

1 Introduction
Due to the high surface to volume ratio, the incorporation of micro- and nano-particles can substantially increase the performance of initial materials. Inorganic particles such as gold, silver and silica are commonly used to obtain films having conductive [1], magnetic [2], optical [3], plasmonic [4], or hydrophobic and hydrophilic properties [5]. In particular, silica nanoparticles offer a potential building block to form ordered materials and composite structures. Thus, silica nanoparticles are employed in a wide range of applications, including biomedicine [6], optics [3] and paper industry [7]. However, the formation of films generally requires the use of another component possessing opposite charges. In this case, the two components can be self-assembled if the electrostatic forces are sufficient to overcome the other repulsive forces.
Different strategies have been used to prepare multilayer films incorporating nanoparticles. This includes deposition of natively oppositely charged nanoparticles [8,9] or of oppositely charged nanoparticles obtained by pre-deposition of polyelectrolytes on their surfaces [10–12]. Another interesting approach consists in the assembly of nanoparticles and oppositely charged polyelectrolytes [13,17–19]. This method has been used to prepare thin films with interesting optical [20] or sensing properties [21]. As an example, a multilayer film made of HgTe nanoparticles and a polymer showed strong emission in the near-infrared region allowing potential application in optoelectronics [22]. The photocatalytic response of multilayer films combining TiO2 nanoparticles and polymers led to promising results for environmental applications [14,15]. The encapsulation of silica nanoparticles by polyelectrolytes was also used for the preparation of biocompatible microcapsules [11], antibacterial substrates [24] and superhydrophobic films that can be used as self-cleaning surfaces [23]. Liu et al. [3] reported the formation of nanostructured antireflective surfaces by deposition of monodispersed 120-nm-silica nanoparticles on a glass substrate by electrostatic attraction between negatively charged colloidal particles and positively charged polyelectrolytes.
Taking this literature into account, the present study aims at developing new dense composite thin films, with tunable size and wettability, made of silica nanoparticles and a cationic polyelectrolyte used as the binding agent. Growth of films was achieved by the alternate adsorption of negatively charged colloidal silica particles and positively charged polycations using the layer-by-layer technique [25]. Silica nanoparticles were assembled in alternation using two different polycations, PAH (poly(allylamine hydrochloride)), a weak polyelectrolyte and PDDA (poly(diallyldimethylammonium chloride)), a strong polyelectrolyte. The effect of other parameters including pH of the polycation solution and pH and concentration of the silica suspension, on the build-up of the composite films, was also investigated. These studies were carried out using dissipative quartz crystal microbalance (QCM), contact angle measurements, profilometry, scanning electron microscopy (SEM) and atomic force microscopy (AFM).
2 Experimental
2.1 Particles
Silica nanoparticles Bindzil 30/220 (30 wt% SiO2 suspension in water, an average particle size of 20 nm and a specific surface area of 220 m2/g) were purchased from Eka Chemicals AB. The stock silica suspension was diluted with 0.01 M NaCl up to desired silica concentrations ([SiO2] = 1, 0.1, 0.01 g/L). The pH of SiO2 dispersion (concentrated at 1 mg/mL) was 8.5 at 20 °C. The pH of the silica dispersion was adjusted to pH 4 by the addition of aqueous HCl.
2.2 Characterization of silica particles
Electrophoretic mobility and hydrodynamic diameter measurements were conducted on silica particles dispersed and sonicated in 0.01 M aqueous NaCl solution using a Malvern Zetasizer Nano ZS to determine their zeta potential and size distribution. It is generally considered that there are many silanol groups on the surface of SiO2 nanoparticles, and the point of zero charge (PZC) is 2.1 for SiO2. This makes the surface of SiO2 nanoparticles negatively charged in aqueous solutions at a pH above 2.1 [26]. This was confirmed by our ζ potential measurements as a function of pH (Table 1). A negative ζ potential was obtained at all pH values investigated with the magnitude increasing from a value of −2.4 mV at pH 2.3 to a maximum value of −20.1 mV at pH 8.5. The size distributions of the silica suspensions in Table 1 are presented in percentage of intensity. They revealed the presence of two populations at all pH values investigated. However, the low size particles (Dh = 23 ± 5 nm) are present in majority (Intensity ≥87% at pH 2.3 and 98% at pH 8.5).
Hydrodynamic diameter (Dh) and zeta potential of silica nanoparticles dispersed in 0.01 M NaCl solutions at different pH.
| pH | Dh (nm) (% in intensity)a | Zeta potential (mV) |
| 2.3 | 22 (87.6) 376 (12.4) | −2.4 |
| 4.0 | 26 (95.7) 171 (4.3) | −7.5 |
| 6.0 | 23 (98.2) 193 (1.8) | −19.6 |
| 8.5 | 23 (98.9) 263 (1.1) | −20.1 |
a Measurements uncertainty: ± 5 nm.
2.3 Polyelectrolytes
Poly(diallyldimethylammonium chloride) solution (PDDA, 20 wt% in water, MW 100 000–200 000) and poly(allylamine hydrochloride) (PAH, MW 58 000) were purchased from Sigma Aldrich and used without further purification. Polyelectrolyte solutions were prepared at a concentration of 0.1 g/L with 0.01 M NaCl. The pH values of PDDA and PAH solutions were adjusted to 4 or 10 with dilute HCl or NaOH. PAH is a weak polyamine with a pKa value around 8.7 [27].
2.4 Substrates and self-assembly
Polyelectrolyte/particle films were assembled on silicon wafers or QCM SiO2 chips. SiO2 substrates were cleaned with piranha solution (98 wt% H2SO4/30 wt% H2O2, 2/1) for 15 min, abundantly rinsed with Milli-Q water and blown dry with pure nitrogen gas. Then, the polyelectrolytes, PAH or PDDA, and silica particles were alternately deposited under various experimental conditions. In all cases, the deposition time was set to 10 min and the substrates were rinsed with 0.01 M NaCl for 30 s between each deposition. Assemblies were carried out until reaching 11 adsorbed layers.
2.5 Film thickness
A quartz crystal microbalance with dissipative monitoring E4 equipment supplied by Q-sense AB (Sweden) was used to follow in situ the mass change during the film growth. The film build-up was examined on resonators covered by silica sensors supplied by Q-sense. The equipment detects the adsorption of particles and polymers onto the substrate by measuring simultaneously the change in frequency and the dissipation of the crystal oscillating at the fundamental frequency, 5 MHz, and its overtones. A decrease in frequency is related to a mass increase through the Sauerbrey equation [28]. Film thicknesses were also estimated from profilometry measurements using a stylus-based mechanical probe profiler (Alpha-Step IQ, KLA Tencor). Film thicknesses were measured in five different points for each film and the results were averaged over the five measurements.
2.6 Wettability
To examine the surface energy of the composite films, water contact angles were measured using a Dropscan contact angle analyzer (from Itconcept). The instrument is equipped with a CCD camera, a sample stage and a syringe holder. A 5 μL drop of ultrapure water was formed at the tip of a syringe needle and placed onto the sample surface by raising the sample until contact was made. Then, an image of the drop was captured, and contact angles were determined by drawing the tangent close to the edge of the droplet. At least five drops were deposited after each layer adsorption. The results were averaged over the five measurements.
2.7 SEM
The morphological features of the particle/polymer composite films were examined by using a high-resolution scanning electron microscope (SEM). Once deposited and dried, the samples were studied in a SEM Quanta 450 W from FEI with an electron beam energy of 10 keV.
2.8 AFM
The imaging of the surface topographies was performed with a commercial atomic force microscope (AFM PicoSPM from Molecular Imaging, USA), in contact mode with a gold coated silicon tip. The Si rectangular AFM cantilever was 450 μm – long and its stiffness was about 0.27 N m−1. The experiments were carried out under atmospheric conditions at room temperature.
3 Results and discussion
We first started our experiments using a silica dispersion concentration of 1 g/L in 0.01 M NaCl. The pH of the resulting solution was equal to 8.5. At this pH, silica particles were negatively charged as shown by electrophoretic mobility measurements (see the experimental procedure and Table 1). Indeed, zeta potential of silica particles was measured as a function of pH and was found to be negative for pH >2.3, reaching approximately −20 mV at pH 8.5. Considering the negative charges on the surface of silica particles, their electrostatic assembly is only possible with cationic polyelectrolytes. We chose to assemble silica particles using either PAH, a weak cationic polyelectrolyte, or PDDA, a strong cationic polyelectrolyte. Indeed, it is well known that polyelectrolyte properties of multilayer films depend on many parameters including the nature of the polyelectrolyte, its ionic strength, the adsorption time or rinsing procedure used during film growth.
3.1 Influence of the polyelectrolyte nature and pH of the polycation solutions
It is well known that pH has a strong influence on multilayer assembly when the building blocks contain a weak polyelectrolyte. Therefore, we studied the impact of the pH of PAH solutions on composite assemblies. The pH values of the SiO2 nanoparticle suspension and PAH polycation solution used to build-up this assembly were 8.5 and 4.0, respectively. PAH was first adsorbed onto the QCM SiO2 chip followed by adsorption of silica nanoparticles. These steps were repeated until the formation of a (PAH/SiO2)5-PAH film composed of 11 successive adsorbed layers. Each adsorption step was performed during 10 min Fig. 1a shows the evolution as a function of time of the film thickness deposited on a SiO2 QCM chip. Fig. 1b represents the evolution of thicknesses reached at the end of each adsorption step as a function of the number of adsorbed layers. Fig. 1a and b clearly illustrate that SiO2 nanoparticles can be deposited alternately with a cationic polyelectrolyte. Larger and smaller steps correspond respectively to adsorption of SiO2 particles and PAH. Each PAH adsorption step increases the film thickness by around 5 nm whereas each SiO2 adsorption step increases the film thickness by around 35 nm. It can be noticed that such an adsorption process is governed by the attractive long-range van der Waals forces, repulsive electrostatic interactions between the like-charge SiO2 particles and attractive electrostatic interactions between negatively charged SiO2 particles and cationic polyelectrolytes.
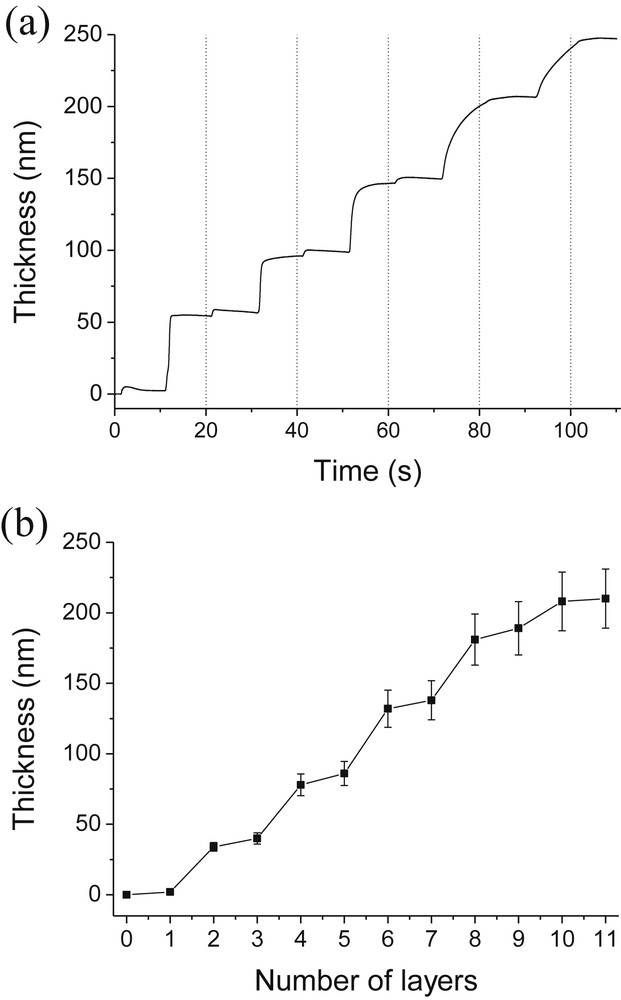
Evolution of thicknesses with time (a) and with the number of layers (b) during the alternate adsorption of PAH and SiO2. [PAH] = 0.1 g/L [SiO2] = 1 g/L; pH(PAH) = 4.0; pH(SiO2) = 8.5.
We thus achieved our main goal which was the formation of films composed mainly of silica using a polycation as the binding agent.
The high ratio adsorbed silica/adsorbed polycation can be related to the presence of long-distance charge pairs between SiO2 and PAH instead of the exclusive formation of contact ion pairs expected for polycation/polyanion multilayer films [16]. In fact, a PAH adsorption step leads to few nanometers thick PAH layers which form contact ion pairs only at the lower surface of the incoming SiO2 particles. A schematic representation of PAH/SiO2 and PDDA/SiO2 assemblies built up in our work is illustrated in Fig. 2.
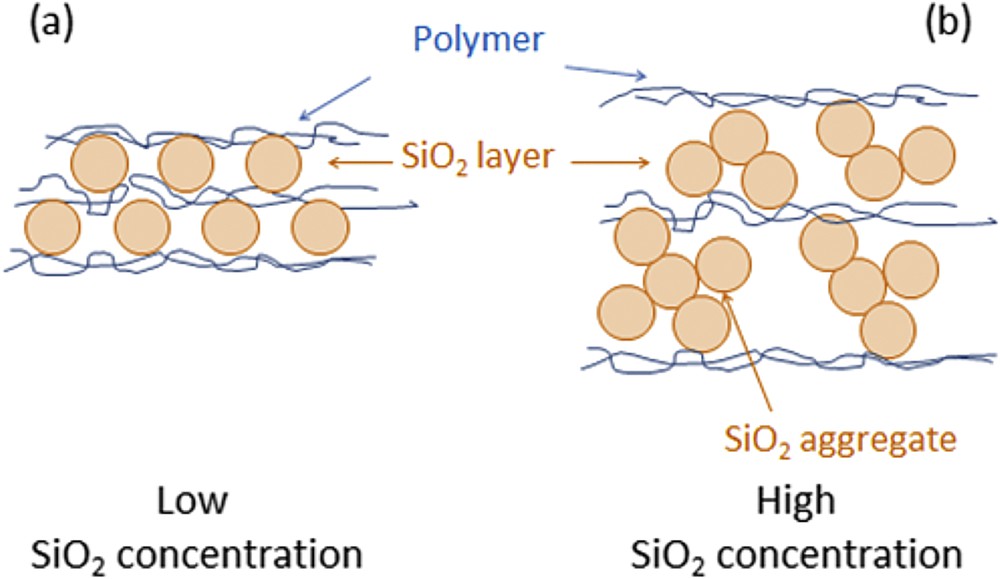
Schematic representation of Polycation/SiO2 assemblies.
We then carried out a similar experiment by increasing the pH value of the PAH solution to 10. At this pH, PAH possesses few positive charges in its backbone. Consequently, it can be supposed that interactions between negatively charged SiO2 nanoparticles and positive or neutral PAH could impact on the PAH/SiO2 growth. However, our results showed that PAH/SiO2 films obtained at pH 4 and pH 10 have similar thicknesses: 210 nm at pH 10 and 206 nm at pH 4 (Table 2). As such, it can be concluded that the pH of the PAH solution has no influence on the film growth. This is probably due to the fact that the film thickness mainly comes from SiO2 nanoparticle adsorption.
Comparison of thicknesses obtained by QCM-D and profilometry under different experimental conditions.
| Experience | Polymer (C = 0.1 g/L) | Particle | Thickness QCM-D measurements | Thickness profilometry measurements | |||
| Nature | pH | Nature | pH | C (g/L) | |||
| 1 | PAH | 4 | SiO2 | 8.5 | 1 | 206 ± 20 nm | 80 ± 15 nm |
| 2 | PAH | 10 | SiO2 | 8.5 | 1 | 210 ± 20 nm | 75 ± 15 nm |
| 3 | PDDA | 4 | SiO2 | 8.5 | 1 | 182 ± 18 nm | 75 ± 12 nm |
| 4 | PDDA | 10 | SiO2 | 8.5 | 1 | 150 ± 15 nm | 65 ± 10 nm |
| 5 | PAH | 4 | SiO2 | 4 | 1 | 430 ± 40 nm | 185 ± 20 nm |
| 6 | PDDA | 4 | SiO2 | 4 | 1 | 393 ± 40 nm | 160 ± 20 nm |
| 7 | PAH | 4 | SiO2 | 4 | 0.1 | 290 ± 30 nm | 125 ± 15 nm |
| 8 | PAH | 4 | SiO2 | 4 | 0.01 | 45 ± 5 nm | <20 nm |
| 9 | PDDA | 4 | SiO2 | 4 | 0.1 | 185 ± 20 nm | 90 ± 15 nm |
| 10 | PDDA | 4 | SiO2 | 4 | 0.01 | 12 ± 5 nm | < 20 nm |
Similar experiments were performed using PDDA as the cationic polyelectrolyte. As for PAH, thicknesses were mainly due to SiO2 nanoparticle adsorption (curves are not shown as they are very similar to Fig. 1b). However, they were lower than those obtained with PAH/SiO2 assemblies (182 nm at pH 4 and 150 nm at pH 10). These results suggest that the chemical structure of the polyelectrolyte may influence the multilayer film thickness. The small difference in film thicknesses observed for PDDA/SiO2 assemblies at pH 4 and 10 was surprising since PDDA is a strong polyelectrolyte whose charge density is not pH-dependent.
Profilometry was used to confirm trends observed by QCM. Before comparing the results obtained by these two methods, it must be emphasized that QCM thicknesses were obtained during film growth in aqueous solutions whereas profilometry measurements were performed after rinsing and drying of polymer films. Thus, profilometry measurements led to lower thicknesses. Nevertheless, profilometry measurements were in accordance with QCM results. Thicknesses of PAH/SiO2 films ranged from 80 nm at pH 4–75 nm at pH 10 (Experiences 1 and 2 in Table 2) while those of PDDA/SiO2 films were estimated to be 75 nm (pH 4) and 65 nm (pH 10) (Experiences 3 and 4 in Table 2). These results confirmed that the pH of the polyelectrolyte weakly impacts film thicknesses for both strong and weak polyelectrolytes. However, slightly thicker films were obtained when PAH was used as the polyelectrolyte.
The water wettability of each layer was monitored during the growth of PAH/SiO2 and PDDA/SiO2 films (Fig. 3). The naked silica was marked as layer 0 and has a contact angle of ∼45°. Contact angle values depend on the nature of the outermost layer. After adsorption of the first polyelectrolyte (PDDA or PAH) layer, the contact angle was increased. This was due to the adsorption of the polyelectrolyte polar head groups onto the negatively charged SiO2 surface with their nonpolar hydrocarbon chains oriented upward toward the bulk portion of the droplet. After the first adsorption of hydrophilic SiO2 nanoparticles, the surface angle decreased. Upon addition of alternating layers, periodic oscillations in contact angle values were observed in all cases. Moreover, as wettability of PDDA/SiO2 films was concerned, only small contact angle layer-to-layer oscillations were observed within a range of about 10° (except during the deposition of the first bilayer) (Fig. 3b). The contact angle values obtained at pH 4 and pH 10 were comparable indicating that the wettability of the film was only slightly pH-dependent.
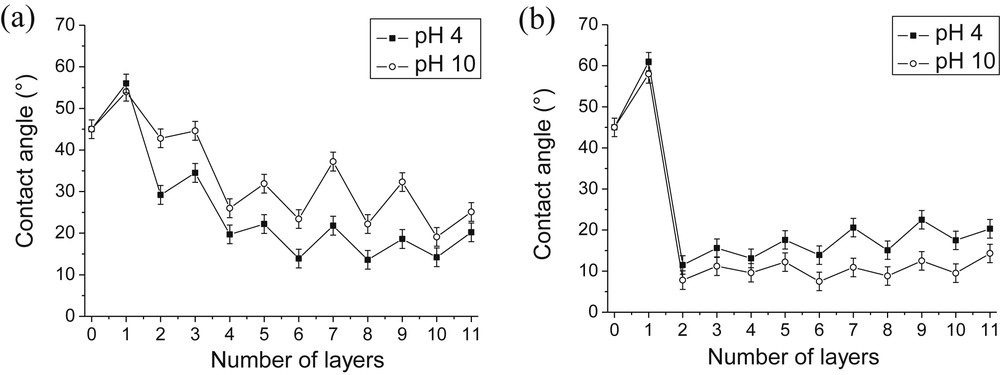
Influence of the pH of the polyelectrolyte solution on the evolution of the wettability of PAH/SiO2 (a) and PDDA/SiO2 (b) films. [PAH] = [PDDA] = 0.1 g/L; pH(SiO2) = 8.5 [SiO2] = 1 g/L.
If a high surface energy (a low contact angle value) was immediately reached with PDDA/SiO2 films, it was not the case for PAH/SiO2 films whose surface energy progressively increased (Fig. 3a). The contact angle values obtained at pH 4 and pH 10 followed the same trend but they differed more significantly for PAH/SiO2 films (around 20°) than for PDDA/SiO2 ones (around 10°). As PAH is a weak cationic polyelectrolyte, this could be due to a decrease of the charge density under basic conditions which modifies the adsorption of PAH onto negatively charged silica particles.
In conclusion, the pH of the polyelectrolyte solution had no significant effect on the thicknesses and wettability properties of PAH/SiO2 and PDDA/SiO2 films. At first sight, the low influence of the pH appeared surprising. This was certainly due to the fact that the films were mainly composed of SiO2 nanoparticles. Hence, silica particles certainly impacted more strongly on the film properties than polyelectrolytes.
3.2 Influence of the pH of the silica solution
As discussed above, the pH of the polyelectrolyte solution had no significant influence on the film properties. We therefore focused our next experiments on the effect of the pH of silica nanoparticle suspensions. Thus, it was chosen to work with: i) PAH and PDDA solutions at pH 4 as thicker films were obtained at this pH; ii) a silica solution at pH 4 in order to compare the results with those obtained at pH 8.5. Fig. 4 shows the evolution of the thicknesses obtained by QCM for PAH/SiO2 and PDDA/SiO2 films at pH 4 and pH 8.5.
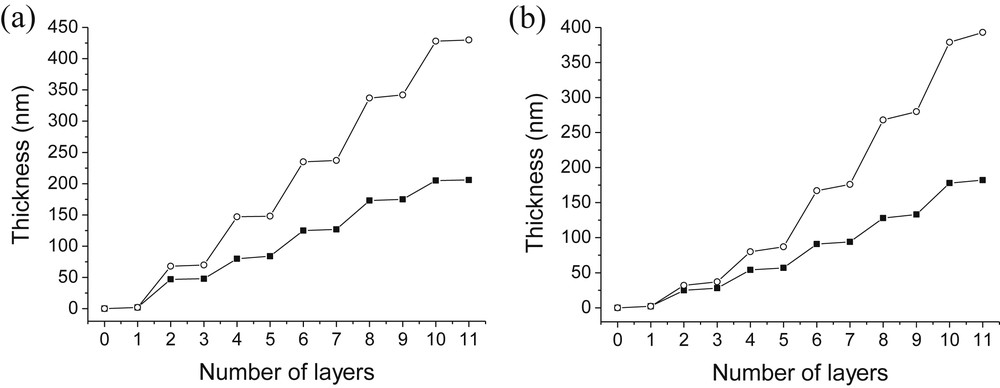
Influence of the pH of the SiO2 solution on the thickness of PAH/SiO2 (a) and PDDA/SiO2 (b) films. (○): pH(SiO2) = 4.0; (■): pH(SiO2) = 8.5 [SiO2] = 1 g/L [PAH] = [PDDA] = 0.1 g/L; pH(PAH) = pH(PDDA) = 4.0.
Once again, this figure exhibits larger steps corresponding to the adsorption of SiO2 particles and smaller ones corresponding to the adsorption of PAH or PDDA. In addition, Fig. 4 shows that thicker films (approximately twice the size) are obtained for both polycations when the pH of particles is lowered to pH 4. According to QCM measurements, PAH/SiO2 films obtained at pH 4 and 8.5 were 430-nm- and 206-nm-thick and 185 nm and 160 nm-thick, respectively, from profilometry measurements. A comparison of the amount of polylectrolytes and silica adsorbed at pH 4 and 8.5 clearly illustrated that the formation of thicker films at pH 4 was due to higher adsorption of silica nanoparticles. The amount of PAH adsorbed increased the film thickness by around 5 nm at both pH whereas the amount of silica adsorbed at pH 4 was twice the amount absorbed at pH 8.5. Clearly, the loss of charge stabilization at lower pH led to the formation of more silica particle aggregates which contributed to the increase of the film thickness. Another important parameter is the effect of time on the film thicknesses. We should stipulate that the same silica dispersion was used throughout the experiments. As illustrated in Fig. 4, the upper silica layers are thicker (90 nm) than the first silica layers (50 nm) showing slow silica aggregation with time. Very similar trends were observed for PDDA/SiO2 films. Thicknesses obtained by QCM measurements were 393 nm at pH 4 and 182 nm at pH 8.5. Our results are in agreement with those reported by Cohen and collaborators. They demonstrated that an optimized pH solution of nanoparticles close to 3 led to maximum film growth [29].
The morphological features of the composite films (PAH/SiO2 and PDDA/SiO2) obtained at pH 4 for both components were investigated. Fig. 5 shows SEM images of the resulting PAH/SiO2 and PDDA/SiO2 coatings. Clearly, isolated domains of aggregated silica nanoparticles are observed but their distribution on the substrate surface is regular and homogeneous.

SEM images of PAH/SiO2 (a) and PDDA/SiO2 (b) films. pH(SiO2) = 4.0; [SiO2] = 1 g/L; pH(PAH) = pH(PDDA) = 4.0; [PAH] = [PDDA] = 0.1 g/L.
The wettability of PAH/SiO2 and PDDA/SiO2 films was also studied at pH 4 for comparison with the measurements performed at pH 8.5 (Fig. 6). The silica layer systematically increased the free surface energy regardless of the experimental conditions used. The results obtained at pH 4 and 8.5 in the case of PDDA/SiO2 films were comparable. A high surface energy was reached immediately after the adsorption of the first bilayer (Fig. 6b). However, in the case of PAH/SiO2 films a high surface energy was reached more rapidly at pH 4 than at pH 8.5 (Fig. 6a). This difference might be explained by a higher quantity of silica nanoparticles adsorbed onto the first layer of the polycation at pH 4 than at pH 8.5 resulting in the formation of a more hydrophilic film.
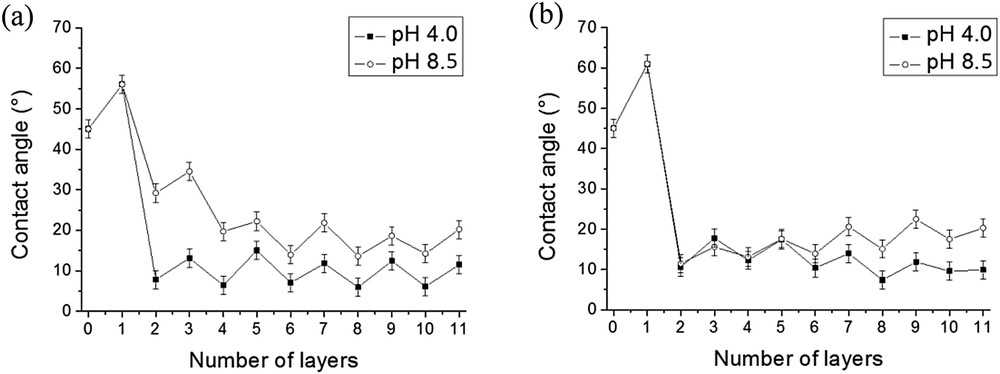
Influence of the pH of the SiO2 solution on the wettability of PAH/SiO2 (a) and PDDA/SiO2 (b) films. [PAH] = [PDDA] = 0.1 g/L; pH(PAH) = pH(PDDA) = 4.
In conclusion, a change in the pH of silica nanoparticle suspensions affects the growth of PAH/SiO2 and PDDA/SiO2 films. In particular, film thicknesses strongly increased when the pH of the silica suspensions was decreased from pH 8.5 to pH 4.0. This was due to the reduction of the repulsive interaction between negatively charged silica nanoparticles and the pre-adsorbed layers of polymers. Thus, it was possible to easily tune the thickness of the composite films by modifying the pH of the silica suspension.
3.3 Influence of the silica solution concentration
Another important deposition parameter is the concentration of silica suspensions. Therefore, we prepared PAH 4.0/SiO2 4.0 and PDDA 4.0/SiO2 4.0 assemblies starting from different concentrations of silica suspensions ([SiO2] = 1, 0.1, 0.01 g/L). Fig. 7 shows the evolution of the PAH 4.0/SiO2 4.0 and PDDA 4.0/SiO2 4.0 film thicknesses during their growths for the different silica concentrations tested.
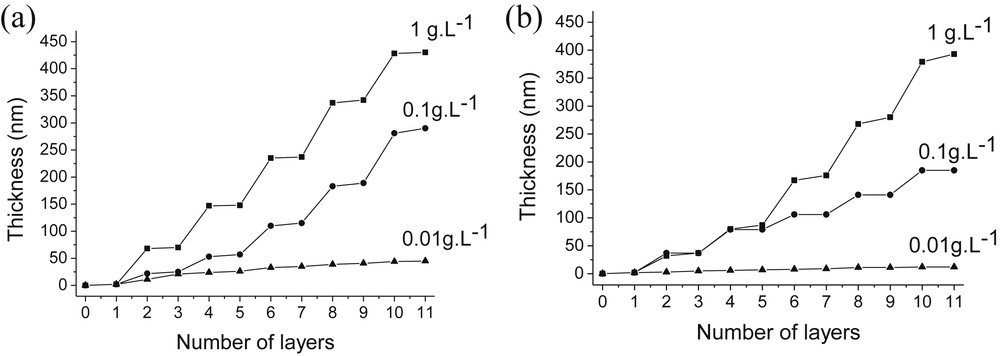
Influence of the SiO2 particle concentration on the thickness of PAH/SiO2 (a) and PDDA/SiO2 (b) films. [PAH] = [PDDA] = 0.1 g/L; pH(PAH) = pH(PDDA) = pH(SiO2) = 4.
A constant increase of the film thickness was observed for all SiO2 concentrations. Once again, larger and smaller steps correspond respectively to adsorption of SiO2 nanoparticles and PAH or PDDA. The magnitude of the growth steps during silica adsorption as well as the overall film thicknesses increased with SiO2 concentrations. The thicknesses of PAH/SiO2 multilayer films increased from 45 nm for the lowest silica concentration ([SiO2] = 0.01 g/L), to 290 nm ([SiO2] = 0.1 g/L) and finally to 430 nm at the highest silica concentration ([SiO2] = 1 g/L) (Fig. 7a). Similarly, PDDA/SiO2 films obtained at the same silica concentrations were respectively 12, 185 and 393 nm-thick (Fig. 7b). Consequently, the growth of PAH/SiO2 and PDDA/SiO2 assemblies was strongly dependent on the SiO2 concentration. Two hypotheses can explain the strong influence of the silica concentration on the film growth. First of all, there might be an increase of the ionic strength when the SiO2 concentration is increased resulting in an enhanced adsorption of the silica particles. Secondly, larger aggregates of SiO2 particles can be formed at higher SiO2 concentrations (see Fig. 2b), leading to the adsorption of heavy SiO2 aggregates on the polycation.
AFM images (Fig. 8) and AFM roughness measurements have been performed to study the influence of silica concentration on the morphological features of the PAH/SiO2 films. At a silica concentration of 0.1 g/L, the surface of the film appears flat and homogeneous even though peaks, probably due to the presence of aggregates, can be observed (Fig. 8b). In addition, a low roughness was obtained (Rrms = 12.3 ± 1.2 nm). At higher silica concentration (1 g/L), a higher roughness (Rrms = 27.8 ± 9.3 nm) was obtained due to the formation of a thicker film. This film consists of mountains and valleys regularly distributed on the substrate (Fig. 8a). At the lowest silica concentration (0.01 g/L), the roughness of the film was high (Rrms = 34.3 ± 6.7 nm) but this might be due to the incomplete coverage of the surface by silica particles. In this latter case, the roughness was measured by the height difference between the uncoated substrate and the silica particles or aggregates (Fig. 8c).

AFM images of PAH/SiO2 films. pH(SiO2) = 4.0; pH(PAH) = 4.0; [PAH] = 0.1 g/L. [SiO2] = 1 g/L (a), 0.1 g/L (b) or 0.01 g/L (c).
The influence of the silica concentration on the film wettability was also studied (Fig. 9). Fig. 9a shows oscillations of the contact angle values for all PAH/SiO2 films. As expected, when the silica particle concentration was increased from 0.01 to 0.1 g/L and then from 0.1 to 1 g/L the surface energy of the resulting PAH/SiO2 films increased due to the insertion of a larger amount of hydrophilic silica particles into the composite films. Moreover, at a silica concentration of 0.01 g/L, the contact angle values remained around 45–60° even after adsorption of several SiO2 and PAH layers. This demonstrates that it was possible to tune the hydrophilic/hydrophobic state of the multilayer films by varying the silica particle concentration. Concerning PDDA/SiO2 films, an increase of the particle concentration from 0.01 to 0.1 g/L led to lower surface energy. However, further increase of the silica concentration to 1 g/L had no significant effect on the surface energy (Fig. 9b).
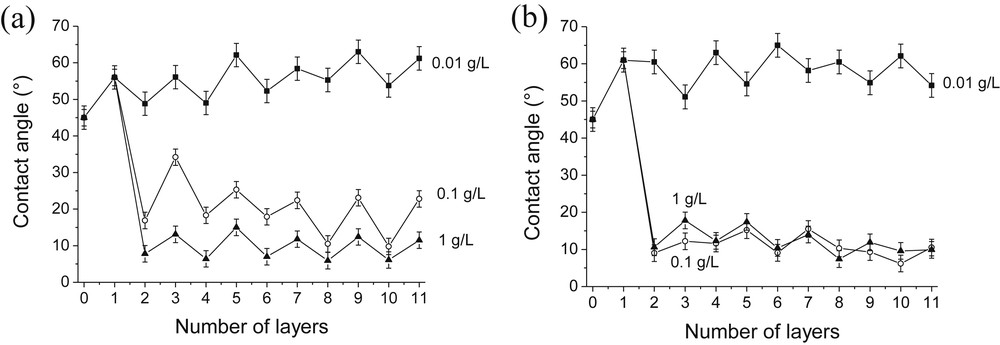
Influence of the SiO2 particle concentration on the wettability of PAH/SiO2 (a) and PDDA/SiO2 (b) films. [SiO2] = 1 g/L (▴), 0.1 g/L (○) or 0.01 g/L (■). [PAH] = [PDDA] = 0.1 g/L; pH(PAH) = pH(PDDA) = pH(SiO2) = 4.
4 Conclusion
Fundamental aspects of the elaboration of thin silica films incorporating polycations, used as binding agents, and negatively charged silica nanoparticles were investigated. Whatever be the conditions used, the contribution of the silica nanoparticle adsorption to the overall thickness was far higher than that of polycations. This has been attributed to the formation of long-distance charge pairs between silica nanoparticles and polycations. QCM, SEM and AFM demonstrated the formation of regular films showing dense and non-porous structures due to the polymer layer between the silica layers which acts as a barrier and prevents porosity of the samples.
The influence of the pH of the polycation solution was shown to be negligible as we obtained similar film properties at different pH. In contrast, a reduction of the pH of silica particle suspensions led to an increase of the composite film thicknesses while the wettability properties remained the same. As expected, film growth was strongly dependent on the SiO2 nanoparticle concentrations. Indeed, both thickness and surface energy of the films increased with silica concentration.
To conclude, in this work we have shown that it is possible to easily tune the thickness and wettability of SiO2/polyelectrolyte composite films. This can be achieved by varying either the pH or the concentration of silica particles. Research on such multicomponent composite assemblies is an interesting topic to pursue as the combination of nanoparticles and polymers is expected to enhance other properties (mechanical, optical, and electrical) of the composite films. This will open the way to their development in the fields of optics, materials, chemistry or biology.
Acknowledgments
This work was supported by a grant of the French National Agency (ANR), project number: ANR-13-JS08-0009-01.