

1 Introduction
Sustainability challenges of today and the future have drawn considerable efforts toward the development of greener alternatives for synthetic chemical reactions. As we celebrate the year of light in 2015, the emphasis on sunlight to drive chemical reactions is receiving greater attention [1–5]. Already at the turn of the 20th century, Prof. Ciamician envisioned the use of sunlight energy to meet global needs with his pioneering work on photochemical organic chemistry [6]. In nature, plants have long mastered the ability to harness light and efficiently convert it into chemical energy through photosynthesis [7]. In the current context of light-driven alternatives, our research group has focused on hybrid P450 enzymes to harness the synthetic potential of P450 enzymes upon visible light excitation [8–13].
The large superfamily of cytochrome P450 heme-thiolate enzymes utilizes two reducing equivalents and molecular dioxygen to perform regio- and stereoselective functionalizations of unactivated C–H bonds in a wide range of substrates [14–16]. They have attracted a lot of interest from deciphering their mechanism to their importance in pharmaceutical and biotechnological applications [17]. The necessary electrons are typically supplied in rate-limiting steps by redox-partner proteins, reductases, from the NAD(P)H cofactor [18, 19]. Due to the limitations associated with the reductase and its cofactor [20], alternative approaches are being developed to power P450 enzymes.
With the hybrid P450 enzymes, we proposed that a Ru(II)-diimine photosensitizer covalently attached to P450 heme domain enzymes will be able to deliver the necessary electrons to the heme active site upon visible light irradiation and sustain photocatalytic activity [11]. This approach stemmed from the wealth of electron transfer studies in Ru(II) functionalized metalloproteins from the Gray group [21–23] and the recent examples of Ru(II) complexes used to power various metalloenzymes leading to the activation of small molecules [24–29]. The first Ru(II)-diimine functionalized heme domain was used to investigate intramolecular electron transfer and the formation of elusive reactive intermediates in the P450 catalytic cycle [8]. We showed that a photogenerated Ru(III) species is able to rapidly oxidize the ferric aquo heme center and generate a high-valent ferryl species, namely Compound II. We have since then used a photoreductive approach to inject electrons into the heme active site and perform P450 reactions upon visible light irradiation.
The first part of this report will introduce the hybrid P450 enzymes with some background on cytochromes P450 and Ru(II)-diimine complexes. We will then highlight the work to date on the optimization of their photocatalytic activity in the hydroxylation of long chain fatty acids and on the expansion of the light-driven approach to another member of the cytochrome P450 superfamily.
2 Cytochrome P450 enzymes
This superfamily of heme-thiolate enzymes utilizes molecular dioxygen and two electrons to perform the selective oxidation of unactivated C–H bonds in a variety of organic substrates. The consensus mechanism involves two successive one-electron transfer steps from the electron-providing reductase to the heme center with the formation of several well-characterized intermediates [30]. Activation of molecular dioxygen at the heme center generates a high-valent Fe(IV)-oxo porphyrin radical species, namely Compound I, capable of performing the desired C–H bond functionalization via a rebound mechanism [31, 32].
To date, the superfamily of P450 enzymes comprises more than 18,000 identified genes with a conserved tertiary fold of their heme domain despite low sequence homology [17]. The cytochrome P450s are often categorized in four major classes depending on their electron-providing reductases [18]. One unique class includes systems where the reductase is fused to the heme domain in a single polypeptide chain, of which cytochrome P450 BM3 or CYP102A1 from Bacillus megaterium is the most well known member [33]. P450 BM3 displays the highest catalytic hydroxylation rate ever measured for P450 enzymes. This high catalytic activity is attributed to the fused reductase, which leads to efficient coupling between the delivery of electrons and activation of oxygen at the heme center.
The dependence on the reductase domain and the use of NAD(P)H to initiate the ET cascade are still limitations in the development of P450 enzymes as biocatalysts [20]. To address these limitations, alternative approaches to deliver electrons have emerged, which include the use of artificial fusion P450 proteins to mimic the P450 BM3 system [34, 35], NAD(P)H regeneration systems [36], terminal oxidants [37], direct chemical [38] and electrochemical [39] reductions, as well as light-activated approaches [40–43].
3 Hybrid Ru(II)-diimine functionalized P450 enzymes
As an alternative approach to perform light-driven P450 reactions, we have developed hybrid P450 BM3 enzymes consisting of a Ru(II)-diimine photosensitizer covalently attached to P450 BM3 heme domain mutants (Fig. 1B,C) [8–13]. This approach takes the advantage of the unique photochemical properties of Ru(II)-diimine complexes, which have been extensively reviewed elsewhere [44–46]. Briefly, these complexes of general formula [Ru(LL)3-n(LL′)n]2+, with LL,LL′ = diimine ligands such as bipyridine or phenanthroline and their derivatives, possess valuable photochemical properties amenable to light-induced electron transfers. They often display a broad absorption band in the visible range centered around 450 nm attributed to a metal-to-ligand charge transfer transition (MLCT). Excitation into this band followed by rapid intersystem crossing due to the heavy atom effect leads to a long-lived triplet excited state with unusual redox properties [46]. This [Ru]2+* excited state can decay back to the ground state via a radiative process or encounter other solute molecules called quenchers. Participation in these bimolecular processes results in energy transfer and either reductive or oxidative electron transfer. Depending on the nature of the quencher, a highly reductive, [Ru(LL)3-n(LL′)n]+ abbreviated [Ru]+ (E1/2 = −1.28 V vs NHE), or oxidative, [Ru(LL)3-n(LL′)n]3+ abbreviated [Ru]3+ (E1/2 = +1.26 V vs NHE), species can hence be photogenerated (Fig. 1).
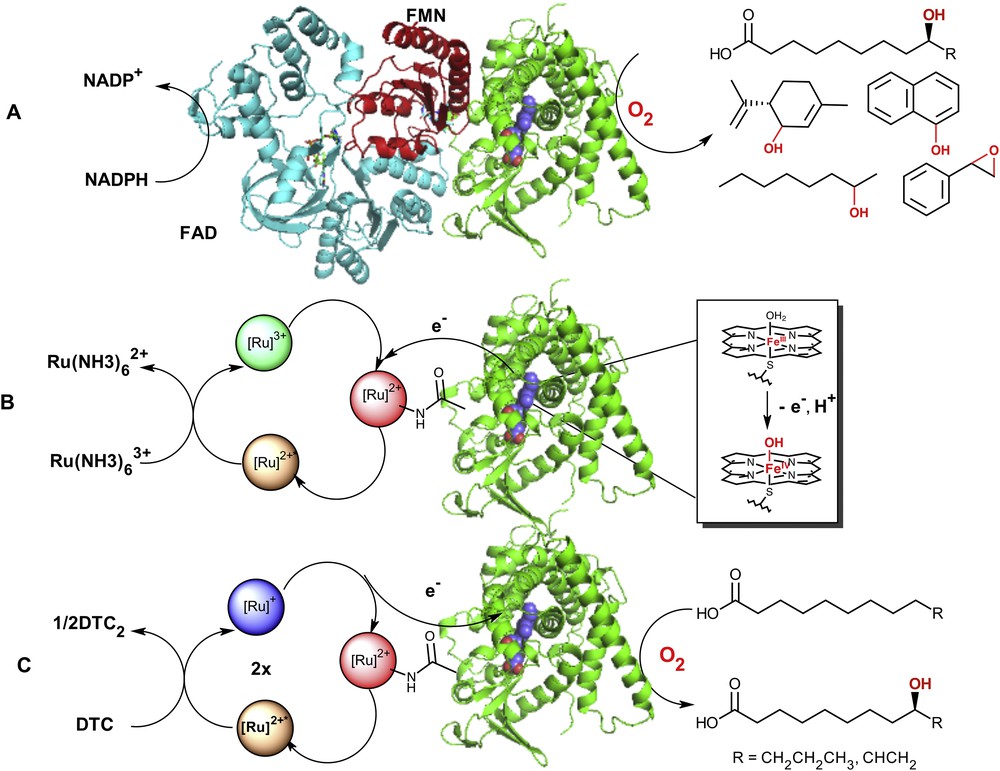
A) Postulated model of the native P450 BM3 enzyme [56, 57] containing the reductase (FAD (blue) and FMN (red) binding domains) and heme domain (green) adapted from X-ray crystal structures (PDB = 4DQK (FAD) [57] and 1BVY (FMN/heme domain) [58]). The electrons are shuttled to the heme active site (blue) from the NAD(P)H cofactor to activate molecular dioxygen and carry on the selective C–H bond functionalization in a wide range of substrates; B) oxidation of the resting ferric-aquo species using a photooxidative flash quench approach with [Ru(NH3)6]3+ as a quencher; C) Photoreductive strategy (DTC = diethyldithiocarbamate) in the hybrid P450 BM3 enzymes leading to the formation of hydroxylated fatty acids at subterminal positions upon visible light excitation.
Covalent attachment of the photosensitizer was established early on to be crucial for electronic communication with the buried heme center in P450 enzymes [8] and for photocatalytic activity of the hybrid enzymes [9]. Introduction of an iodoacetamide-1,10-phentanthroline (PhenIA) [47] or more recently of the 5,6-epoxy-5,6-dihydro-1,10-phenanthroline [12] ligands into the Ru(bpy)2(LL) framework leads to the selective covalent attachment of the photosensitizers to non-native single cysteine mutants of the P450 BM3 heme domain.
Various techniques have been used to confirm the covalent attachment of the photosensitizer including mass spectrometry, UV–vis and fluorescence spectroscopy, X-ray crystallography as well as LC/MS analysis after chymotrypsin digestion of the hybrid enzymes [10–12].
3.1 Electron transfer studies in the hybrid enzymes
Based on the wealth of data on electron transfer studies in Ru(II)-functionalized metalloproteins, we initially investigated the use of Ru(II)-diimine complexes to form elusive reactive intermediates in the P450 catalytic cycle. Using an oxidative flash quench approach (Fig. 1B), we showed that a covalently attached Ru(II)-diimine complex is able to rapidly oxidize the ferric aquo heme center and generate a high-valent ferryl species, Compound II [8]. Using transient absorption measurements and global fitting of the traces, we mapped out the kinetic parameters and intermediates in the formation of the high-valent Fe(IV) species. The photogenerated Ru(III) species initially oxidized the porphyrin ring consistent with other studies in microperoxidase [48] and horseradish peroxidase [49]. Subsequent reorganization of the radical leads to a high-valent Fe(IV)-OH species within 100 μs. In addition, pH dependence studies confirmed the protonated nature of the ferryl species in agreement with the proposed basicity of cys-ligated ferryl species [50–52].
3.2 Light-driven selective hydroxylation of organic substrate C–H bonds
Since Ru(II)-diimine complexes could promote electron transfers in metalloproteins upon visible-light excitation, there has been a strong motivation to drive catalytically competent enzymes with visible light and capitalize on their unique synthetic ability [24–28]. We have hence used a photoreductive approach with the covalently attached Ru(II)-diimine photosensitizer to inject electrons to the heme center and perform P450 reactions upon light activation. Initially, low total turnover numbers in the hydroxylation of the model substrate, lauric acid, were obtained with the first generation of hybrid enzymes (Fig. 2) [9]. Nevertheless, the total turnover numbers were higher than those observed in the controls where each of the individual components was systematically removed. Later on, varying the position and nature of the photosensitizer led to a notable increase in formation of hydroxylated products (Fig. 2) [10]. Eventually, a third generation was developed which includes the highly efficient sL407C-2 mutant containing a Ru(II) photosensitizer with electron donating groups covalently attached to an engineered cysteine residue at position L407C. This hybrid P450 BM3 mutant showed efficient O2 activation with enhanced activity in the light-driven hydroxylation of the C12 fatty acid [11].
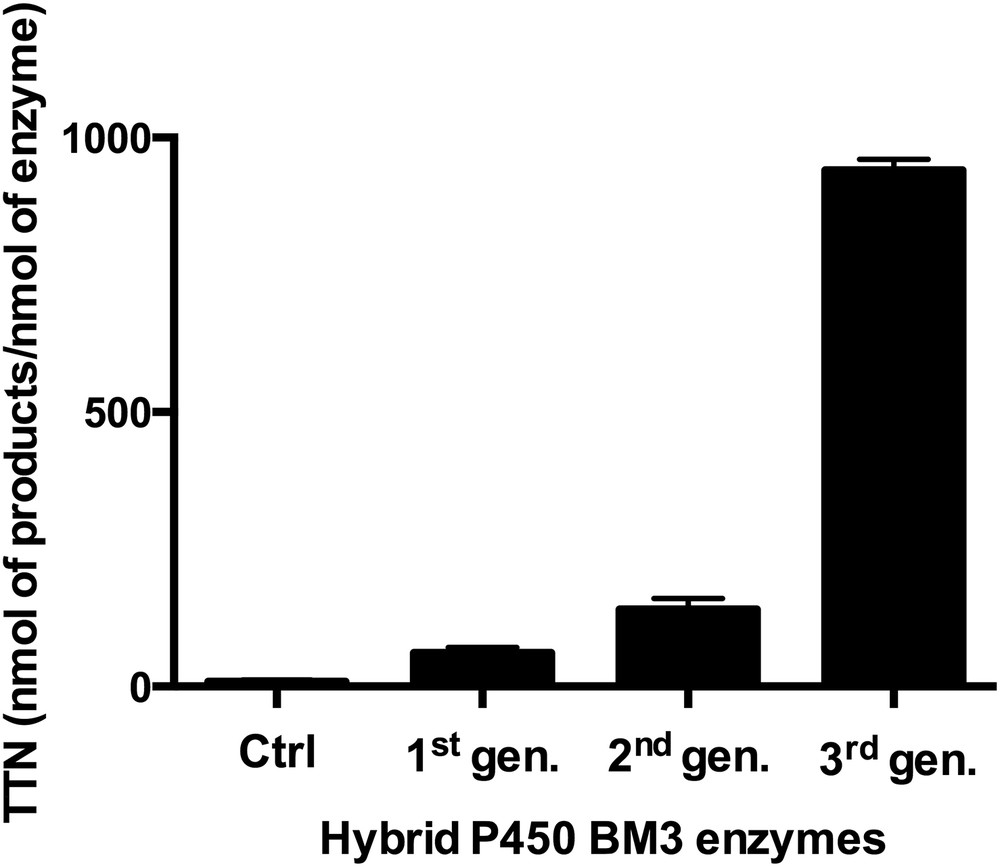
Optimization of the total turnover numbers (TTN, nmol of product/nmol of enzyme) in the selective hydroxylation of lauric acid using several generations of hybrid P450 BM3 enzymes. The first generation consisted of triply mutated enzymes where the two native cysteines were mutated and non-native single cysteines were engineered [9]. In the second generation, non-native single cysteine residues were introduced at the proximal heme site and the nature of the photosensitizer was changed [10]. The third generation includes the singly mutated sL407C hybrid enzyme labeled with various photosensitizers [11]. The controls consist of the systematic removal of each parameter necessary for the light-driven process.
More than 900 total turnover numbers were obtained with a fast initial reaction rate of 120 eq/min. It is worth noting that the nature and composition of the hydroxylated products obtained in the photocatalytic enzyme reaction are identical to that observed with the natural system confirming that the heme domain is responsible for the observed hydroxylation at subterminal positions in long chain fatty acids (Fig. 1). This light-driven P450 biocatalyst was also used in the one-step synthesis of a valuable synthon en route to the synthesis of several natural products [13]. The high photocatalytic activity established that the photogenerated reductive [Ru]+ species is able to inject electrons into the heme active site and sustain photocatalytic activity. In addition, this approach circumvents the use of the natural electron transfer pathway (Fig. 1), reductase and NAD(P)H cofactor, and can therefore be more applicable to other members of the large family of cytochrome P450 enzymes.
4 Expanding the light-driven approach to other P450 heme domain enzymes
One of the striking features of cytochrome P450s is their conserved tertiary fold despite low sequence homology of their heme domains [17]. This prompted us to expand the light-driven approach to another soluble cytochrome P450, P450cam or CYP101 [53]. P450cam performs the selective hydroxylation of camphor to yield 5-exo-hydroxy-camphor. In contrast with P450 BM3, its redox partners (PdR and PdX) are not covalently attached to the heme domain and lower hydroxylation rates are typically observed.
We followed the same general procedures to generate hybrid mutants of P450cam and engineered a single non-native cysteine mutant for the covalent attachment of the photosensitizer. Because P450cam contains seven cysteines in addition to the heme-ligated cysteine (C357), it was first necessary to remove the surface exposed cysteines. This process was facilitated thanks to a generous donation from Prof. David Goodin of a plasmid containing five of the surface exposed cysteines already mutated (C334A, C58S, C85S, C136S, and C285S) [54]. We mutated an additional cysteine to serine (C148S) to obtain our starting platform plasmid. We then introduced a non-native cysteine at position 102 as well as the L358P mutation, which has been shown to induce structural changes observed upon reductase binding [55].
We were then able to successfully express, purify and label the P450cam mutant following reported protocols [54] to yield the first hybrid P450cam mutant, CAM-H1. Labeling of the P450cam mutant was achieved with the Ru(bpy)2PhenIA photosensitizer and confirmed using the above-mentioned techniques used for the hybrid P450 BM3 enzymes. As noted in Fig. 3, the mass spectrum of CAM-H1 shows the characteristic shift of 649 amu upon labeling of the protein with Ru(bpy)2PhenA and the UV–vis spectrum shows the MLCT band from the photosensitizer at 450 nm. It is worth noting that the hybrid CAM-H1 mutant is still able to bind camphor as shown in Fig. 3B. Under visible light irradiation following our established flash quench reductive conditions, hydroxylation of camphor was detected and the nature of the hydroxylated product was confirmed by GC–MS (Fig. 3C). The total turnover numbers are low but our lab is currently working on optimization following the same strategy developed for P450 BM3. Overall, these findings support that the light-driven approach can be expanded to other members of the P450 superfamily.

A) Characteristic mass shift upon covalent attachment of the photosensitizer (649 amu) in Cam-H1 with theoretical protein masses in parenthesis; B) UV–vis spectra of the substrate-free (SF, blue) and camphor-bound (SB, red) hybrid P450cam mutant containing the Ru(bpy)2PhenA label (black); C) GC chromatograms of the light-driven reaction and control after dichloromethane extraction and trimethylsilylation, which show the presence of the desired 5-exo-hydroxy-camphor. (Borneol as internal standard, I.S.).
5 Conclusions
Adding to the list of Ru(II)-diimine functionalized metalloproteins, the results obtained with the hybrid P450 BM3 enzymes establish the ability of the photosensitizer to efficiently deliver electrons to the heme domain and sustain photocatalytic activity. This approach circumvents the use of the redox partner proteins and the NAD(P)H cofactor and is therefore applicable to other members of the P450 superfamily as shown with the hybrid P450cam study. The hybrid P450 enzymes therefore present a valuable alternative to perform selective light-driven functionalization of various unactivated C–H bonds in a wide range of substrates in response to the current sustainability challenges.
Acknowledgments
This work was supported by the National Institute of General Medical Science of the National Institutes of Health under award number SC3 GM095415. L.C. and J.H. would like to thank the National Institute of Health Research Initiative for Scientific Enhancement (RISE II, R25 GM071383) program. The authors would like to thank Prof. David Goodin for the generous donation of the P450cam plasmid.