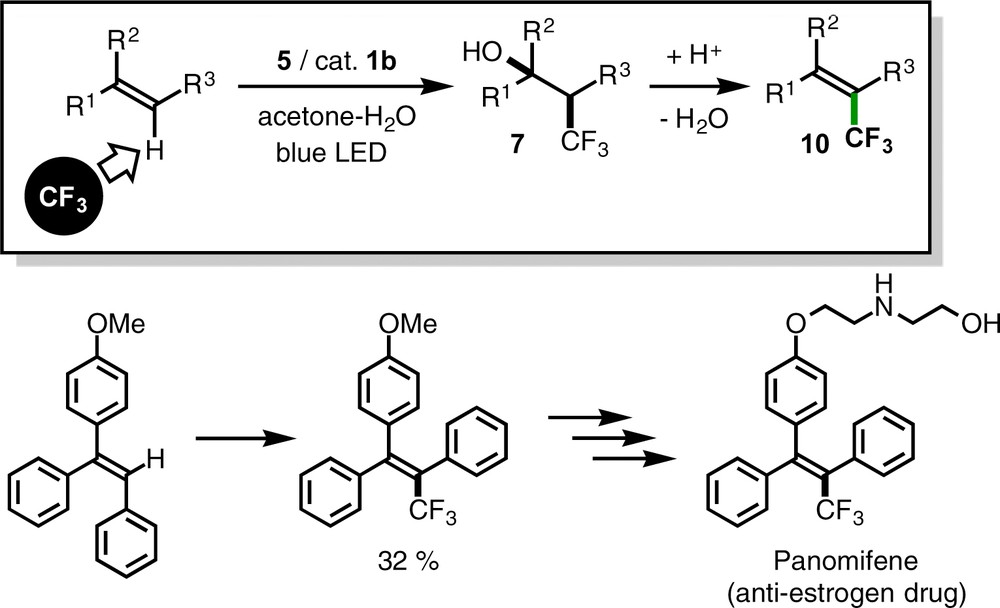
Formal substitution of a vinylic hydrogen atom by a CF3 group.
4.2 Aminotrifluoromethylation of olefins giving amides of 1-aryl-3,3,3-trifluoropropylamine [10h]
When the oxytrifluoromethylation of olefins was carried out in the presence of nitrile and H2O, aminotrifluoromethylation occurred to give amides of 1-aryl-3,3,3-trifluoropropylamines 11 (Scheme 10). The reaction mechanism can be interpreted in terms of a Ritter-type mechanism, where the carbocationic intermediate 9 is first trapped by nitrile and the resultant adduct is hydrolyzed by H2O.
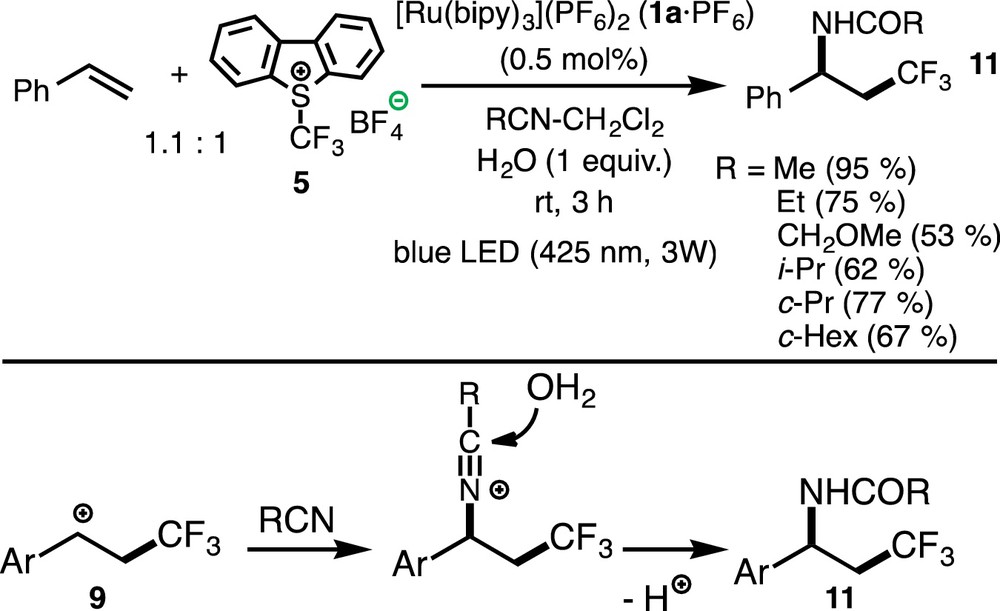
(Color online.) Aminotrifluoromethylation of olefins.
The present aminotrifluoromethylation turns out to be tolerable with many functional groups such as halogens, carbonyl and amino groups, and even Bpin group and, therefore, can serve as a late-stage trifluoromethylation method for bioactive compounds with many functional groups and a vinyl group, i.e. a trifluoromethyl group can be introduced to the vinyl part at a final stage of a sequence of transformations.
4.3 Ketotrifluoromethylation of olefins giving 1-aryl-3,3,3-trifluoropropan-1-one [10i]
During the course of our study, we found an intriguing solvent effect for the trifluoromethylation of olefins, i.e. ketone derivatives are obtained when the reactions were carried out in DMSO. It turned out that the reaction depended not only on the photocatalysts, but also on the trifluoromethylating reagents.
When a DMSO solution of styrene and Togni's reagent 6a was irradiated in the presence of Ir(ppy)3 1b (5 mol%) by blue LED lamps for 10 min (in an NMR tube), 3,3,3-trifluoropropiophenone 12 was formed in 75% yield as a sole product (entry 1; Scheme 11). But when Umemoto's reagent 5 was used in place of 6a, DMSO-adduct 13 (35%) was formed in addition to 12 (45%) (entry 2). Further irradiation of the mixture for 20 min caused complete conversion of 13 into 12 (86%) (entry 2’). Furthermore, when [Ru(bipy)3](PF6)2 1a·PF6 was used in place of 1b, the DMSO-adduct 13 was formed exclusively in 92% yield, but the reaction was slow. It took 2 h for the complete conversion of styrene (entry 3). Further irradiation of the reaction mixture caused conversion of only a part of 13 into 12 (entry 3’). However, when NEt3, a base, was added to the reaction mixture of entry 3 containing 13 as the dominant component, 13 was converted to 12 spontaneously and quantitatively (entry 3”). Quantitative conversion of 13 to 12 was also observed, when a catalytic amount of Ir(ppy)3 1b was added to the reaction mixture of entry 3 and then the mixture was further irradiated for 1 h (entry 3”’). The reaction with 1a and 6a was slow but selective (entry 4).
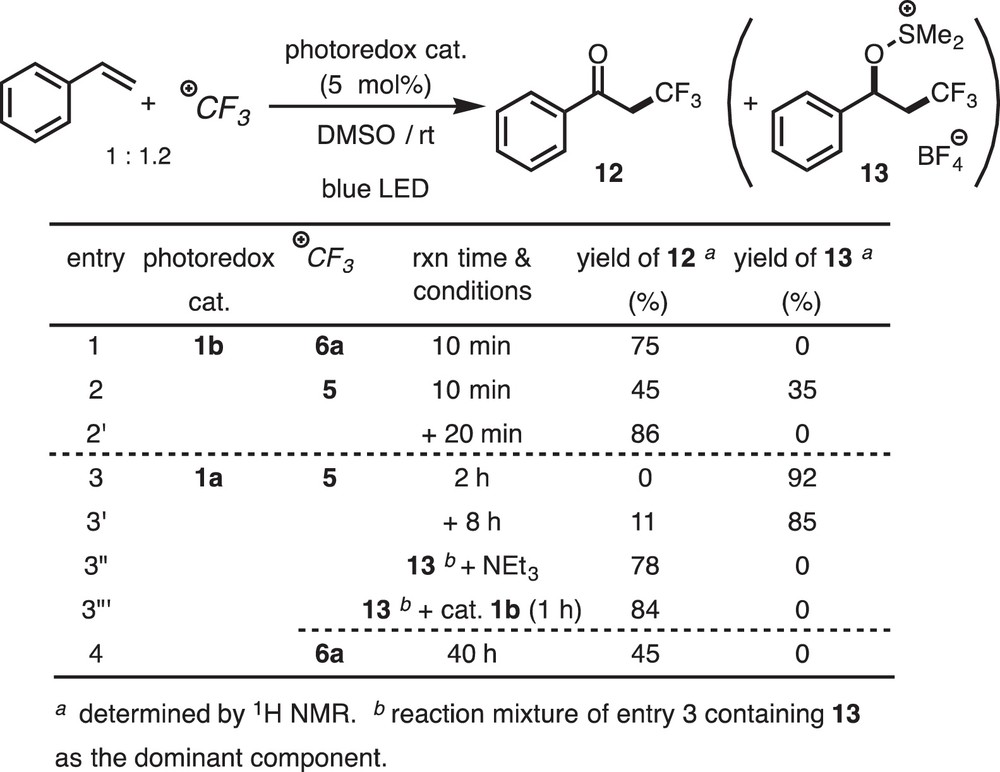
Ketotrifluoromethylation of styrene.
The following features are noted for the present reaction. (i) For the trifluoromethylating reagents, Togni's reagent 6a is superior to Umemoto's reagent 5. (ii) For the photosensitizers, Ir(ppy)3 catalyst 1b is superior to [Ru(bipy)3]2+ catalyst 1a·PF6. (iii) The DMSO-adduct 13 is an intermediate for 12. (iv) Two mechanisms are operating for the conversion of 13 into 12; one is a base-promoted Kornblum oxidation-type mechanism and the other is photoredox catalysis.
β-Alkyl-substituted styrenes were also converted into the corresponding 2-alkyl-3,3,3-trifluoropropiophenone 12′. To our surprise, α-alkyl-substituted derivatives were converted to 12 via a C–C bond cleavage process, and the eliminated alkyl groups were detected as sulfonium salts 14 (Scheme 12).
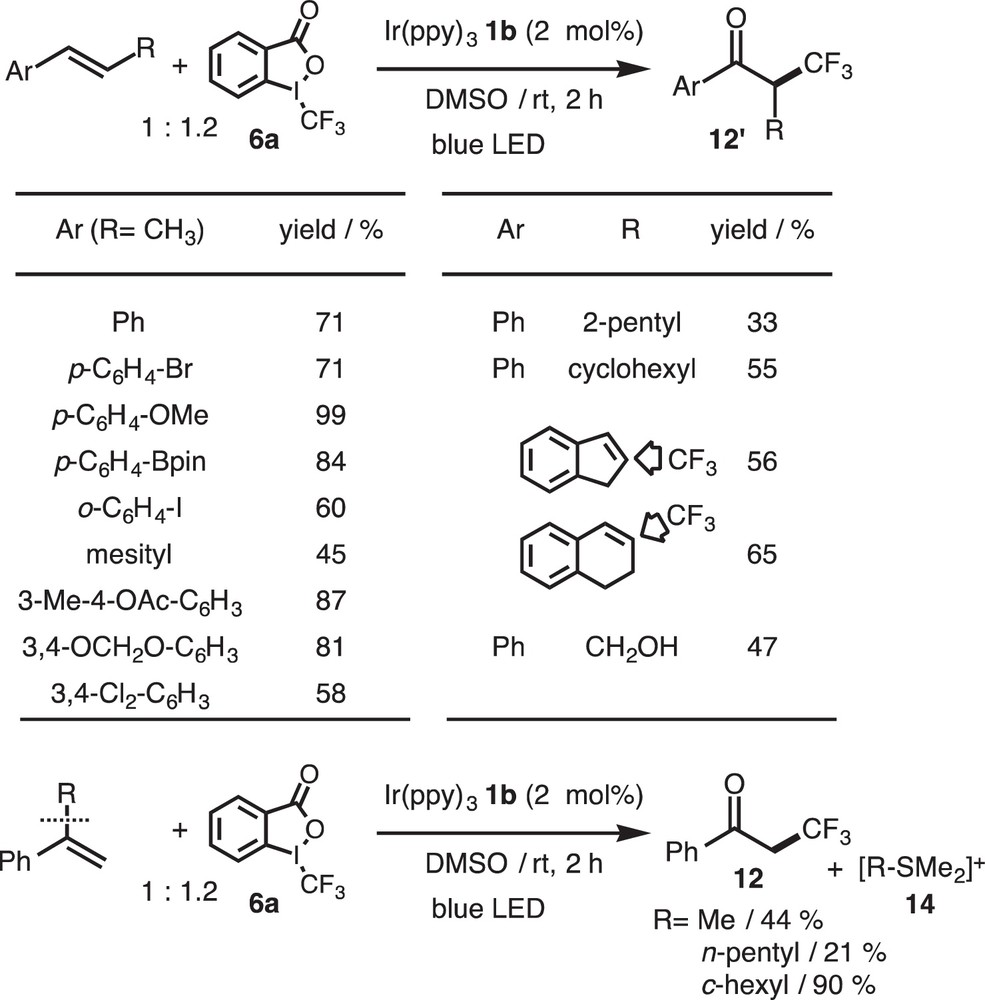
Ketotrifluoromethylation of olefins.
The above-mentioned features are explained by the reaction sequence shown in Scheme 13. The photoredox catalysis first generates the benzyl cation-type intermediate 9 (Scheme 8), which is captured by DMSO to form the adduct 15.

(Color online.) Plausible mechanisms for ketotrifluoromethylation.
In the presence of a base, 15 is converted to 12 via a Kornblum oxidation type-mechanism (path i), i.e. deprotonation from the methyl group in the SMe2 part, followed by intramolecular deprotonation of the benzylic hydrogen atom associated with the elimination of SMe2, leads to 12. This reaction pathway is supported by the observation that, when the reaction was carried out in DMSO-d6, CD3SCD2H was formed as confirmed by the characteristic quintet 1H NMR signal for the CD2H group. Because the reaction proceeds in the absence of a base and in the presence of the iridium catalyst (entry 3”’), some photocatalytic mechanism—path (ii)—should also work (see below).
The extremely fast reaction observed when Togni's reagent 6a is employed as the CF3-source (entry 1) can be ascribed to o-iodobenzoate, the byproduct (Y) of the initial OQC cycle, which works as a base to accelerate path (i). In the case of the reaction of α-alkylstyrenes, the alkyl group at the benzylic position in 15 cannot be removed by the action of a base—path (i)—and, therefore, some other mechanism should operate.
For the alternative photoredox mechanism—path (ii)—, we have no experimental evidence. Our proposed mechanism involves a 1e-reduction of 15 by the photo-excited Ir* species followed by a sequence of S–O bond cleavage, C-to-O migration of the R group, and oxidation by Ir+, giving the cationic intermediate 16, which is converted into the product 12 via O–H/Me bond heterolysis induced by SMe2 (generated in the step of the S–O bond cleavage). It is noteworthy that, in the case of α-alkylstyrene, some photoredox catalysis operates for the C–C bond cleavage process.
Thus we observed remarkable solvent effects for the present solvolytic trifluoromethylation of olefins. It should be noted that, by simply changing the solvent, olefins undergo oxytrifluoromethylation (reactions 2b,c in Scheme 2), aminotrifluoromethylation (reaction 2d), and ketotrifluoromethylation (reaction 2e) to give the functionalized trifluoromethylated derivatives.
4.4 Trifluoromethylation of olefinic substrates via substitution reactions [10j,k]
The above-mentioned mechanisms of the solvolytic trifluoromethylation reactions involving the cationic intermediates prompted us to examine substitution reactions of olefinic substrates via deprotonation from the cationic intermediate 9.
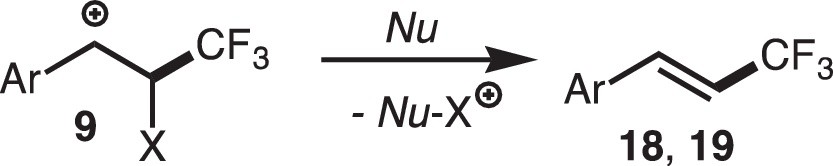
We first examined direct trifluoromethylation of olefins with various CF3 sources and photoredox catalysts, but it turned out to be unsuccessful. Then we designed a coupling reaction between the electron-deficient·CF3 radical and the electron-rich alkenylborates 17, K[R–CH=CH–BF3], which proceeded efficiently to give the substituted products 18 (Scheme 14) [10j]. We also found that 1,1-diarylethene derivatives underwent direct trifluoromethylation without activation by the borate auxiliary (Scheme 15) [10k]. The cationic intermediate is formed by following the processes explained above, and subsequent deboronation or deprotonation from the β-position of 9 should furnish the substituted products 18 and 19 (Scheme 16). The reaction of borates 17 should be promoted by the attractive electrostatic interaction between the two reagents as designed above, and the driving force for the 1,1-diarylethene should be formation of the very stable diarylmethyl radical.

Trifluoromethylation of alkenylborates.
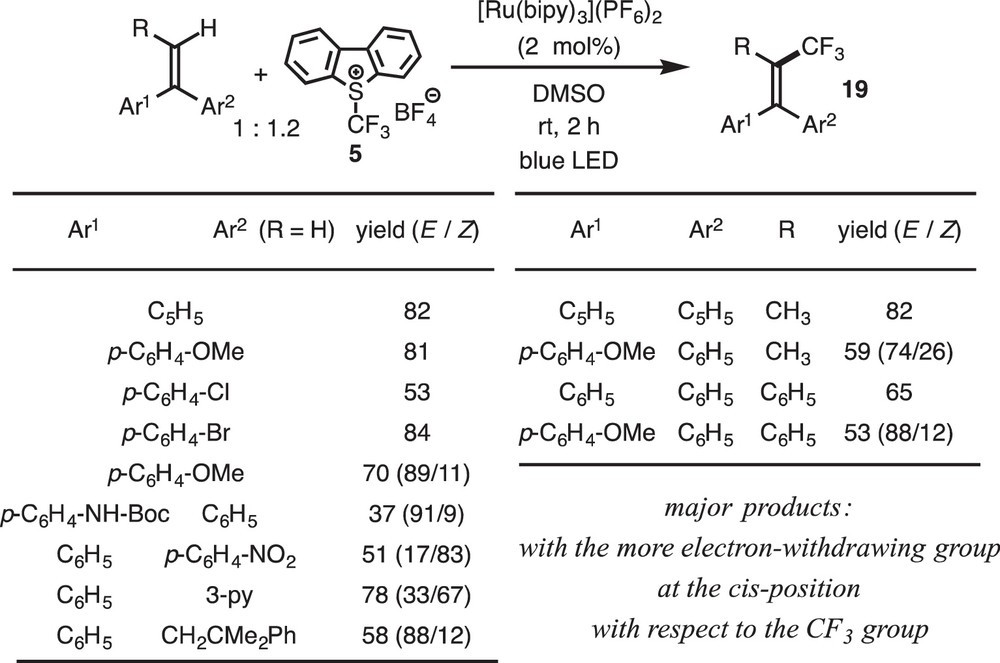
Trifluoromethylation of 1,1-diarylethenes.
A mechanism for the formation of the substitution products.
4.5 Summary of reaction mechanisms
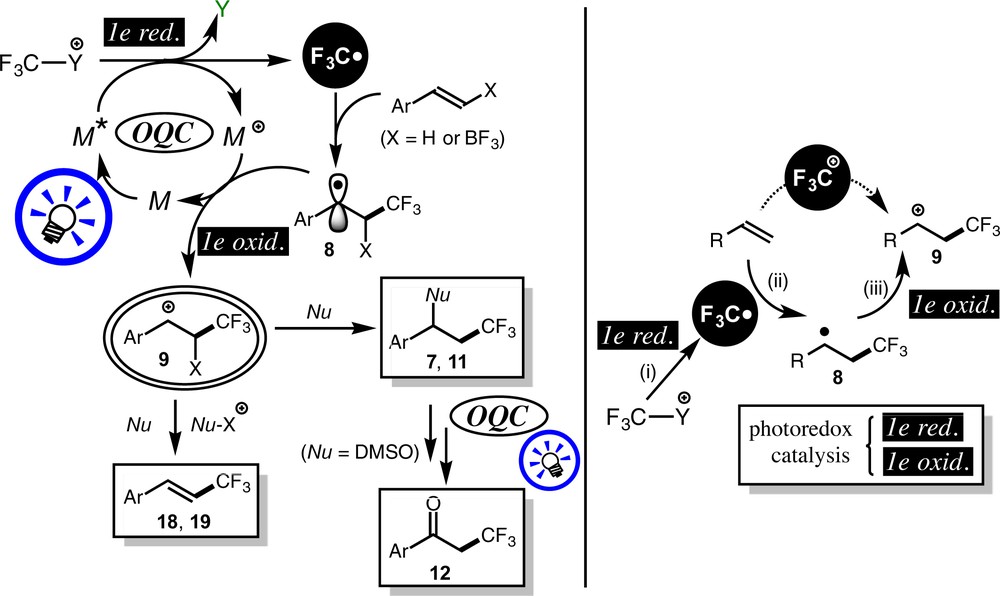
The mechanisms of the solvolytic and substitution reactions discussed above are summarized in Scheme 17. Trifluoromethyl radical·CF3, the key intermediate of these transformations, is generated by 1e-reduction of the electrophilic trifluoromethylating reagents 5 or 6 via OQC and adds to olefins to form the benzyl radical intermediate 8, which is converted into the benzyl cation intermediate 9 by the action of the latter part of OQC. Subsequent processes are dependent on the reaction conditions. In the presence of a nucleophile, it adds to the cationic center to afford the solvolysis products 7 or 11, which may undergo further transformation (e.g., an oxidation giving the ketone product 12). In the other cases, the nucleophile does not attack the cationic center, but the boron-functional group or the proton at the β-position is eliminated to furnish the substituted products 18 and 19.
(Color online.) Trifluoromethylation of olefins.
The β-CF3-substituted cationic species 9 may be generated by the interaction of olefin with trifluoromethyl cation +CF3 (Scheme 17), but such a reaction has not been reported so far, presumably because of the low reactivity of the cationic +CF3 species to unsaturated bonds [13] (See also [11c]). Instead, in the present system, the β-CF3-substituted cationic species 9 is generated via a sequence promoted by photoredox catalysis, i.e. the CF3 radical generated by the action of OQC—step (i)—adds to olefin to generate the benzylic radical intermediate 8—step (ii)—, which is oxidized by the action of OQC—step (iii)—to give the cation 9. As mentioned in the introductory part, photoredox catalysis can be a powerful synthetic tool when it is combined with organic transformations demanding such a pair of redox processes. Recently, an increasing number of photoredox-catalyzed trifluoromethylation reactions have been reported [5], and this technique can be used complimentarily together with, for example, transition metal catalysis [14] and electrocatalysis.
5 Photoredox catalysis, a green method for radical generation: future prospects
Organic radicals are usually generated by thermolysis, photolysis, and redox reactions of appropriate precursors having weak bonds, which usually contain heavy elements (mostly harmful; e.g., Sn) or are potentially explosive (e.g. O-O bond). Additionally, all processes require fossil fuel as an energy source to drive the reactions.
In sharp contrast to the classical radical chemistry, photoredox catalysis turns out to be superior to the classical methods and green from the point of view of the above-mentioned aspects. All you need are reagents, catalyst, flask, and sunlight.
Photoredox catalysis is, of course, not almighty. It becomes powerful only when the target reaction meets the above-mentioned requirements. But the scope of photoredox catalysis will be surely expanded by, for example, the following approaches. One is development of appropriate precursors. We revealed that electrophilic trifluoromethylating reagents are powerful sources for ·CF3 radical when combined with photoredox catalysis (OQC). For generation of R·(R = any organic fragment), there should be numerous combinations for R, X, and Y (Scheme 5). Another approach is combination with other catalytic systems. The group of MacMillan, the leading chemist of this research field, reported very unique photoredox catalytic systems coupled with asymmetric organocatalysis [15a] and organometallic catalysis [15b].
From the viewpoint of photocatalysis, visible light irradiation cannot activate organic substrate directly, but it does activate the photoredox catalyst, which shows unique redox properties upon excitation as described above. In the present case, as typically exemplified by Scheme 16, the difficult thermal reaction pathway is circumvented by the photoredox pathway.
As shown in Fig. 2, the numbers of published items and citations relevant to photoredox catalysis have grown rapidly during the last several years. We believe that photoredox catalysis will be extended to many organic transformations. We would be pleased if readers recognized the great utility of photoredox catalysis. Just leave your flask under sunlight, and you will obtain the desired product in a good yield!!
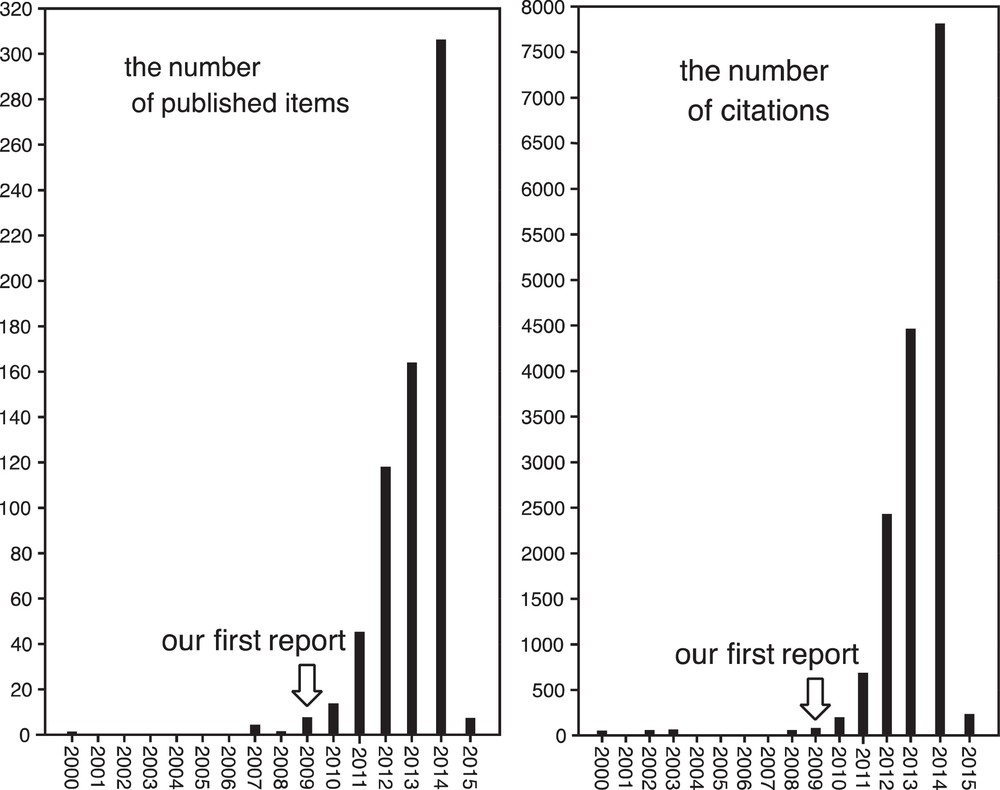
The growing numbers of published items and citations relevant to “photoredox catalysis” as of 24 January 2015.
Acknowledgement
The present research is supported by a Grant-in-Aid for Scientific Research from Japan Society for the Promotion of Science (No. 26288045).