

1 Introduction
The increase in the environmental impact of a high CO2 concentration on the atmosphere through the greenhouse effect has been of acute concern to the global community. Many efforts have been made to reduce CO2 emissions and to improve the climate conditions using various procedures such as removal, recovery, and storage [1–8]. However, it is desirable to develop a technique whereby carbon dioxide recovered can be converted into valuable materials [2,3]. Therefore, catalytic conversion of CO2 into useful chemicals like methanol or hydrocarbons is one of the efficient ways not only to curtail the growing amount of carbon dioxide emission, but also to improve economic benefits [1,6,9–11]. Notwithstanding an extensive literature concerning the production of hydrocarbons via syngas hydrogenation [8–19], only few papers deal with hydrocarbons synthesis via CO2 hydrogenation [5,20–29].
Fundamentally, there are two routes for the production of hydrocarbons through CO2 hydrogenation:
- • a two-step process, in which methanol is initially produced in the first reactor over a metallic catalyst site, followed by methanol conversion into hydrocarbons via DME according to an MTG or MTO process over the acid catalyst located in a second reactor [2,3,14,15];
- • a one-step process, in which a bifunctional catalyst prepared by physically mixing metallic and acidic functionalities are used in only one reactor [30–32].
Methanol is initially produced on a metallic catalyst and then dehydrated into DME on an acidic site of the zeolite. Thus, the DME formed in turn is converted into ethene and then higher into hydrocarbons through oligomerization and isomerization reactions [17]. After that, ethene or other alkenes, which are transported to the metallic catalyst, are hydrogenated into the corresponding alkanes, preventing carbon homologation and restraining the formation of heavier hydrocarbons [17]. Fujimoto and Shikada [2] claimed that the hydrogenation of alkene by the metallic catalyst plays an important role in hydrocarbon synthesis through CO2 hydrogenation.
The main advantage of the single-step process is the elimination of the thermodynamic restriction of methanol synthesis.
We can assume that the transport step between the metallic and the acidic sites of the bifunctional catalysts plays a significant role in the catalytic performances and in the hydrocarbons’ selectivity (see Fig. 1).
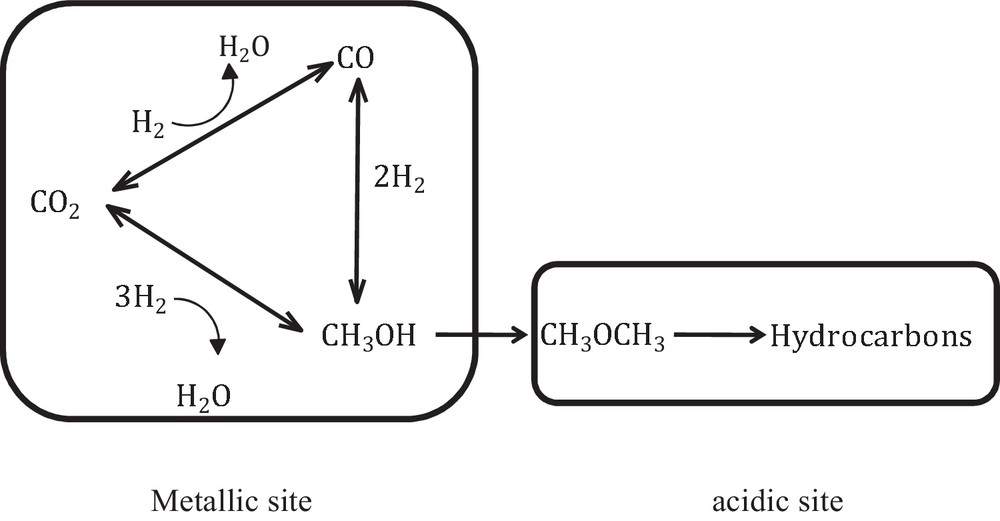
CO2 hydrogenation into hydrocarbons in one single step using a bifunctional system.
Catalysts for the direct CO2-to-HC process should be able to efficiently catalyze both methanol and hydrocarbon synthesis reactions, while the yield of CO formed via the RWGS side reaction [27] should be kept low and/or be transformed into methanol. Typically, the methanol synthesis reaction is carried out on Cu–ZnO–Al2O3 catalysts, while the hydrocarbon reaction occurs mainly over a solid acid component, as a zeolite.
Indeed, our challenge in this study is to achieve CO2 hydrogenation into hydrocarbons in a one-step process using a bifunctional system constituted by a methanol synthesis catalyst, Cu–ZnO–Al2O3 (CZA), and a zeolite (HZSM-5).
In order to evaluate the effect of the proximity between metallic and acidic sites on CO2 activation, hydrogenation and methanol dehydration into hydrocarbons, three different combination dispositions of the two components have been adopted.
2 Experimental
2.1 Catalyst preparation
A copper-based catalyst Cu–ZnO–Al2O3 (CZA) with nominal composition Cu:Zn:Al = 51.0:22.0:3.5 wt. %, determined by ICP-AES, was prepared according to a procedure described in detail elsewhere [30].
To evaluate the effect of the contact between the two catalytic functions, metallic-acid sites, on hydrocarbons synthesis, the copper-based catalyst CZA was admixed to a commercial HZSM-5 microporous zeolite (Zeolyst Int.CBV3024E). The physicochemical properties of the acidic catalysts (HZSM-5) are listed below: framework Si/Al ratio = 40, surface area = 452 m2·g−1, pore volume = 0.3 cm3·g−1. The acidity of the zeolite (number of Brønsted acid sites able to retain pyridine at 150 °C, infrared spectroscopy) was 297 μmol·g−1.
Three different dispositions of the metallic and the acidic catalysts (see Fig. 2) adopted during the catalytic tests are described in details in the following section:
- • M1 is used to load the two catalysts in a dual-bed arrangement; the contact between the catalysts was avoided by a glass wool between the catalysts layers;
- • M2 is used to grind, pressure mold, crush and sieve each component CZA and HZSM-5, respectively, to prepare particles of 0.2–0.4 mm. Then, the particles of each component were thoroughly mixed;
- • M3 is to grind the fine particles of CZA and HZSM-5 catalysts in a porcelain mortar for sufficient time so that the two kinds of catalysts could be further uniformly mixed and then the mixture is pelletized, crushed, and sieved until a 0.2–0.4-mm particle size.
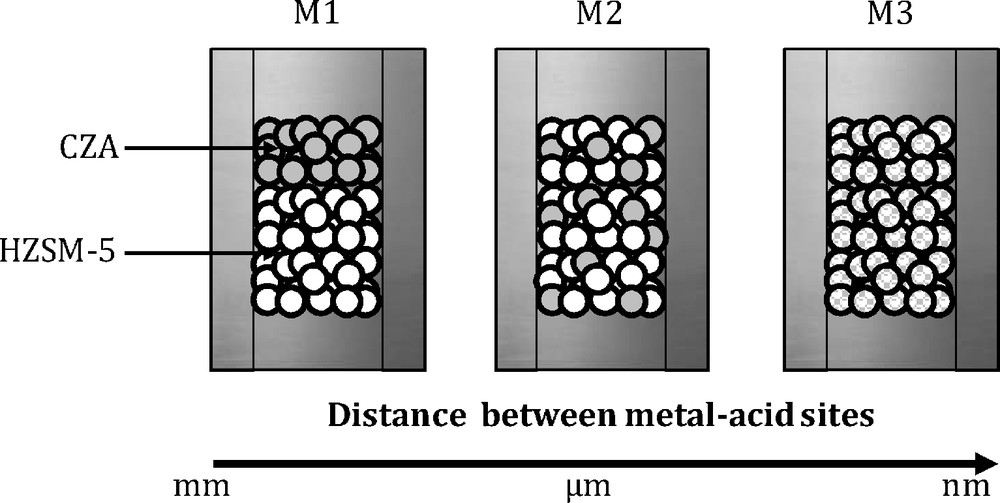
Combination procedures between methanol and acid catalysts [: Cu–ZnO–Al2O3 (CZA); : a zeolite (HZSM-5)].
Considering the results obtained previously in our laboratory dealing with the effect of zeolite loading within the hybrid catalyst on the activity and the yield of hydrocarbons, a weight ratio of 1/3 (oxide/zeolite) was used to prepare the different dispositions.
2.2 Characterization of the catalysts
2.2.1 Nitrogen adsorption–desorption (BET)
The surface areas of the oxide, hybrid and HZSM-5 catalysts were determined by adsorption–desorption of nitrogen at the temperature of liquid nitrogen (−196 °C) using a Micromeritics Tristar and ASAP 2000 instruments. Before measuring, the samples (oxide and hybrids) were degassed at 250 °C for 4 h and the zeolite HZSM-5 was heated at 350 °C overnight. The surface areas were calculated according to the method of Brunauer, Emmet and Teller (BET).
2.2.2 In situ X-ray diffraction
In situ XRD measurements were collected using an ADVANCE D8 diffractometer with a Cu Kα radiation source (λ1 = 1.54056 Å, λ2 = 1.54439 Å) equipped with a high-temperature reaction cell to enable the temperature-driven in situ reduction of the catalysts.
The catalysts were exposed to 3%H2/He at 30 °C for 1 h and then the temperature of the oven was raised from 50 to 350 °C at a rate of 5 °C/min.
The sample was kept 1 h in a reducing atmosphere at each temperature, except at 350 °C, at which it was kept for 4 h. The diffraction pattern was recorded every 50 °C in the 2θ range from 20° to 90° with a step size of 0.024° and a time/step of 1 s.
After subtraction of the background and correction for instrumental broadening, the peak width and the diffraction angle 2θ were used to calculate the average crystallite size dhkl [18]:
2.2.3 H2 adsorption
Hydrogen adsorption measurements were performed at 30 °C using a Micromeritics Autochem 2920II apparatus. The samples were reduced for 3 h at 350 °C in a hydrogen flow (30 mL/min), which was subsequently evacuated using an argon flow (30 mL/min) at the same temperature for 1 h. The reactor was cooled down to room temperature using argon and then pulses of 10% H2/Ar were injected until saturation. After that, the sample was degassed with argon for 10 min to evacuate the H2 physisorbed, which was followed by another series of H2 pulse injections.
2.3 Catalytic testing
Carbone dioxide hydrogenation was performed in a continuous-flow fixed-bed reactor (12.5 mm id) made of stainless steel, using 0.5 g of oxide catalyst or 2 g of hybrid catalyst, diluted with silicon carbide to avoid thermal effects (hot spots) (SiC, d = 1.9 mm) at a weight ratio of 4:1. The temperature in the tubular reactor was controlled with a coaxially centered thermocouple in contact with the catalytic bed.
The catalysts used in this reaction were loaded in the reactor according to the three ways presented in Fig. 2. Prior to the reaction, the sample was reduced in situ at 350 °C at a heating rate of 5 °C/min under a flow of H2 (30 mL/min, 99.99% H2) for 4 h at atmospheric pressure. After that, a gas mixture (H2/CO2 = 3) was introduced into the reactor at 350 °C, and the pressure was then raised up to 3.0 MPa. The liquid products were separated from the gas flow in the gas liquid separator and the condenser. The effluent products from the heated line were analyzed by an online gas chromatograph system, which was composed of three gas chromatographs:
- • Porapak Q, TCD for CO and CO2;
- • AT-Aquawax (FID) for methanol and dimethylether (CH3OH, CH3OCH3);
- • with a capillary column HP-Plot Q, FID for hydrocarbons (C1-C6).
All analysis lines and valves were heated at 150 °C to prevent a possible condensation of the products before entering the gas chromatograph.
3 Results and discussion
3.1 XRD diffraction
In situ XRD powder patterns recorded for different temperatures of the hybrid catalyst M3 are shown in Fig. 3. The presence of alumina and HZSM-5 phases in the hybrid catalyst is clearly confirmed by the Bragg diffraction peaks at 2θ = 44.4° (JCPDS 34-0493) for alumina and at 23–25° for HZSM-5, whatever the temperature. However, heating the sample between RT and 200 °C does not induce changes in the crystal structure, and the XRD powder patterns are fully indexed using mainly two phases: CuO at 2θ = 35°, 38.8° (JCPDS 00-045-0937) and to ZnO at 2θ = 31°, 36° (JCPDS 00-036-1451).
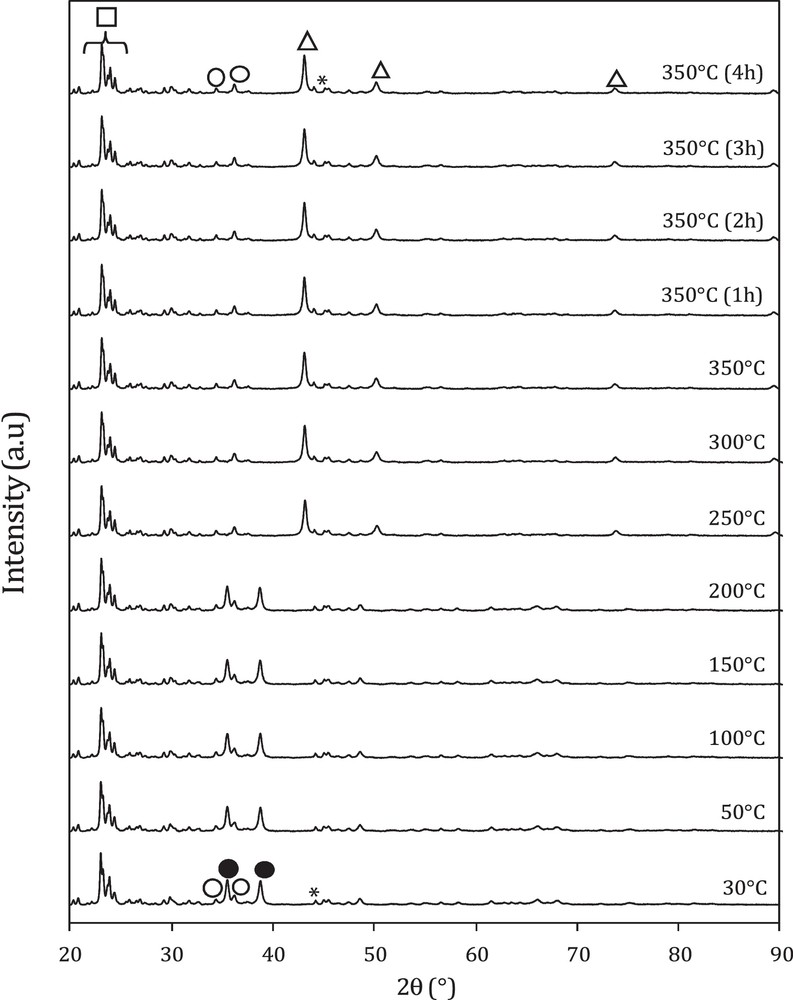
In situ X-ray diffraction patterns of hybrid catalyst (M3) recorded during reduction in 3% H2 in He. ●: CuO; : ZnO; Δ: Cu; : Al2O3; □: a zeolite (HZSM-5).
Beyond 200 °C and up to 350 °C, we noticed a full disappearance of the peaks relative to copper oxide CuO at the expense of new peaks related to metallic copper peaks at 2θ = 43.3°, 50° and 73° (JCPDS 01-089-2838), which indicates a complete reduction of CuO into Cu. However, no changes were observed for zinc oxide and the peaks relative to ZnO are still remaining in the structure. The particle sizes of CuO and Cu in the oxide and the hybrid catalysts determined using the Scherrer equation are 28 nm and 34 nm, respectively. This result means that the addition of zeolite had no effect on the particle size (Table 1).
CuO and Cu particles size of the catalysts.
| Catalyst | d (nm)a | |
| Before reduction (CuO, 38.8°) | After reduction (Cu, 43°) | |
| CZA | 28 | 34 |
| M3 | 28 | 34 |
a Particle sizes determined by the Scherrer equation.
3.2 Catalytic results
Carbon dioxide hydrogenation over oxide and hybrid catalysts were carried out using a stainless steel reactor under the following reaction conditions: H2/CO2 = 3, T = 350 °C, and P = 3.0 MPa; the results are listed in Table 2.
Effect of the contact between oxide and zeolite on carbon dioxide hydrogenation over the hybrid catalysts.
| Test | CO2 conv. (%) | Yield (%, C-mol) | O/(O + P)b | Coke (%) | ||
| CO | MeOH | HCa | ||||
| MeOH synthesisc | 30.3 | 29.6 | 0.7 | – | – | – |
| CZA | 32.0 | 29.0 | 2.4 | 0.6 | 0.0 | 0.4 |
| CZA + HZSM5(M1) | 36.4 | 24.6 | 0.0 | 11.8 | 1.0 | 1.8 |
| CZA + HZSM5(M2) | 37.3 | 21.6 | 0.3 | 15.3 | 0.1 | 1.2 |
| CZA + HZSM5(M3) | 39.8 | 21.3 | 1.6 | 16.8 | 0.0 | 0.5 |
a O/(O + P) = C mol (olefin C2 to C6)/[(olefin C2 to C6) + (paraffin C2 to C6)].
b HC: hydrocarbons.
c Thermodynamic equilibrium.
As shown in Table 2, carbon monoxide CO is the main product formed over the copper-based catalyst according to the reverse water gas shift reaction widely known from the literature. The high yield of carbon monoxide can be also attributed to methanol decomposition at high-temperature, as already reported in the literature [31], and this explains the small yield of MEOH formed at 350 °C (0.7%). A small amount of hydrocarbons was also detected (mainly CH4 and C2H6). Over the hybrid catalysts, CO remains predominant together with the formation of a good yield of hydrocarbons. In fact, mixing HZSM-5 with methanol synthesis catalysts improves significantly CO2 conversion and the yield of hydrocarbons. The formation of hydrocarbons is explained by the transformation of methanol and dimethylether intermediates formed over copper-based catalysts into hydrocarbons over zeolite acid sites.
The proximity between the metallic and the acidic sites is one of the main keys in the formation of hydrocarbons through CO2 hydrogenation. In fact, the investigation of the effect of the contact between the two catalytic functions on the hydrocarbons distribution and the selectivity is an important factor to be studied. This can bring a lot of clarifications on the mechanism of hydrocarbons formation over the hybrid catalysts. We plotted in Fig. 4 the hydrocarbons selectivity versus the number of carbons for the different catalytic bed arrangements.
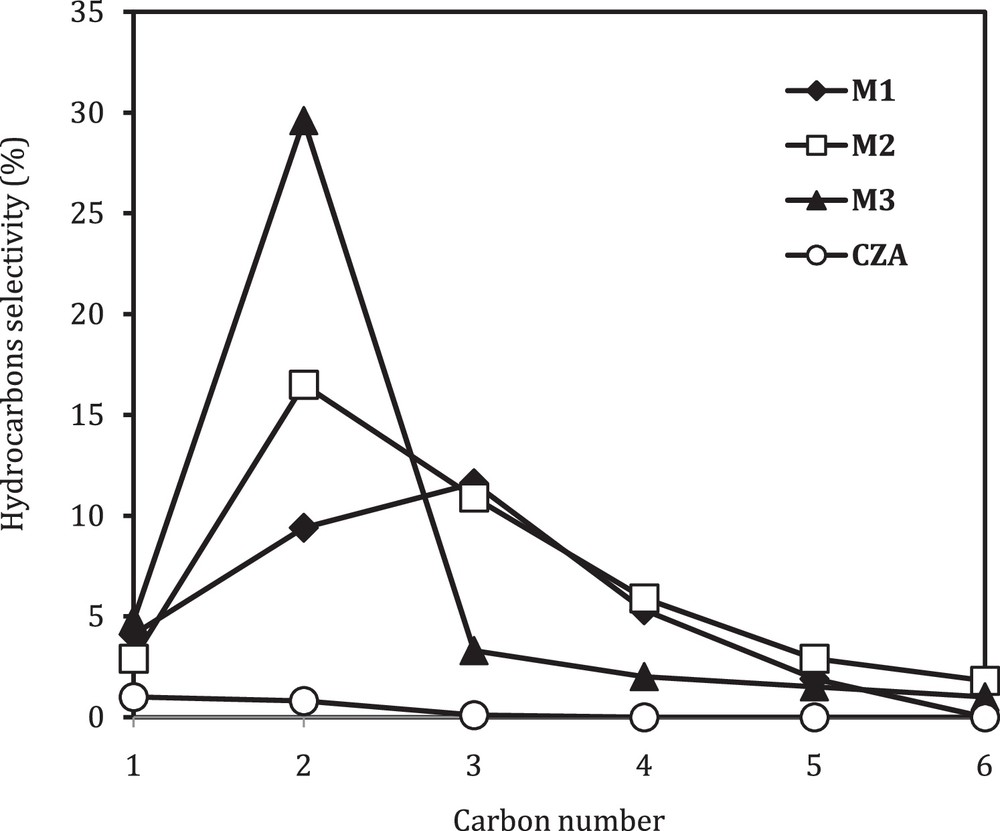
Effect of the contact between oxide and zeolite on hydrocarbon selectivity.
Whatever the proximity between metallic and acidic sites (M1, M2 or M3), the selectivity to methane over the hybrid catalysts was low, and the distribution of hydrocarbons was shifted toward higher carbon numbers, selectively C2. This behavior is pronounced with the increase of the proximity between oxide and zeolite, and the same results were previously reported by Jeon et al. [16]. According to them, the formation of ethane appears to be due to the abundance of strong Brønsted acid sites that have a high hydrogenating ability to reduce alkenes into alkanes.
The product distribution over the follow-bed arrangement (M1) and the single-bed system (M2) are quite similar. We can assume that the equilibrium constraints of methanol formation can be avoided by the removal of methanol from the gas phase through its conversion into hydrocarbons when the hybrid catalyst in the single-bed system (M2) is used. This equilibrium shift in the methanol synthesis mechanism enables a larger yield of hydrocarbons than the thermodynamically limited methanol formation from carbon dioxide. However, in the follow-bed arrangement (M1), the methanol formed over the copper catalyst in the first stage was converted into hydrocarbons over HZSM-5 in the second stage. The thermodynamic limits of methanol formation could not be overcome in this arrangement and the distribution of hydrocarbons follows the non-Anderson–Schultz–Flory distribution.
These results, therefore, indicate that, in the case of the composite catalysts, C2+ hydrocarbons were not formed directly from carbon dioxide, but through a MTG reaction of methanol obtained from carbon dioxide. The yields of C4+ hydrocarbons were extremely poor, and alkenes such as ethene and propene were not produced. The formation of higher hydrocarbons (C4+) is enhanced while decreasing the proximity between the two catalysts (oxide–zeolite), and the highest yield is obtained with the two-bed arrangements (M1). These results may be explained by hydrogen spillover, which does not promote the chain growth and the formation of heavier hydrocarbons. Consequently, the carbon deposit was lower on the hybrid catalysts loaded in the single-bed system compared to the duel bed arrangements, as we can see in Table 2. The same explanation has been already used by several researchers [29,32,33] and, according to them, hydrogen spillover from oxide catalyst to the zeolite surface enhances the reducibility of oxide catalysts and prevents chain growth.
To confirm these conclusions, hydrogen adsorption over the hybrid catalysts prepared through the three ways presented in Fig. 2 was used and the results are presented in Fig. 5.
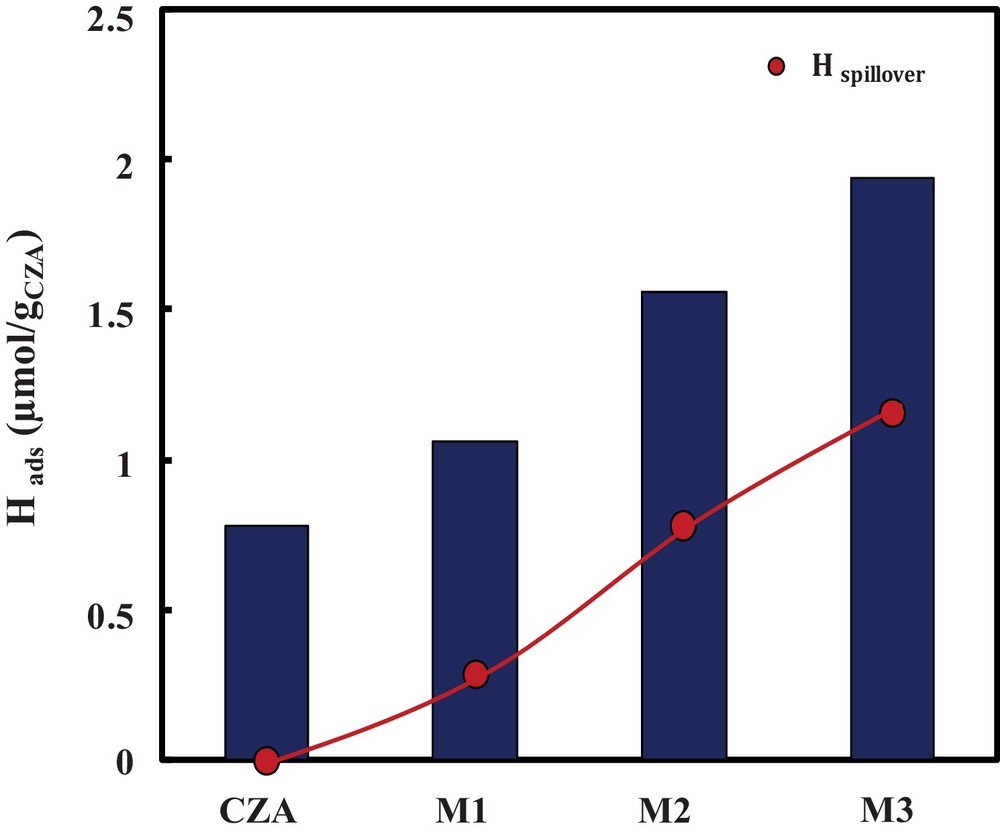
(Color online.) H2 adsorption together with Hspillover over hybrid catalysts for M1, M2, and M3.
Over oxide catalyst CZA, 0.8 μmol/gCZA of H2 was adsorbed; however, mixing the oxide catalyst with a zeolite induces a significant change in hydrogen adsorption over the hybrid catalysts. In fact, over the hybrid catalysts, the amount of hydrogen adsorbed increased significantly, and this feature is more pronounced when the proximity between oxide and zeolite increases, as shown in Fig. 5.
In the case of the hybrid catalyst M1, an amount of 0.3 μmol·g−1 was obtained, which then increases with the increase of the proximity oxide–zeolite to reach 1.1 μmol·g−1 in the case of M3.
This increase is explained by hydrogen spillover (Fig. 6), and its value increases in the proximity between oxide and zeolite.
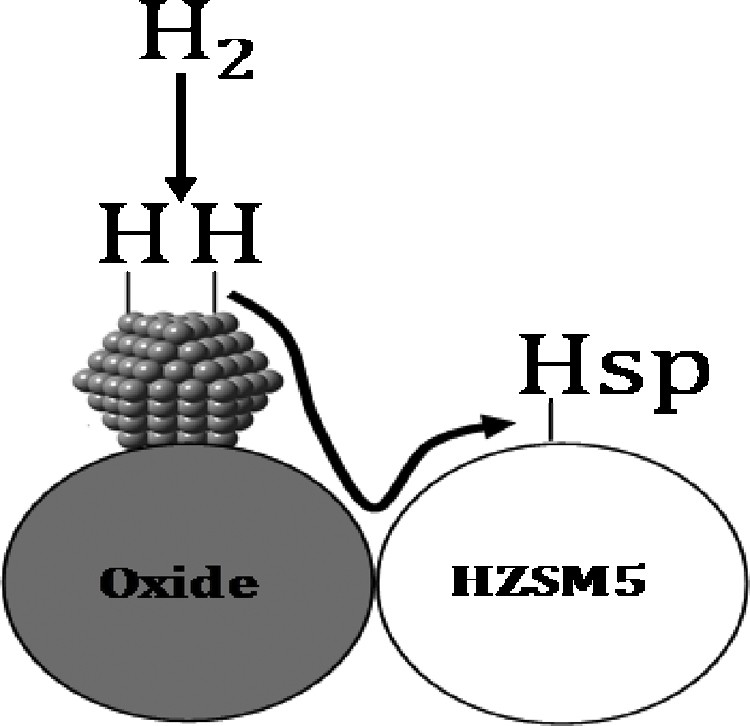
Hydrogen spillover over hybrid catalysts.
The amount of the hydrogen spillover was calculated using the following formula:
This results are in good agreement with the hydrocarbon distribution, where the higher yield of C2 is obtained with catalytic bed M3, in which hydrogen spillover is the most important; consequently, the carbon chain growth is avoided.
4 Conclusion
Hybrid catalysts prepared by physically mixing a copper-based catalyst with HZSM-5 were used for hydrocarbon synthesis through carbon dioxide hydrogenation. It was shown that the addition of HZSM-5 does not affect the copper particles size, whereas the hydrogenation of CO2 into hydrocarbons was found to be strongly influenced by the proximity between oxide and zeolite. Indeed, the highest conversion and the best yield of hydrocarbons (mainly C2) were obtained when the proximity between oxide and zeolite was low. The formation of C2 is explained by hydrogen spillover, which does not promote chain growth.
Acknowledgements
H. Ahouari thanks the Institute of Chemistry of Media and Materials in Poitiers (IC2MP) and the “Agence universitaire de la Francophonie” (AUF) for financial support.