

1 Introduction
High-energy density materials (HEDMs) have attracted considerable attention owing to their superior explosive performances over conventional energetic compounds [1–7]. Organic-cage compounds were widely investigated due to their large strain energies, high heat of formation, and compact structures as an important category of HEDMs [8]. Typical examples of these compounds are CL-20 and octanitrocubane (ONC). Octanitrocubanes, which were synthesized in 2000 from heptanitrocubane [9], are derivatives of the strained cubane molecule, have a high nitrogen content and have been found to have a high density, ranging from 1.98 to 2.06 g/cm3, depending on the nitro group's orientation and to be shock insensitive, as it has been confirmed by hitting the compound with a hammer. However, it is a pity that it cannot be used widely in practical applications because of its production cost nowadays. Furthermore, another two types of organic cage compounds, nitro derivatives of hexaazaadamantane (HAA) and adamantane series, have also been investigated as promising candidates for HEDMs [10,11]. Recently, two novel and super-high energy cage explosives, dodecanitrohexaprismane and hexanitrohexaazaprismane, have also been studied by Zhu et al. [12]. The results indicate that dodecanitrohexaprismane has much higher energetic properties than those of ONC, which may be the most powerful nonnuclear explosive known so far. These investigations further indicate that cage skeleton is a good parent structure for HEDMs.
We are interested in another cage structure: prismane, which is a hydrocarbon with a large ring-strain energy of 606.9 kJ/mol [13], and has been synthesized and fully characterized [14,15]. In addition, many prismane derivatives have also been studied in detail with quantum chemistry methods. Such as, azaprismanes were studied by Politzer et al. in 1989 [16]. Their results show that in each set of azaprismane isomers, the most stable is the one having the fewest N–N bonds. Lately, polyisocyanoprismanes and polyazidoprismanes have been studied by Xu et al. [17]. These results indicate that these prismane derivatives have large and positive heat of formation values. However, the detonation performances of derivatives have not been studied in their paper. Otherwise, the nitroprismane derivatives have been calculated in detail at the B3LYP/6-311G** level by Li et al. [18]. The results show that polynitroprismanes with five and six nitro groups meet the demands of practical HEDMs. Furthermore, we also studied the stability and detonation performance of polydinitroaminoprismanes by using density function theory, and found that tridinitroaminoprismanes and tetradinitroaminoprismanes have excellent detonation properties [19].
Recently, a larger number of nitrate ester derivatives were synthesized, which showed high detonation performance and low melting point [20,21]. These works provided an effective strategy for designing energetic materials. In the present work, the H atoms in prismane were systematically replaced with a nitroester group, generating the series of molecules C6Hn–6(ONO2)n (n = 1–6). The main difference between the nitroester (ONO2), nitro (NO2), isocyano (CN), and azido (N3) functional groups is the amount of oxygen presented. As far as combustion is concerned, the nitroester group has remarkable advantages.
2 Computation methods
In our previous studies, we had studied the properties of energetic compounds at the B3LYP and MP2 levels, and the similar calculated results had been obtained at two different levels [22,23]. Therefore, we think that reliable results can be obtained by using B3LYP function. In the present work, structure optimization and frequency calculations were performed with Gaussian 03 package at the B3LYP/6-311G** level [24], and no constraints were imposed on the molecular structure during the optimization process. Heats of formation, detonation performances, and impact sensitivity were studied at the same level, and bond dissociation energies were calculated at the UB3LYP/6-311G** level. All optimization molecular structures are shown in Fig. 1.

Structures of prismane and its derivatives. 1- denotes 1-nitroesterprimane, 1,2-binitroesterprimane, and so on.
The heats of formation in the gas state (HOFgas) of all molecules were evaluated by using an isodesmic reaction.
| (1) |
| (2) |
Since the condensed phases of most energetic compounds are solid, calculating detonation properties requires knowledge of the solid-phase heat of formation (HOFsolid). According to Hess’ law of constant heat summation, HOFgas and the heat of sublimation (ΔHsub) can be used to evaluate HOFsolid:
| (3) |
| (4) |
The empirical Kamlet–Jacobs equations [26] are employed to estimate the detonation velocity and pressure for the energetic materials containing carbon, hydrogen, oxygen and nitrogen atoms:
| (5) |
| (6) |
D is the detonation velocity (km/s), P is the detonation pressure (GPa). N is the number of moles of gas produced per gram of explosive, and is the mean molecular weight of the gaseous detonation products. Q is the heats of detonation (cal/g). ρ0 is molecular density (g/cm3), which was calculated from the molar weight (M) divided by the average value molar volume (V). V is defined as a contour of 0.001 electrons/Bohr3 density that was evaluated using a Monte Carlo integration. We performed 100 single-point calculations for each molecule to get an average volume.
But the procedure used to estimate densities can lead to significant errors. Politzer et al. [27,28] considered that the solid molecular density can be adjusted for the electrostatic potential (Eqs. (7)–(11)):
| (7) |
| (8) |
| (9) |
| (10) |
| (11) |
The strength of bonding, which could be evaluated using the bond dissociation energy (BDE), is fundamental to understanding chemical processes [30]. The energy required for bond homolysis at 298 K and 1 atm corresponds to the enthalpy of reaction A – B → A + B, which is the BDE of the molecule A – B by definition. Therefore, at 0 K, the homolytic BDE can be given using Eq. (12):
| (12) |
BDE(A–B) is the BDE of A–B, , and are the total energies of the parent molecule and of the corresponding radicals, respectively.
The characteristic height (H50), which can also reflect the impact sensitivity and stability of the compounds, was estimated using Eq. (13), suggested by Pospíšil et al. [31]:
| (13) |
3 Results and discussion
3.1 Heats of formation (HOFs)
For experimental researchers, the heat of formation (HOF) is the most important parameter in the determination of the energetic properties and the energy content of a chemical system. Previous studies [32–35] have shown that the HOF theoretically predicted using the isodesmic reaction approach was in good agreement with the experiments. Table 1 lists the total energy (E0), the zero-point energy (ZPE), the thermal corrections (ΔHT) and the HOFsolid values for the reference compounds used in isodesmic reactions. In Table 2 are summarized E0, ΔHT, HOFgas and HOFsolid of all the designed molecules at the B3LYP/6-311G** level.
Relevant data of reference compounds at the B3LYP/6-311G** level.
| Compounds | E (a.u) | ZPE (kJ/mol) | HT (kJ/mol) | HOF (kJ/mol) |
| CH4 | –40.53374 | 117.09 | 10.03 | –74.60 |
| CH3ONO2 | –320.28274 | 142.24 | 15.81 | –110.57a |
a Value obtained at the G-2 level.
Calculated heats of formation and related parameters of all compounds at the B3LYP/6-311G** level.
| Compounds | E (a.u) | ZPE (kJ/mol) | HT (kJ/mol) | HOFgas (kJ/mol) | Hsub (kJ/mol) | HOFsolid (kJ/mol) |
| C6H6 | –232.11118 | 253.89 | 13.22 | 567.70 | 67.32 | 500.42 |
| 1- | –511.87070 | 269.81 | 22.73 | 498.61 | 79.31 | 419.30 |
| 1,2- | –791.61857 | 285.36 | 32.59 | 460.09 | 107.02 | 353.07 |
| 1,4- | –791.62636 | 283.95 | 32.84 | 438.47 | 111.72 | 326.75 |
| 1,5- | –791.62165 | 286.01 | 32.33 | 452.39 | 104.65 | 347.74 |
| 1,2,3- | –1071.36488 | 299.77 | 43.05 | 425.12 | 127.92 | 297.20 |
| 1,2,4- | –1071.37624 | 300.13 | 43.05 | 395.65 | 128.59 | 267.06 |
| 1,2,5- | –1071.37329 | 300.61 | 42.68 | 403.51 | 108.91 | 294.60 |
| 1,2,3,4- | –1351.11682 | 314.68 | 44.11 | 366.47 | 144.82 | 221.65 |
| 1,2,4,5- | –1351.12234 | 314.35 | 53.67 | 361.21 | 145.56 | 215.64 |
| Penta- | –1630.87490 | 328.69 | 62.34 | 307.97 | 170.76 | 137.21 |
| Hexa- | –1910.61230 | 342.97 | 74.90 | 298.37 | 201.57 | 96.80 |
As is shown in Table 2, the HOFsolid values of all molecules are large and positive, which is favorable to high-energy materials. To show the change trend of HOFsolid with the number of substituents (n) more clearly, the plot giving the calculated HOFsolid values versus n is presented in Fig. 2. It can be seen that there is a good linear relationship between n and HOFsolid. The correlation coefficient is 0.997, which indicates a good group additivity of HOFsolid for prismane derivatives, and each nitroester group addition will decrease the value of HOFsolid by 65.65 kJ/mol. It should be pointed out that average HOFsolid values were used for isomers. The space orientations of the –ONO2 group also affect the HOFsolid value of the compounds. As usual, the shorter the distance between the –ONO2 groups in a compound is, the larger the HOFsolid of the compound is. For instance, as for the isomers with two –ONO2 groups, the HOFsolid value of 1,4-binitroesterprismane is smaller than that of 1,2-binitroesterprismane because the two –ONO2 groups in 1,4-binitroesterprismane are farther away from each other than those in 1,2-binitroesterprismane. A similar change trend is also true for other isomers series.
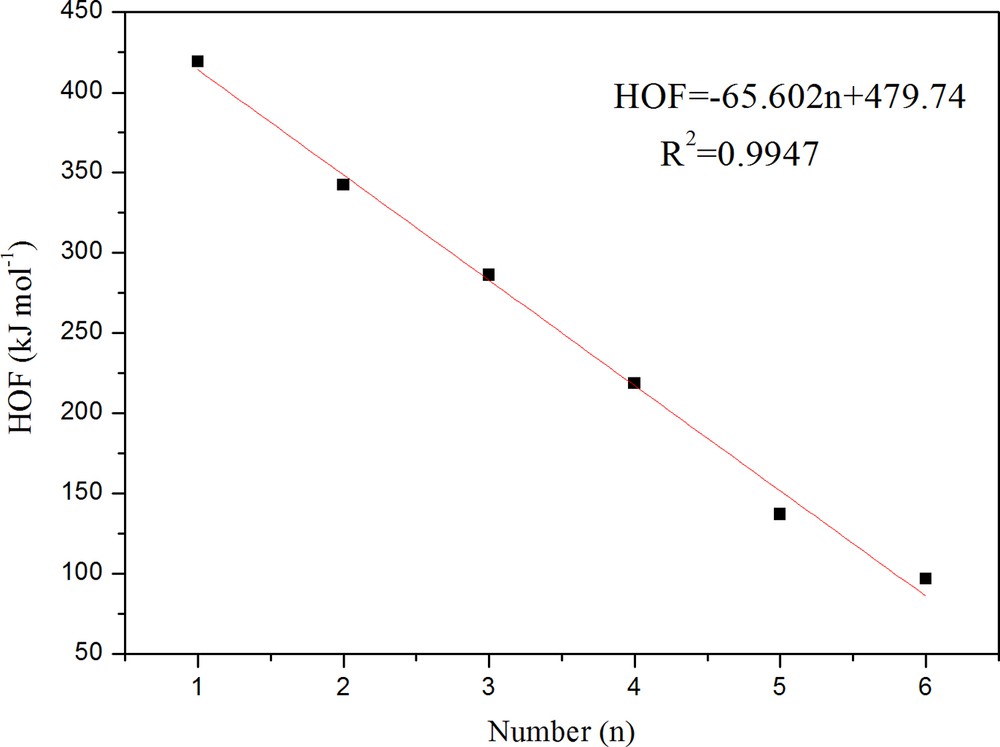
(Color online.) Correlations between HOF values and the number of ONO2 groups (n) for all molecules.
3.2 Detonation performance
Detonation velocity and detonation pressure are two important detonation parameters for an energetic material. They refer to the stable velocity of the shock front that characterizes detonation and the stable pressure that is developed behind the front, respectively [36]. Table 3 presents the calculated molecular density, the heats of detonation, the detonation velocity and pressure for all molecules. For comparison, the experimental detonation performance of RDX (1,3,5-trinitro-1,3,5-triazinane) is also listed in Table 3 together with another known explosive HMX (1,3,5,7-tetranitro-1,3,5,7-tetrazocane). Comparing the theoretical density (ρ) with crystal densities (ρ0), a change trend is found: the theoretical densities are slightly smaller or equivalent to the crystal density for mononitroesterprismane and dinitroesterprismane. However, when the numbers of substituent groups are above two, crystal densities are smaller than molecular density by about 0.02–0.09 g/cm3. Inspecting the Q values, when the numbers of substituent are from 1 to 4, the change in Q is negligible. For pentanitroesterprismane and hexanitroesterprismane, the Q values decrease; this is caused by the highly positive oxygen balance. The fact that a large number of extra oxygen is produced in the combustion reaction or the explosive reaction contributes negatively to the energy released. In addition, it should also be noticed that the positions of the substituent group affect not only HOFsolid, but also Q. The closer the distance to the substituent is, the bigger Q is. Analyzing D and P in Table 3, except for molecule 1-mononitroesterprismane, all molecular detonation velocities are over 8.0 km/s, and that of 1,2,3,4-tetrnitroesterprismane and 1,2,4,5-tetrnitroesterprismane are over 9.0 km/s. The detonation pressures of all molecules are above 30.0 GPa, except for mononitroesterprismane and dinitroesterprismane. Considering the request as HEDM (molecular density > 1.9 g/cm3, detonation velocity > 9.0 km/s, and detonation pressure > 40 GPa), 1,2,3,4-tetrnitroesterprismane can be selected as the outstanding candidate of HEDMs.
Predicted theoretical densities (ρ), correctional densities (ρ0), heat of detonation (Q), detonation pressures (P) and detonation velocities (D) of all compounds.
| Compounds | ρ (g/cm3) | ρ0 (g/cm3) | Q (cal/g) | P (GPa) | D (m/s) |
| 1- | 1.51 | 1.52 | 1929.62 | 17.10 | 6.55 |
| 1,2- | 1.71 | 1.72 | 1940.42 | 27.99 | 8.05 |
| 1,4- | 1.72 | 1.74 | 1908.97 | 28.41 | 8.09 |
| 1,5- | 1.75 | 1.75 | 1934.04 | 28.92 | 8.14 |
| 1,2,3- | 1.88 | 1.86 | 1955.63 | 35.00 | 8.80 |
| 1,2,4- | 1.82 | 1.80 | 1928.03 | 32.55 | 8.57 |
| 1,2,5- | 1.85 | 1.78 | 1952.75 | 32.03 | 8.53 |
| 1,2,3,4- | 1.94 | 1.87 | 1950.46 | 40.05 | 9.28 |
| 1,2,4,5- | 1.93 | 1.86 | 1946.00 | 38.59 | 9.24 |
| Penta- | 1.97 | 1.88 | 1634.45 | 36.47 | 8.95 |
| Hexa- | 2.08 | 1.97 | 1323.05 | 36.12 | 8.79 |
| RDX | 1.82 | 1591.03 | 34.00 | 8.75 | |
| HMX | 1.91 | 1633.90 | 39.00 | 9.10 |
3.3 Power index
In an explosive reaction, heat and gases are liberated. The volume values of gas (V) and the heat of detonation (Q) can be calculated independently, but these values can be combined to give the value for the explosive power by the following equation.
Explosive power = Q × V
The values of V are calculated in standard conditions (273 K, 1 atm).
The value for the explosive power is then compared with the explosive power of a standard explosive (picric acid) resulting in the power index by using the following equation:
Power index = 100 × Q × V/Q(picric acid) × V(picric acid), where the values of Q(picric acid) and V(picric acid) are 1379.07 kJ/mol and 0.831 dm3/g [37], respectively.
The decomposition products are obtained according to modified Kistiakowsky–Wilson rules [38]:
- • hydrogen atoms are converted into water;
- • if any oxygen remains, then carbon is converted into carbon monoxide;
- • if any oxygen still remains, then carbon monoxide is oxidized into carbon dioxide;
- • all the nitrogen is converted into nitrogen gas, N2.
Table 4 shows the decomposition products and power index values of the compounds. As displayed in Table 4, it is found that power index values increase when the numbers of substituent groups are greater than or equal to 3, but the power index values will decrease when the numbers of substituent groups continue to be increased. The power index values of binitroesterprismanes, trinitroesterprismanes and tetrnitroesterprismanes are over 100%, which indicates that these molecules have better detonation performance than picric acid. Compared with the famous materials, TNT (116%), TATB (101%), and HNS (109%), the most investigated molecules have superior high-powered property (see Fig. 3), and furthermore, the conclusion drawn from power index is consistent with that drawn from K–J equations.
Power index values of the all compounds.
| Compounds | Quantity of decomposition products/mol | ||||||
| N2 | H2O | CO | CO2 | C | O2 | Power index (%) | |
| 1- | 0.5 | 2.5 | 0.5 | 0 | 5.5 | 0 | 94.96 |
| 1,2- | 1 | 2 | 4 | 0 | 2 | 0 | 132.74 |
| 1,4- | 1 | 2 | 4 | 0 | 2 | 0 | 130.59 |
| 1,5- | 1 | 2 | 4 | 0 | 2 | 0 | 132.30 |
| 1,2,3- | 1.5 | 1.5 | 4.5 | 1.5 | 0 | 0 | 131.80 |
| 1,2,4- | 1.5 | 1.5 | 4.5 | 1.5 | 0 | 0 | 129.94 |
| 1,2,5- | 1.5 | 1.5 | 4.5 | 1.5 | 0 | 0 | 131.61 |
| 1,2,3,4- | 2 | 1 | 1 | 5 | 0 | 0 | 106.55 |
| 1,2,4,5- | 2 | 1 | 1 | 5 | 0 | 0 | 106.31 |
| Penta- | 2.5 | 0.5 | 0 | 6 | 0 | 2.5 | 75.07 |
| Hexa- | 3 | 0 | 0 | 6 | 0 | 3 | 52.42 |
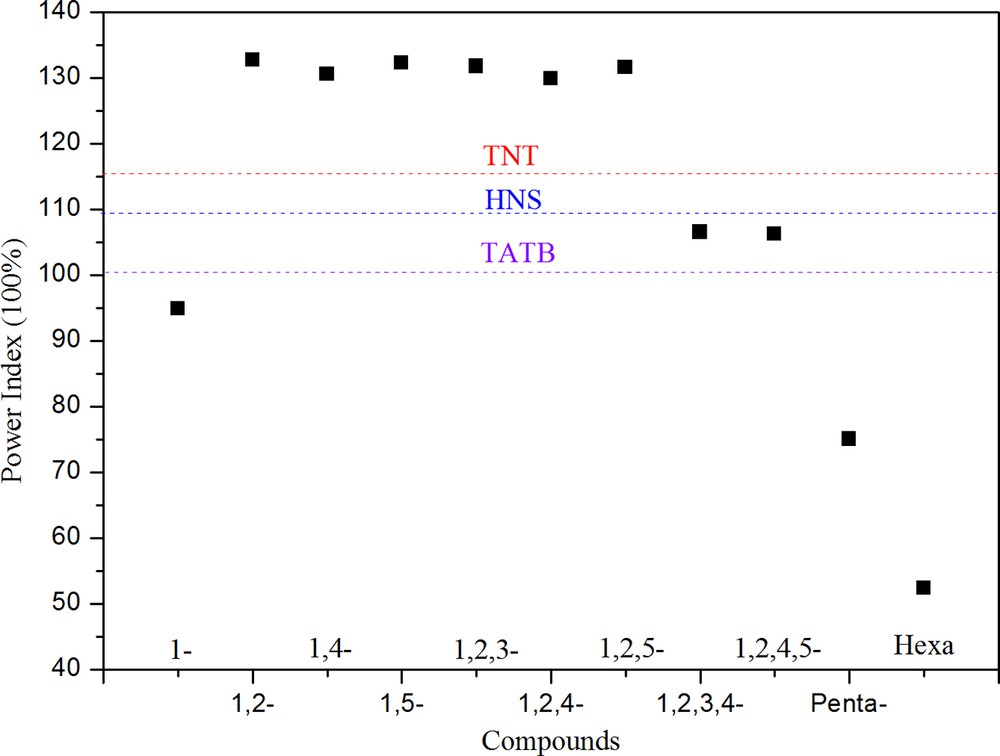
Power index of prismane derivatives together with three known explosives TNT, HNS, and TATB. The purple, blue, and red lines denote the power index of TATB, HNS, and TNT, respectively. (For interpretation of the references to color, see the online version of this article.)
3.4 Bond dissociation energies (BDE)
To elucidate the pyrolysis mechanism and thermal stability of all molecules, we calculated the bond dissociation energies for the possible initial steps in the pyrolysis route. It should be pointed out that we select the weakest C–ONO2 and O–NO2 bonds based on the principle of smallest bond order [39,40]. A smaller Wiberg bond index (WBI) generally indicates a weaker bond, and the predicted bond order and BDE are listed in Table 5. From Table 5, all bond order values of C–ONO2 and O–ON2 bond are smaller than 1.0, which indicates that the strength of these bonds is weak. And the bond order of C–ONO2 is larger than that of O–NO2. The calculated BDEs agree with the change trend of the bond order, which shows that the O–NO2 bond can be considered as the triggering one in the explosive reaction. The BDEs of trigger bonds are over 80 kJ/mol. It is noteworthy that the calculated BDEO–N values of O–NO2 bonds are almost not affected by the position and the number of substituent group. Previous studies [41,42] have reported that a compound could be considered as a practical energetic material if it has BDEs above 80 kJ/mol. According to this, it can be deduced that the designed molecules have a sufficient energy barrier against the removal of the −NO2 group and are stable enough for use.
Predicted bond order (BC-O and BO-N) and bond dissociation energies (BDEC-O and BDEO-N) for C–ONO2 and O–NO2 bonds of all compounds at the UB3LYP/6-311G** level.
| Compounds | BC–O | BDEC–O (kJ/mol) | BO–N | BDEO–N (kJ/mol) |
| 1- | 0.9579 | 417.43 | 0.7653 | 91.66 |
| 1,2- | 0.9319 | 391.83 | 0.7289 | 84.46 |
| 1,4- | 0.9790 | 413.15 | 0.7376 | 88.72 |
| 1,5- | 0.9050 | 365.08 | 0.7636 | 99.48 |
| 1,2,3- | 0.9890 | 398.42 | 0.7285 | 81.34 |
| 1,2,4- | 0.9654 | 396.37 | 0.7190 | 83.33 |
| 1,2,5- | 0.9398 | 388.89 | 0.7267 | 91.45 |
| 1,2,3,4- | 0.9630 | 383.48 | 0.6911 | 82.61 |
| 1,2,4,5- | 0.9518 | 384.53 | 0.6865 | 88.58 |
| Penta- | 0.9728 | 389.18 | 0.6290 | 82.68 |
| Hexa- | 0.9749 | 417.43 | 0.6322 | 87.40 |
3.5 Impact sensitivity
Predicting the impact sensitivity of HEDMs is a very important step for developing new high-energy compounds. Impact sensitivity is usually characterized through a drop hammer test. It is measured by the height H50 from which a given weight falling upon the compound has a 50% probability of producing an explosion. This is because the greater H50 value a compound has, the less sensitivity the compound has. Table 6 lists the H50 values of all molecules at the B3LYP/6-311G** level. From Table 6, there is a legible relationship between the decrease in H50 values and the increase in the number of substituent groups. The H50 values of all molecules lie in the range between 9.06 and 53.66 cm. Compared with the H50 values of the famous explosives CL-20 (12 cm), it is found that all compounds have a lesser sensitivity, except hexanitroesterprismane. Furthermore, the conclusion drawn from the H50 values is not very consistent with that drawn from BDEs. For example, the BDE of O–NO2 cannot be affected by the number and the position of the nitroester group, but it is not the case for H50, which supports the proposal of Politzer that the correlation between bond strength and impact sensitivity is not general, but limited to certain classes of molecules [43].
Calculated H50 and energy gaps (ΔE) for all compounds.
| Compounds | H50 (cm) | ΔE (a.u) |
| 1- | 53.66 | 0.17586 |
| 1,2- | 51.89 | 0.17179 |
| 1,4- | 51.28 | 0.17715 |
| 1,5- | 48.97 | 0.18025 |
| 1,2,3- | 37.77 | 0.17705 |
| 1,2,4- | 33.42 | 0.18012 |
| 1,2,5- | 35.36 | 0.18485 |
| 1,2,3,4- | 19.25 | 0.17902 |
| 1,2,4,5- | 18.78 | 0.17372 |
| Penta- | 12.92 | 0.18709 |
| Hexa- | 9.06 | 0.18514 |
To further estimate molecular sensitivity, the energy gaps of all molecules were calculated; they are listed in Table 6. The energy gap between the highest occupied molecular orbitals (HOMOs) and the lowest unoccupied molecular orbitals (LUMOs) determines the kinetic stability, the chemical reactivity, the optical polarizability, and the chemical hardness/softness of a molecule [44]. The energy gap is also closely related to the molecular orbital energy level in a single molecule and the intermolecular interaction in solids. This is the reason explaining the belief that energy gap closing may be the initiation step for the detonation of explosives [45]. Inspecting the energy gaps, we can find that the change trend is inconspicuous with the increase in the numbers of substituents and the change of position of ONO2. The result is very consistent with the analysis drawn from BDE. Moreover, the energy gaps of the molecules are larger than that of TATB (0.1650 a.u), which indicates that the nitroester prismane derivatives may well have thermodynamic stability.
4 Conclusions
By the theoretical investigation on the nitroester prismane derivatives, the following conclusions can be summarized: Most compounds have a large and positive heat of formation in the solid and gas states. The HOFsolid values are affected by the number and position of the nitroester group. There is a good linear relationship between the number of nitroester groups and HOFsolid. Each addition of a nitroester group will decrease HOFsolid by 65.65 kJ/mol. Most molecules have large molecular density, heat of detonation, power index, and detonation performance, which indicates that the nitroester group into prismane is helpful for enhancing its detonation properties and, according to the calculated BDE and bond order, the O–NO2 bond can the one triggering the explosive reaction. In addition, the BDE values are not affected by the number of nitroester groups and the BDE of all molecules are above 80.0 kJ/mol; based on the calculated H50 and band gap, most molecules show good stability. By considering their detonation properties and stability, we think that 1,2,3,4-tetrnitroesterprismane might be a good candidate for explosives. We hope that the calculated results can provide useful and instructive information for the molecular design of highly energetic materials.
Acknowledgements
This work is financially supported by the Major State Basic Research Development Programs of China (2011CBA00701), the National Natural Science Foundation of China (21473010, 20933001), and the opening project of the State Key Laboratory of Explosion Science and Technology (Beijing Institute of Technology) (ZDKT12-03).