

1 Introduction
The selective oxidation of alcohols to carbonyl compounds is an important reaction in organic synthesis with many applications in the fine chemical industry [1]. A number of contributions, including recent ones, have shown that manganese dioxide efficiently carries out this transformation as a stoichiometric reagent, especially for benzylic, allylic and propargylic alcohols [2]. However, a large excess (2–25 equiv) is generally necessary to obtain high conversions. In addition, the use of active forms of MnO2 is often mandatory to achieve high yields of the ketone or aldehyde product [3]. Only recently has the oxidation of alcohols to carbonyl compounds been carried out with a catalytic amount of MnO2 using dioxygen or benzoylperoxide as an oxidant [4]. Here again, however, activated forms of manganese dioxide (nanoparticles, colloidal solutions or dispersed on the surface of inorganic substrates) must be used [4]. In this communication, we present a new catalytic system for the selective oxidation of alcohols into the corresponding carbonyl compounds based on commercially available plain MnO2. The results presented here are also relevant with respect to the common use of MnO2 to quench oxidants such as H2O2 or tert-butylhydroperoxide (TBHP) in oxidation processes carried out with other catalysts.
2 Results and discussion
2.1 Catalytic studies
The present study was motivated by the accidental discovery that commercially available technical activated MnO2 (Aldrich: activated, >90%, reference 63548-250G-F) affects the determination of the conversions during kinetic monitoring of alcohol oxidation by TBHP. A frequently used procedure to quench the excess oxidant, typically H2O2 [5] but also TBHP [6], involves the treatment of the withdrawn reaction mixture aliquots with excess MnO2 before gas-chromatographic or NMR quantification of conversion. In an experiment where 1-phenylethanol (1a) was oxidized with 2 equivalents of TBHP in the presence of a molybdenum catalyst, we obtained erratic and non-reproducible conversions as a function of time. These results interrogated us on the role of MnO2 as a stoichiometric oxidant or as an oxidation catalyst during the TBHP quenching phase. We therefore proceeded to study the same oxidation process in the presence of various amounts of the above-mentioned, commercially available MnO2 (Table 1). When using one equivalent of MnO2 (entry 1), a high substrate conversion was observed after 7 h at room temperature. The amount of MnO2 could be limited to 20 mol%, proving its catalytic role in the reaction, but longer reaction times were needed to obtain high conversions (entries 2–3). The amount could be further decreased to 5 mol% but the reaction was rather slow even at 40 °C (entries 4–5). At 80 °C, good conversions and yields could be obtained limiting the amount of MnO2 to 10 mol% and the reaction time to 7 h (entry 6). We also checked another MnO2 source, more precisely a purer product although non-activated (ReagentPlus® Sigma-Aldrich, ≥ 99%) (see Table 1, entry 7). This ReagentPlus® MnO2 exhibited a very low catalytic activity. We checked also that both the catalyst and oxidant are essential to the reactivity. Negligible conversion was observed when using TBHP/decane in the absence of MnO2 (entry 8). However, MnO2 alone was able to slowly consume 1-phenylethanol as a stoichiometric oxidant and transform it into acetophenone with good selectivity (entries 9–10). The use of TBHP as the commercially available aqueous solution also gave good results (entry 11). This shows that the presence of water in the system does not negatively affect the reaction. Finally, we attempted to use hydrogen peroxide, a greener oxidant generating only water as a by-product [7], in place of TBHP but neither substrate conversion nor acetophenone formation were observed (entry 12). In this experiment, we presume that the catalase activity (disproportionation of H2O2 to H2O and O2 catalyzed by manganese compounds [8], including MnO2 [9]) is faster than O atom transfer to the alcohol substrate. Finally, we carried out a catalyst recycling experiment. At the end of the reaction, MnO2 was collected at the bottom of the reaction vessel by centrifugation, carefully washed with the reaction solvent (acetonitrile/toluene mixture) after the removal of the organic solution, and reused by adding a fresh acetonitrile/toluene solution containing 1-phenylethanol and TBHP (see entry 13). Indeed, MnO2 was still active in the recycle run but with a significant decrease of the catalytic activity.
Oxidation of 1-phenylethanol (1a).а
| Entry | Substr./MnO2/TBHP | T/°C | time/h | Conversion/%b | Yield/%b |
| 1 | 1/1/2 | RT | 7 | 96 | 84 |
| 2 | 1/0.2/2 | RT | 7 | 75 | |
| 3 | 1/0.2/2 | RT | 16 | >99 | c |
| 4 | 1/0.05/2 | RT | 16 | 35 | |
| 5 | 1/0.05/2 | 40 | 7 | 39 | 33 |
| 6 | 1/0.1/2 | 80 | 7 | 76 | 67 |
| 7d | 1/0.2/2 | RT | 7 | 5 | |
| 8 | 1/0/2 | 40 | 24 | 6 | 0 |
| 9 | 1/1/0 | 40 | 7 | 20 | 14 |
| 10 | 1/1/0 | 40 | 24 | 33 | 30 |
| 11 | 1/0.05/2e | 40 | 7 | 38 | 34 |
| 12 | 1/0.05/2f | 40 | 5 | 1 | 0 |
| 13g | 1/1/2 | RT | 7 | 65 | 57 |
a 1.2 mmol alcohol in 3 mL of acetonitrile and 1 mL of toluene, 453 μL of 5.3 M TBHP solution in decane (2.4 mmol), MnO2.
b Determined by GC.
c 70% isolated yield.
d ReagentPlus® MnO2 was used instead of technical activated MnO2 (see the experimental part).
e TBHP (70%) in water instead of TBHP in decane.
f H2O2 (30%) in water instead of TBHP in decane.
g Catalyst recycle after run 1 (see text).
The substrate scope of this catalytic system was subsequently explored (the investigated substrate transformations are summarized in Scheme 1 and the results are collected in Tables 2 and 3). In all the studied cases, the presence of MnO2 substantially increases the reaction rate (cf. the experiments run at 40 °C with the corresponding blank reactions run in the absence of MnO2, i.e. entries 18, 24, 29, 35, 44 and 47). We first investigated primary benzylic alcohols (2a) and (3a) (Table 2). These alcohols proved a little more difficult to oxidize than the secondary benzylic alcohol (1a) (entries 16–17 and 22–23 versus entry 5 in Table 1) but their transformation into the corresponding aldehydes was efficient using 1 equivalent of the catalyst at RT (entries 14–15 and 19–21). On the other hand, the aldehyde product yield decreased at long reaction times due to further oxidation under these conditions. 4-methoxybenzaldehyde (3b) was consumed much faster than 4-methylbenzaldehyde (2b). Similar results were obtained for the oxidation of the allylic alcohol (4a) (entries 25–26), the rate of consumption of the cinnamaldehyde product (4b) being similar to that of (2b). The withdrawn aliquots for entries 25 and 26, after removing the solid MnO2 by filtration but remaining in the presence of residual TBHP, showed a decrease of the (4b) product upon reinjection into the GC after 24 days. This decrease, however, is much slower than in the reaction in the presence of solid MnO2 and is probably attributed to traces of soluble manganese compounds. Non-activated substrates like cyclohexanol, (5a), can also be oxidized (entries 30–34), although more slowly than the activated ones. Better results could be obtained at a slightly higher temperature (60 °C, entry 34), while using one equivalent of MnO2 at room temperature results in low conversions (entries 30 and 31).
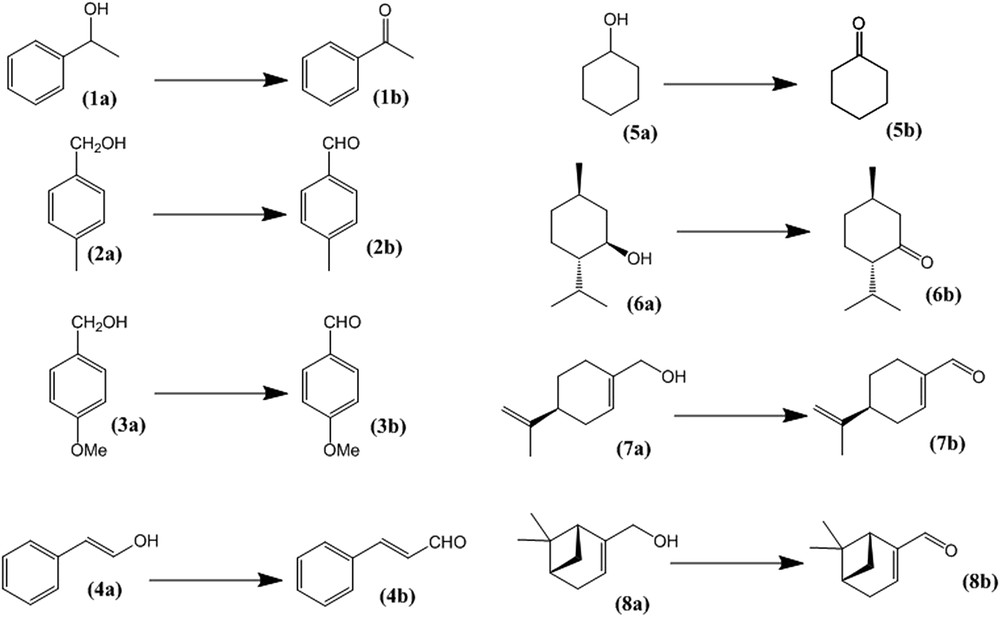
Transformations presented in this study.
Oxidation of other alcohols.а
| Entry | Substr. | Substr./MnO2/TBHP | T/°C | time/h | Conversion/% | Yield/%b |
| 14 | (2a) | 1/1/2 | RT | 7 | 94 | 51 |
| 15 | (2a) | 1/1/2 | RT | 24 | 100 | 39 |
| 16 | (2a) | 1/0.05/2 | 40 | 7 | 26 | |
| 17 | (2a) | 1/0.05/2 | 40 | 16 | 39 | |
| 18 | (2a) | 1/0/2 | 40 | 24 | 54 | 33 |
| 19 | (3a) | 1/1/2 | RT | 2.5 | 86 | 50 |
| 20 | (3a) | 1/1/2 | RT | 7 | 99 | 20 |
| 21 | (3a) | 1/1/2 | RT | 24 | 100 | 0 |
| 22 | (3a) | 1/0.05/2 | 40 | 7 | 35 | |
| 23 | (3a) | 1/0.05/2 | 40 | 16 | 47 | |
| 24 | (3a) | 1/0/2 | 40 | 24 | 11 | 7 |
| 25 | (4a) | 1/1/2 | RT | 7 | 100 | 60c |
| 26 | (4a) | 1/1/2 | RT | 24 | 100 | 36c |
| 27 | (4a) | 1/0.05/2 | 40 | 5 | 12 | |
| 28 | (4a) | 1/0.05/2 | 40 | 16 | 22 | |
| 29 | (4a) | 1/0/2 | 40 | 24 | 29 | 5 |
| 30 | (5a) | 1/1/2 | RT | 7 | 28 | 28 |
| 31 | (5a) | 1/1/2 | RT | 24 | 36 | 35 |
| 32 | (5a) | 1/0.05/2 | 40 | 7 | 23 | |
| 33 | (5a) | 1/0.05/2 | 40 | 16 | 40 | |
| 34 | (5a) | 1/0.1/2 | 60 | 7 | 48 | |
| 35 | (5a) | 1/0/2 | 40 | 24 | 5 | 0 |
a 1.2 mmol alcohol in 3 mL of acetonitrile and 1 mL of toluene, 453 μL of 5.3 M TBHP solution in decane (2.4 mmol), MnO2.
b Determined by GC.
c A new injection of the withdrawn aliquots after 24 days gave a yield of 36 for 7 h and 26 for 24 h of reaction.
Oxidation of alcohols from biomass.а
| Entry | Substr. | Substr./MnO2/TBHP | T/°C | time/t | Conv./% | Yield/%b |
| 36 | (6a) | 1/0.05/2 | 40 | 5 | 5 | |
| 37 | (6a) | 1/0.05/2 | 40 | 16 | 10 | |
| 38 | (6a) | 1/0.1/2 | 40 | 7 | 7 | |
| 39 | (6a) | 1/0.1/2 | 80 | 7 | 11 | |
| 40 | (6a) | 1/0.1/2 | 80 | 16 | 12 | |
| 41 | (6a) | 1/1/2 | 40 | 5 | 5 | |
| 42 | (7a) | 1/1/2 | RT | 7 | 64 | 22 |
| 43 | (7a) | 1/1/2 | RT | 24 | 81 | 20 |
| 44 | (7a) | 1/0/2 | 40 | 24 | 5 | 4 |
| 45 | (8a) | 1/1/2 | RT | 7 | 74 | 28 |
| 46 | (8a) | 1/1/2 | RT | 24 | 89 | 25 |
| 47 | (8a) | 1/0/2 | 40 | 24 | 9 | 9 |
a 1.2 mmol alcohol in 3 mL of acetonitrile and 1 mL of toluene, 453 μL of 5.3 M TBHP solution in decane (2.4 mmol), MnO2.
b Determined by GC.
The catalytic system was then tested for the oxidation of alcohols from biomass (Table 3). For the oxidation of menthol (6a), whatever conditions were tried, no satisfactory yields of menthone could be obtained (entries 36–41). In particular, increasing the amount of catalyst from 0.05 to 1 equivalent or raising the temperature from 40 °C to 80 °C did not significantly improve the substrate conversion. The more activated perillyl alcohol (7a) and myrtenol (8a), on the other hand, were oxidized with relatively good conversions using 1 equivalent of MnO2 at room temperature (entries 42–43 and 45–46) but afforded the corresponding aldehydes (7b) and (8b) with moderate selectivities and with slow erosion of the yield at longer reaction times, like for the other aldehyde products, (2b), (3b) and (4b), discussed above.
Since one possible cause for the poor performance of this system for less reactive alcohols is the competing MnO2-catalyzed TBHP disproportionation (see next section), we have also investigated the oxidation of menthol with slow addition of the substrate via a syringe pump (complete addition of 2.4 equiv of TBHP in 15 h), under conditions otherwise identical to those of run 36–37 (40 °C, substr./MnO2 = 1/0.05). The results of this experiment, see Fig. 1, demonstrate that this stratagem does not significantly improve the system performance, since a conversion of only 13% was measured after 48 h.
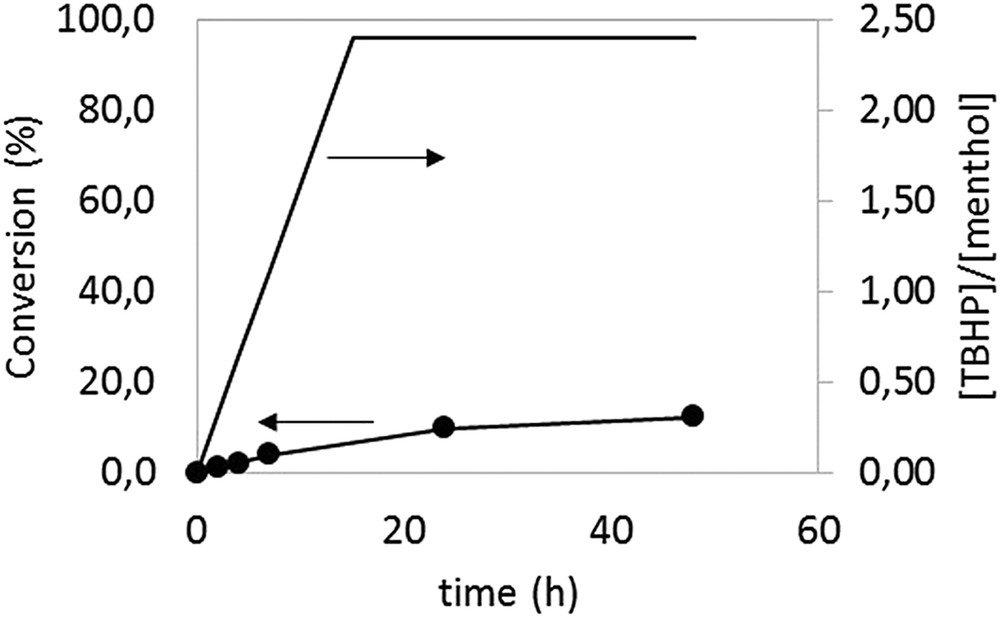
Conversion vs. time of menthol (6a) for the slow TBHP addition experiment. T = 40 °C; [6a] = 0.3 M; [MnO2]/(6a) = 0.05:1.
2.2 Mechanistic investigations
From the catalytic results presented above, it is clear that MnO2 is able to catalyze the oxidation of alcohols to carbonyl products at rates that are competitive with those of the TBHP disproportionation. It was therefore important to evaluate the rate of TBHP disproportionation under the conditions used for the alcohol oxidation. From the results shown in Fig. 2, it is clear that the TBHP disproportionation process is catalyzed by MnO2 (faster decay in the presence of a greater MnO2 amount) but is indeed relatively slow, in agreement with the observed parallel catalytic action for alcohol oxidation. However, the catalase activity is relatively important. Thus, this system becomes interesting for the practical application to the catalytic oxidation of organic substrates only if the substrates can be oxidized extensively in a relatively short time, efficiently competing with the TBHP disproportionation process. This justifies the better results obtained with the benzylic and allylic alcohols.
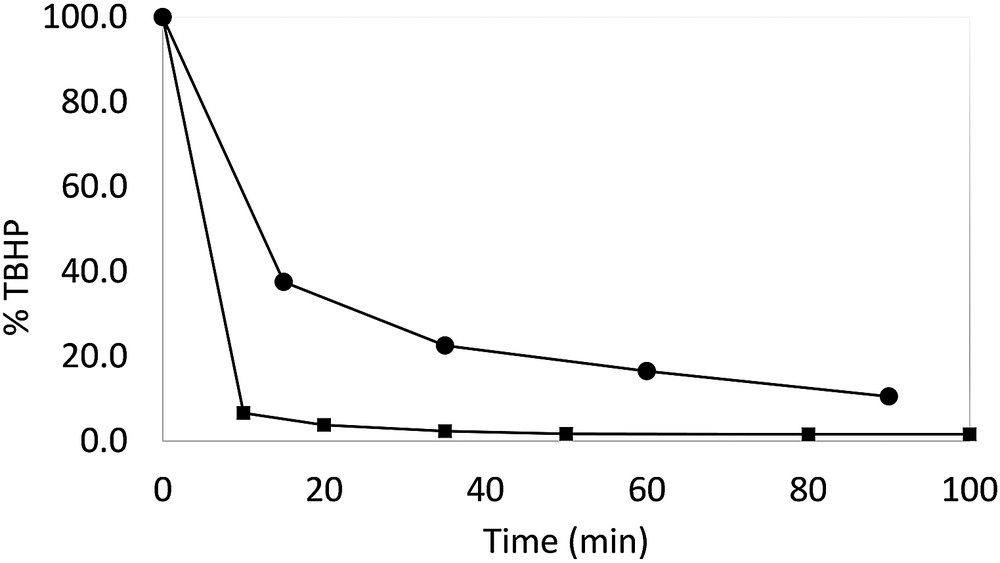
Time evolution of the TBHP concentration in the presence of MnO2 at room temperature. [TBHP] = 0.65 M in acetonitrile. MnO2/TBHP = 0.5:1 (circles); 5:1 (squares).
3 Conclusions
We have shown that manganese dioxide is an efficient catalyst for the oxidation of various alcohols into the corresponding carbonyl compounds by TBHP in acetonitrile, especially the activated benzylic and allylic alcohols but also some non-activated alcohols like cyclohexanol. MnO2 also catalyzes the decomposition of hydroperoxides by disproportionation [10], which proceeds at competitive rates in parallel to the catalyzed alcohol oxidation. However, the procedure described herein is efficient for the conversion of sufficiently reactive substrates, even certain non-activated and non-hindered alcohol such as cyclohexanol, even though an overstoichiometric amount of oxidant and a large amount of catalyst are required to achieve good conversions. The relatively low cost of MnO2 and its efficiency without requiring any special additional activation procedure made this procedure of potential interest for many applications.
Another point of interest is that the MnO2-catalyzed TBHP decomposition is not as fast as many people think. We, as several other investigators, have used this procedure to eliminate potentially dangerous peroxides from the reaction mixture at the end of oxidation reactions. One should be aware that for substrates bearing an alcohol function, this work-up step can yield overoxidation to corresponding carbonyl compounds. Furthermore, this relatively slow quenching process does not justify using MnO2 when carrying out detailed kinetic studies. The use of other more efficient quenchers such as aqueous Na2S2O3 [11] should be preferred.
4 Material and methods
4.1 - General considerations
MnO2 was purchased from Aldrich (activated, >90%, reference 63548-250G-F) and used as received without any special precaution or further activation procedure. A second batch of MnO2 (Sigma–Aldrich, ReagentPlus®, reference 243442, ≥ 99%) was also tested (Table 1, entry 7). The tBuOOH solutions in decane (5.3 M, Aldrich) and in water (70%, Acros) and the H2O2 solution (30 wt % in water, Aldrich) were used as received. The substrates were purchased from commercial sources and used as received: phenylethanol (Aldrich, 98%), 4-methyl benzyl alcohol (Aldrich, 98%), 4-methoxy benzyl alcohol (Acros, 98%), trans-cinnamyl alcohol (Acros, 98%), cyclohexanol (Aldrich, 99%), menthol (Aldrich, 99%), perillyl alcohol (Aldrich, 96%), and myrtenol (Aldrich, 95%). The solvents (MeCN, toluene) were of analytical grade and were dehydrated on molecular sieves (4A) prior to use. Conversions for the oxidation experiments were determined by GC, using a Fisons 8130 instrument equipped with a DB-1MS capillary column (30 m × 0.32 mm × 0.25 μm) and with a flame ionization detector.
4.2 - General procedure for the oxidations of alcohols
The alcohol (1.2 mmol) of interest was dissolved in a mixture of acetonitrile (3 ml) and toluene (1 ml) for the experiments with TBHP/decane (or in 4 ml of acetonitrile for the experiments with TBHP/H2O) and the desired amount of MnO2 (see Tables) was added to the solution. Then tert-butyl hydroperoxide (TBHP, as a 5.3 M solution in decane, 453 μL, 2.4 mmol) was added to the reaction mixture, which was set at the desired reaction temperature with magnetic stirring in air. Aliquots (0.2 ml) were withdrawn at different time intervals, diluted with 1 ml of diethyl ether and filtered through a Pasteur pipette filled up with silica to eliminate the residual MnO2. The silica was washed with 2 ml of diethyl ether. The resulting organic phase was analyzed by gas chromatography using diethyleneglycol dibutyl ether as an external standard.
Acknowledgment
We are grateful to the CNRS (Centre National de la Recherche Scientifique) and the IUF (Institut Universitaire de France) for financial support and the “Conseil Régional Midi-Pyrénées” for a post-doctoral fellowship to CB.