

1 Introduction
In laboratories where the working environment does not allow the preparation and purification of acid halides and the handling of reagents such as PCl3, PCl5, SOCl2, or POCl3, a larger number of possible synthetic methodologies of carboxylic acids to the respective anhydrides, amides, and esters are inapplicable. It is in these cases that the use of Appel-type reagents to prepare acyl halides in situ presents itself, where the acyl halides are reacted further with carboxylates, amines, alcohols or oximes.
The versatile combination of triphenylphosphine (PPh3) and tetrachlorocarbon (CCl4) is the original Appel reagent, which can be used in a larger number of transformations [1]. Originally utilized in the reaction of alkanols with chloroalkanes [1,2], PPh3-CCl4 has been shown to be effective in the reaction of carboxamides [3] and aldoximes [4] with nitriles, in esterification and amidation reactions [5] and in the preparation of dichlorovinyl derivatives from the corresponding aldehydes and ketones [6]. Additionally, phosphoric acid derivatives can be transformed to respective amides and phosphoric anhydrides. However, CCl4 is known to be an extremely hepatotoxic substance. Also, because of its long life-span in the atmosphere once released (as it does not oxidize or photodegrade in the troposphere), its involvement in the deterioration of the stratospheric ozone layer has been noted [7]. Furthermore, due to the provisions of the Montreal protocol and its subsequent amendments on substances that deplete the ozone layer, CCl4 has been replaced for many applications. Thus, CCl4 is used no longer as a solvent for chemical reactions, and scientists have started looking for substitutes. In Appel-type reactions, CBr4 instead of CCl4 has been used, but it is expensive. In more recent times, the exchange of CCl4 for BrCCl3 has been investigated to some degree. The latter reagent is preferred over CCl4 since it is of less environmental concern, is not listed as an ozone depletor and because its higher polarity does not reach the stratosphere so easily. Therefore, there is less concern about its photolysis in the stratosphere [8]. The employment of PPh3-BrCCl3 in amidation reactions [9], in the reaction of aldehydes with dihalovinyl compounds [10] and of aldoximes and amides to nitriles [11] has been published already. In this paper, the use of BrCCl3-PPh3 in Appel type transformations of acids to esters and anhydrides and of keto oximes to acyloximes is discussed.
2 Results and discussion
The authors' initial interest in the use of Appel-type transformations of acids to their derivatives stemmed from the need to prepare cinnamides from alkylamidoalkylquinone derivatives [12]. Although the use of BrCCl3/PPh3 had been documented in the conversion of carboxylic acids to carboxamides [13], we were gratified to see that the use of BrCCl3/PPh3 in the reaction of cinnamic acids led exclusively to the corresponding cinnamides and not to 1,4-addition adducts as side products (Scheme 1). Only in some cases of the reaction of alkoxycinnamic acids under these conditions can a small amount of the corresponding (E)-1-bromo-2-(4-alkoxyphenyl)ethene be isolated as a side-product, regardless of the added nucleophile. A typical example is given in the transformation of 4d to 5g, where a small amount of (E)-1-bromo-2-(4-methoxyphenyl)ethene (6) is produced, (Scheme 1).
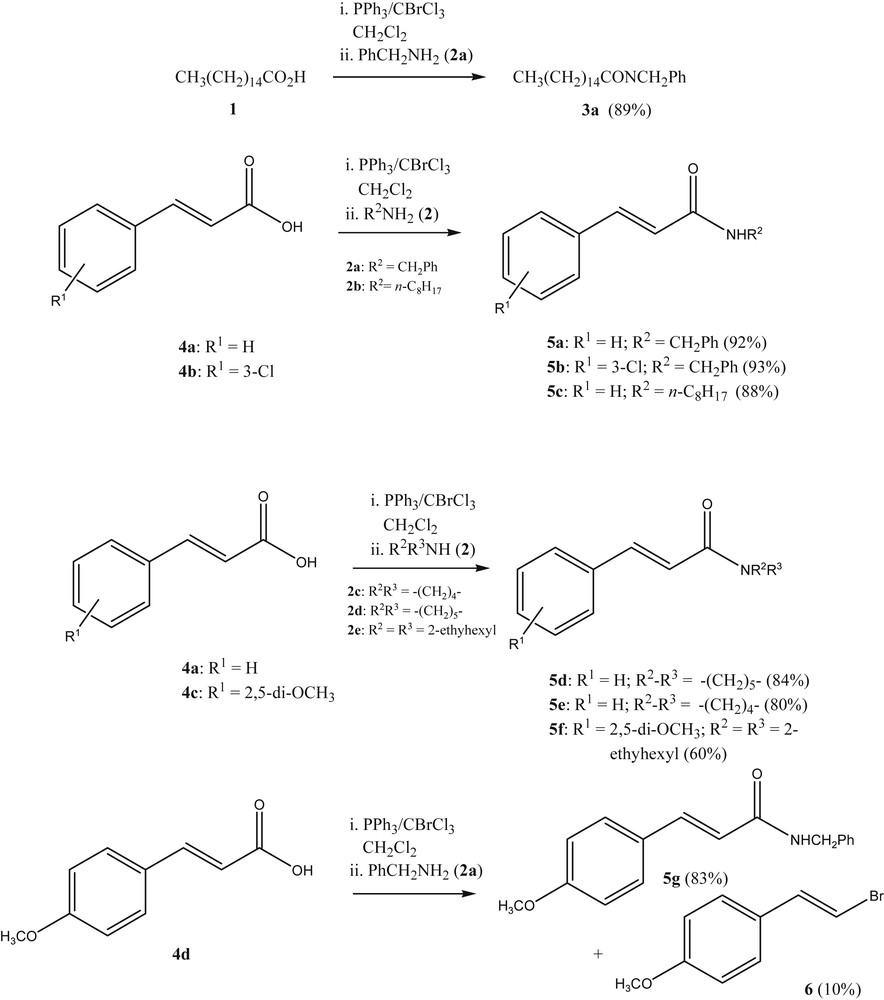
Amidation of cinnamic acids with PPh3-CBrCl3 as a reagent.
Next, we investigated whether the reagent combination BrCCl3/PPh3 could be used in the reaction of carboxylic acids with acid anhydrides. When benzoic acid was treated with BrCCl3/PPh3 in refluxing CH2Cl2, it was realized that acyl halides were formed, where from this reaction benzoyl chloride could be isolated in pure form after flash chromatography. Having established the formation of acyl halides in this transformation, the reaction of the acyl halides in situ with carboxylates as nucleophiles was warranted. Previously, carboxylic acid anhydrides have been prepared from the respective carboxylic acids by using reagents that are both activating and dehydrating such as phosphorus pentoxide [12], carbodiimides [13], isocyanates [14], imidazolinium chlorides [15], phosgene [16] trichlorotrifluoroacetone [17] and triphosgene [18]. Also, carboxylic acids can be transformed into an activated acyl species first, which subsequently is further reacted with an equivalent of the carboxylic acid or the carboxylate to furnish the acid anhydride. Examples of this are the activation with 2,4-dichloro-1,3,5-triazine [19] or the ZnCl2-catalysed reaction of 2-acyl-4,5-dichloropyridazin-3(2H)-ones [20]. Most frequently, however, acid chlorides are used to prepare acid anhydrides. In these cases the acid chlorides can be prepared separately, isolated, and subsequently reacted with the carboxylic acid or its anion under various conditions [21].
We found that the reaction of carboxylic acids with BrCCl3/PPh3 in refluxing CH2Cl2 and the subsequent addition of a further equivalent of carboxylic acid, followed by an equivalent of either dry triethylamine or 1,8-diazabicycloundec-7-ene (DBU), leads to the corresponding acid anhydrides in good yield (Scheme 2). The products were obtained from the reaction mixtures by rapid column chromatography.
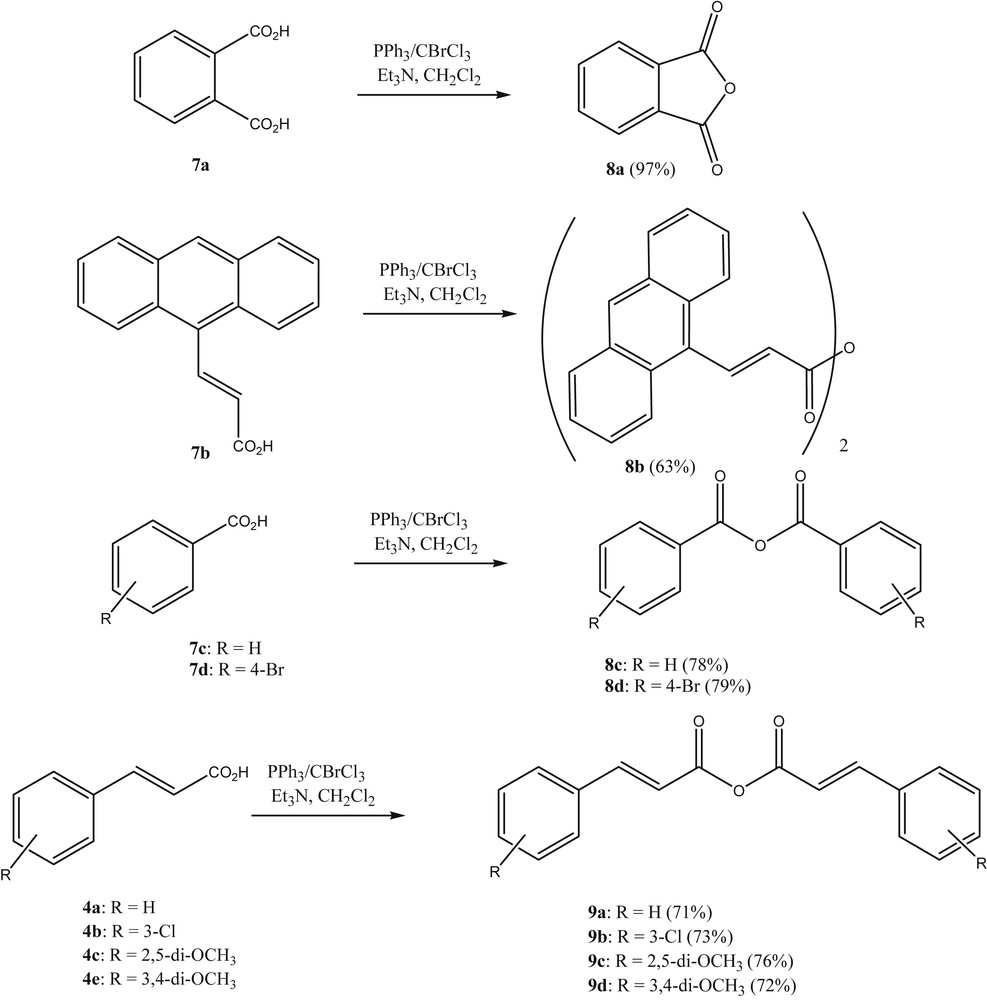
Preparation of anhydrides with PPh3-CBrCl3 as a reagent.
From the above results, it was deemed likely that carboxylic esters should also be accessible through the same strategy, by changing the nucleophile to an alcohol, adding the alcohol after the activation of the acid and/or its in situ transformation to the acyl halide. This indeed proved to be the case as is seen from Scheme 3. The authors preferred venting the hydrogen halide produced as a side product, by submitting the reaction vessel intermittently to vacuum rather than adding a tertiary amine base to the reaction solution. The alkyl carboxylates were produced in acceptable yield and the process can be seen as a viable alternative to other known esterification methods, when used at the laboratory scale.
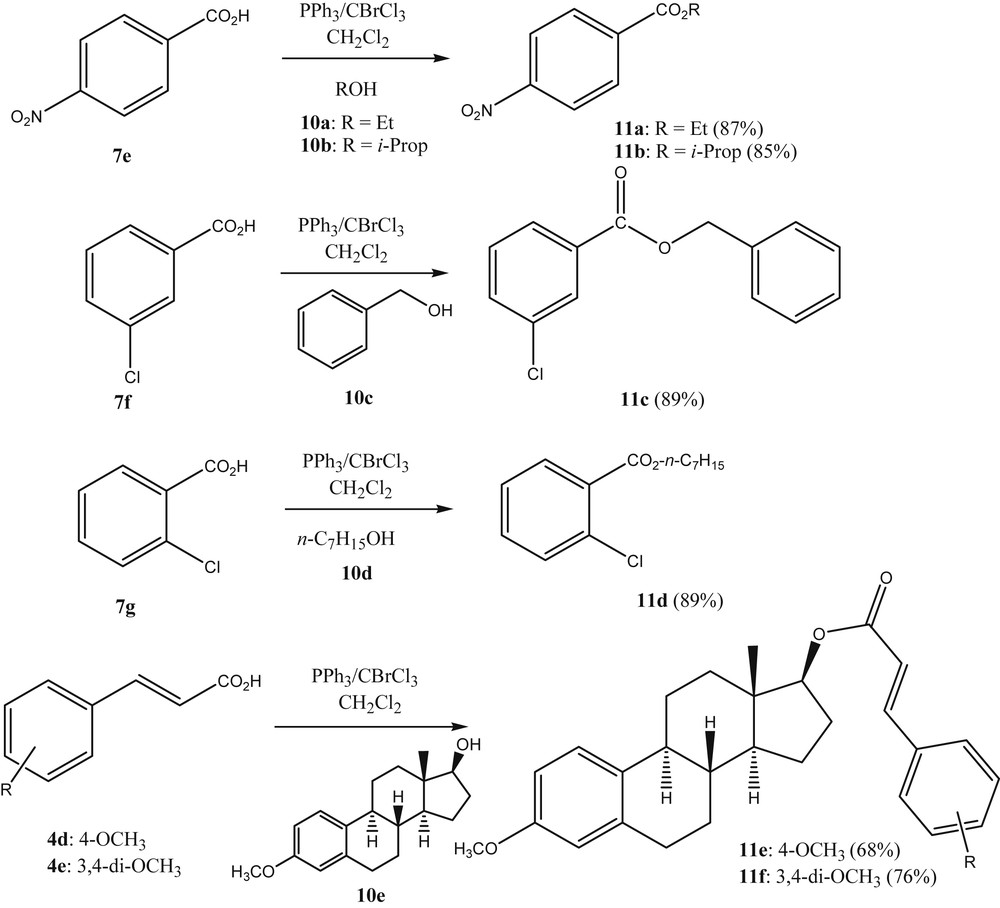
Esterification using PPh3/BrCCl3.
Finally, the authors investigated whether this general strategy of using BrCCl3/PPh3 in the activation of carboxylic acids would give access to acyl oximes as well. Recently, acyl oxime derivatives have attracted considerable attention in medicinal research due to their antiphytoviral, antitumor, antibacterial, and fungicidal bioactivities. A larger number of studies on their synthesis and biological activities have been reported [22]. Some acyl oximes have been found to cleave DNA in a process that is triggered by UV light [23]. The weak N–O bond of the acyl oximes is cleaved selectively to generate the iminyl and carboxy radicals which can then cause the cleavage of the DNA. In our research on cancer-active estrone based molecules, we were interested to prepare O-cinnamoyl estrone-17-oximes such as 13a. Within this context, we succeeded to react estrone-17-oxime 12a with the substituted cinnamic acid 4e in the presence of BrCCl3/PPh3 (Scheme 4). This methodology was found to be quite general to react oximes and acids with O-acyl oximes (Scheme 5).
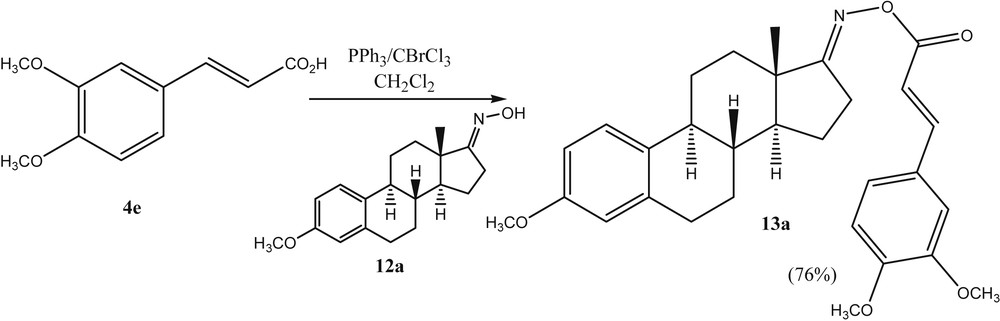
Preparation of a N-cinnamoylestrone-17-oxime.
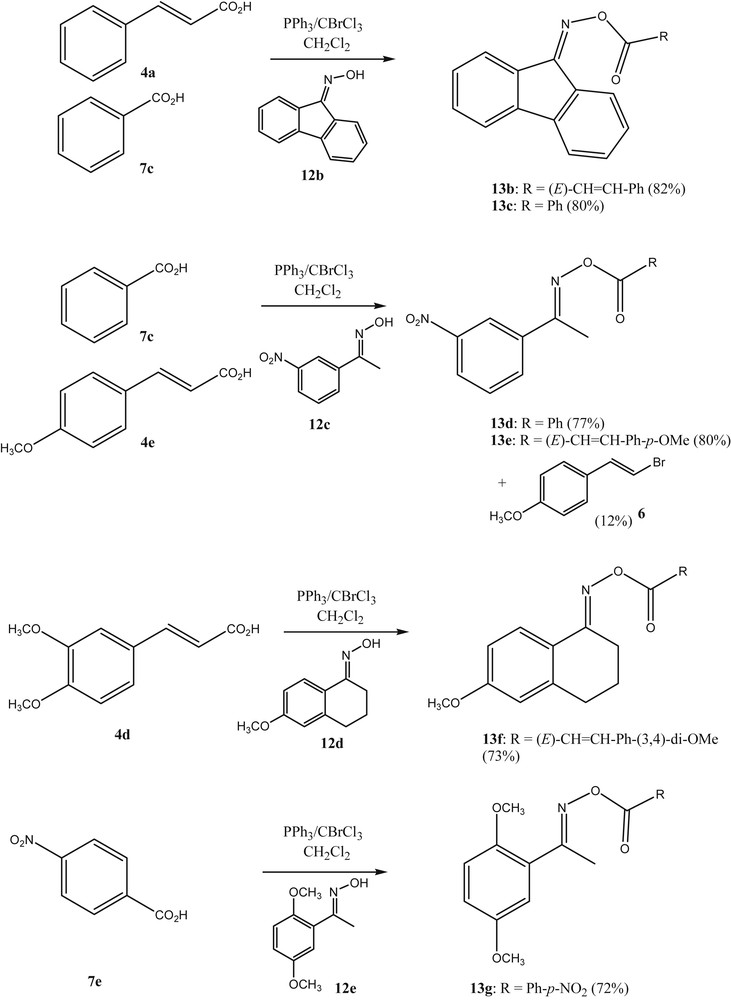
O-acylation of oximes with PPh3/BrCCl3 as the reagent.
Finally, in order to assess the typical molecular structural parameters of aromatic O-acyl oximes, the crystal structure of 2,5-dimethoxyacetophenone oxime 4-nitrobenzoate 13g was studied. The crystallization was carried out in a mixture of CH2Cl2/diethyl ether/hexane (1/1/1–v/v/v) at room temperature, where 13g was isolated as clear light yellow plates. 13g was crystallized in the monoclinic space group P21/C with unit cell parameters: a = 17.1206 (6), b = 8.17107 (19), c = 11.6431 (3) Å, β = 102.779 (3)° (Table 1). Selected bond distances and angles are listed in Table 2. The molecular structure of 2,5-dimethoxyacetophenone oxime 4-nitrobenzoate (13g) is shown in Fig. 1. In the oxime ester moiety, the bond-lengths of C15N1 and C17O4 are 1.288 (2) Å and 1.1977 (19) Å, respectively, which is characteristic of double-bonds. The values of the C17–O3 and N1–O3 bonds are 1.3538 (19) Å and 1.4520 (16) Å, respectively, indicating a single-bond-character. These bond-lengths are in the expected range of oxime-esters [24].
Crystallographic data and structure refinement details.
| CCDC deposit number | 1001765 |
| Chemical formula | C17H16N2O6 |
| Formula weight | 344.32 |
| Temperature (K) | 100 |
| Wavelength (Å) | 0.71073 |
| Crystal size (mm) | 0.75 0.33 0.10 |
| Crystal system | Monoclinic |
| Space group | P21/C |
| a (Å) | 17.1206 (6) |
| b (Å) | 8.17107 (19) |
| c (Å) | 11.6431 (3) |
| β (°) | 102.779 (3) |
| V (Å3) | 1588.45 (8) |
| Z | 4 |
| Dc (g/cm3) | 1.440 |
| F (000) | 720 |
| Absorption coeff. (mm−1) | 0.111 |
| θ range (°) | 3.1–27.5 |
| Index ranges | −19 ≤ h ≤ 21; |
| −6 ≤ k ≤ 10; | |
| −15 ≤ l ≤ 14 | |
| Reflections collected | 7139 |
| Independent reflections | 3515 [Rint = 0.026] |
| Observed reflections | 2726 |
| Data/restraints/parameters | 3515/0/229 |
| Goodness-of-fit on F2 | 1.059 |
| R, wR indices [I > 2σ(°)] | 0.0444, 0.0971 |
| R, wR indices (all data) | 0.0624, 0.1091 |
| Largest diff. peak and hole (e Å−3) | 0.28, −0.28 |
Selected bond lengths (Å) and angles (°).
| Bond lengths | |||
| C(2)–O(2) | 1.3714 (19) | C(17)–O(3) | 1.3538 (19) |
| C(5)–O(1) | 1.3747 (19) | C(17)–O(4) | 1.1977 (19) |
| C(10)–N(2) | 1.477 (2) | O(3)–N(1) | 1.4520 (16) |
| C(13–(1) | 1.428 (2) | O(5)–N(2) | 1.2298 (18) |
| C(14)–O(2) | 1.4348 (19) | O(6)–N(2) | 1.2253 (17) |
| C(15)–N(1) | 1.288 (2) | ||
| Bond angles | |||
| C(5)–O(1)–C(13) | 117.20 (13) | O(5)–N(2)–O(6) | 123.79 (14) |
| C(2)–O(2)–C(14) | 117.03 (13) | N(1)–C(15)–C(1) | 113.67 (13) |
| C(15)–N(1)–O(3) | 108.16 (12) | O(3)–C(17)–O(4) | 125.03 (15) |
| C(17)–O(3)–N(1) | 112.40 (11) |
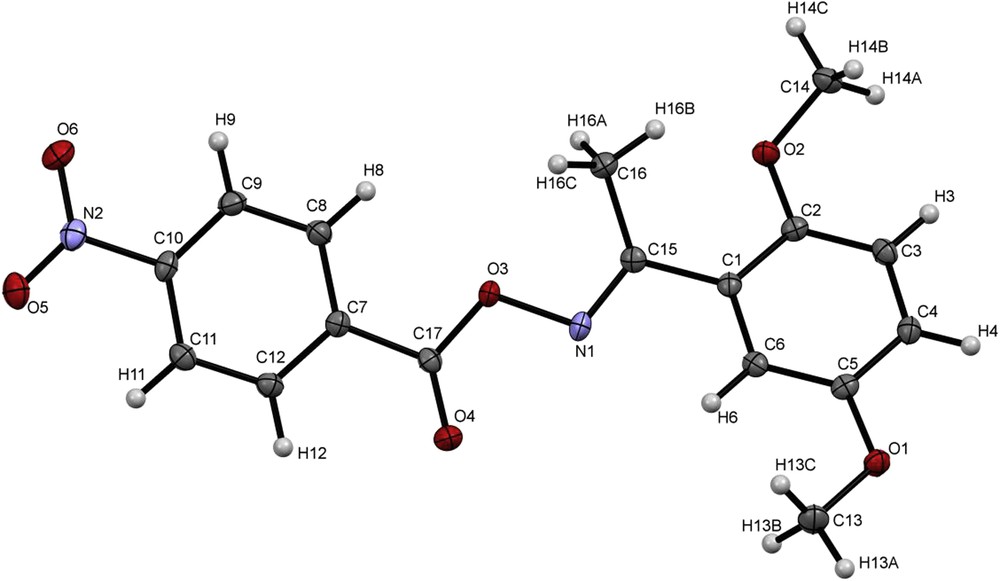
Molecular structure of 2,5-dimethoxyacetophenone oxime 4-nitrobenzoate (13g) with the atomic numbering scheme. Displacement ellipsoids are drawn at the 50% probability level.
The bond angles of C15–N1–O3 = 108.16 (12) °, C17–O3–N1 = 112.40 (11) ° are typical for linear oxime-esters [24].
The nitro-group is slightly tilted with respect to the aromatic ring C7–C12 (torsion-angle: C11–C10–N2–O5 = 4.5 (2) °). The methoxy-groups on the aromatic ring (C1–C6) show a significant difference in torsion angles (C4–C5–O1–C13 = 175.2 (1) °) and C1–C2–O2–C14 of 155.5(2) °). This can be explained with packing-effects. The average two planes of the aromatic rings (non-hydrogen atoms) form an angle of 26.17 (2) ° between each other. This is mainly due to torsions in the bonds O3–N1 and C15–C1 and a further indication of a localized double-bond between C15 and N1 in contrast to an extended π-system.
The molecules of 13g are arranged in two stacks, propagating along the b-axis. Molecules in each stack are related by C-glide operation and are linked to each other through C14–H14A…O5 hydrogen bonding (Fig. 2). Along the c-axis, neighbored stacks are linked by C8–H8…O4, C16–H16C…O4 and C16–H16B…O6 hydrogen bonds (Table 3).
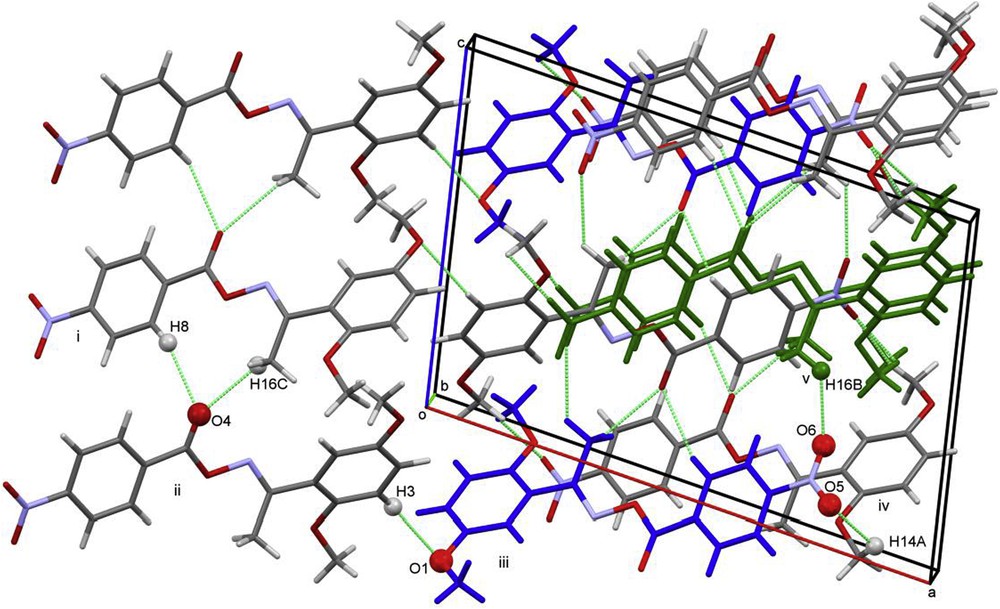
A view of intermolecular interactions between adjacent molecules 13g that lead to the 3D network (colored molecules have the same orientation and layout).
Hydrogen bonds for 13g (Å and °).
| D–H…A | D–H | H…A | D…A | D–H…A |
| C3–H3…O1#1 | 0.950 | 2.496 (1) | 3.375 (2) | 153.9 (1) |
| C8–H8…O4#2 | 0.950 | 2.516 (1) | 3.317 (2) | 142.1 (1) |
| C14–H14A…O5#1 | 0.980 | 2.599 (1) | 3.141 (2) | 115.0 (1) |
| C16–H16B…O6#1 | 0.980 | 2.601 (1) | 3.330 (2) | 131.3 (1) |
| C16–H16C…O4#2 | 0.980 | 2.501 (1) | 3.177 (2) | 125.9 (1) |
Symmetry codes of molecules showing the labeled interactions are shown in Fig. 2: i: −x, −1/2 + y, 1/2 − z; ii: −x, 1 − y, −z; iii: x, 1.5 − y, −1/2 + z; iv: 1 − x, 1/2 + y, 1/2 − z; v: 1 − x, 1 − y, 1 − z.
3 Conclusion
In a modified Appel-type reaction, cinnamic and benzoic acids have been reacted with amines, alcohols, and oximes, using the relatively environmentally friendly reagent combination PPh3-BrCCl3. It was also shown that anhydrides are obtained, when the acids upon activation with PPh3-BrCCl3 are reacted with a further equivalent of acid in the presence of a tertiary amine. An X-ray crystal structural analysis of acyloxime 13g was carried out. It is envisaged that Appel-type reactions as the ones shown here will gain further importance with the advancement of strategies towards the recycling of the by-product triphenylphosphine oxide to triphenylphosphine [25].
4 Experimental
4.1 General
Melting points were measured on a Stuart SMP 10 melting point apparatus and are uncorrected. Infrared spectra were measured with a Thermo/Nicolet Nexus 470 FTIR ESP Spectrometer. 1H and 13C NMR spectra were recorded with a Varian 400 NMR (1H at 395.7 MHz, 13C at 100.5 MHz) and a Varian 200 MHz NMR spectrometer (1H at 200.0 MHz, 13C at 50.3 MHz). The chemical shifts are relative to TMS (solvent CDCl3, unless otherwise noted). Mass spectra were measured with a JMS-01-SG-2 spectrometer. CHN-analysis was performed on a LECO TruSpec Micro instrument. Column chromatography was carried out on a silica gel (60 A, 230–400 mesh, Sigma-Aldrich). Analytical thin layer chromatography (TLC) was carried out on silica on TLC Alu foils from Fluka (with fluorescent indicator at λ = 254 nm). Triethylamine was dried over solid KOH and distilled. 1,8-Diazabicycloundec-7-ene (DBU) was stored over solid KOH. The oximes 12 were prepared by the reaction of the ketones with ammonium hydrochloride (4 mol eq.) in a solvent mixture of ethanol and water [26]. 3-Arylacrylic acids 4b–e and 7b were prepared by a one-pot Wittig olefination – hydrolysis reaction of the respective benzaldehydes and 9-anthranylcarbaldehyde with ethoxycarbonylmethylidenetriphenylphosphorane [27].
4.2 General procedure for the preparation of cinnamides
4.2.1 N,N-Bis(2-ethylhexyl) 3-(1,4-dimethoxyphen-2-yl)propenoic acid amide (5f)
To a solution of PPh3 (980 mg, 3.74 mmol) in dry CH2Cl2 (12 mL) BrCCl3 (1.50 g, 7.66 mmol) was added. The resulting mixture was stirred at rt for 30 min, during which it turned reddish-brown. Thereafter, 2,5-dimethoxycinnamic acid (4c, 700 mg, 3.37 mmol) was added, and the mixture was heated under reflux for 45 min. Thereafter, di(2-ethylhexyl)amine (1.62 g, 6.71 mmol) was added dropwise via a syringe, where the ensuing reaction is exothermic. The reaction mixture was stirred at reflux for 14 h. Then, the cooled mixture was concentrated in vacuo, and the residue was subjected directly to column chromatography on a silica gel to give 5f (875 mg, 2.03 mmol, 60%) as a colorless oil; νmax neat/cm−1) 2961, 2877, 1660, 1622, 1506, 1489, 1047, 772; δH (400 MHz, CDCl3) 1.00 (16H, m), 1.48 (20H, m), 1.69–1.79 (2H, m), 3.28–3.43 (4H, m), 3.77 (3H, s, OCH3), 3.81 (3H, s, OCH3), 6.82 (1H, d, 3J = 8.8 Hz), 6.84 (1H, d, 3J = 8.8 Hz), 7.00 (1H, s), 7.02 (1H, d, 3J = 15.6 Hz), 7.86 (1H, d, 3J = 15.6 Hz); δC (67.8 MHz, CDCl3, DEPT) 10.7 (CH3), 10.8 (CH3), 14.0 (CH3), 14.1 (CH3), 23.1 (CH2, 2C), 23.7 (CH2), 24.0 (CH2), 28.7 (CH2), 28.8 (CH2), 30.5 (CH2), 30.7 (CH2), 37.6 (CH), 40.0 (CH), 50.7 (CH2), 52.0 (CH2), 55.7 (OCH3), 56.0 (CH3), 112.3 (CH), 114.4 (CH), 115.4 (CH), 119.6 (CH), 125.2 (Cquat), 137.5 (CH), 152.8 (Cquat), 153.4 (Cquat), 167.3 (Cquat, CO).
4.2.2 N-Benzylpalmitamide (3a)
As colorless needles, mp 98 °C (Lit. mp 95 °C [28]); IR (KBr) νmax: 3305 (NH), 2918, 2848, 1639, 1549, 1457, 733, 696 cm−1; 1H NMR (400 MHz, CDCl3) δ: 0.81 (t, 3J = 6.8 Hz, 3H, CH3), 1.16–1.26 (m, 14H), 1.57 (m, 2H), 2.14 (t, 3J = 7.6 Hz, 2H), 4.37 (d, 3J = 8.0 Hz, 2H), 5.64 (b, NH, 1H), 7.19–7.29 (m, 5H); 13C NMR (100.5 MHz, CDCl3) δ: 14.1, 22.7, 25.8, 29.3, 29.4, 29.5, 31.9, 36.8, 43.6, 127.5, 127.8 (2C), 128.7 (2C), 138.4, 173.0 (CO); MS (EI, 70 eV) m/e (%) = 345 (M+), 149 (100).
4.2.3 N-Benzyl cinnamide (5a)
Colorless needles, mp 107 °C (Lit. mp 106–108 °C [29]); IR (KBr) νmax: 3280 (br s, NH), 3080, 2921, 1654, 1614, 1541, 1450, 1348, 1230, 1213, 979, 751, 696 cm−1; 1H NMR (CDCl3, 400 MHz) δ: 4.56 (d, 3J = 5.6 Hz, 2H), 6.06 (br s, NH, 1H), 6.42 (d, 3J = 16.0 Hz, 1H), 7.25–7.30 (m, 3H), 7.47–7.48 (m, 2H), 7.66 (d, 3J = 16.0 Hz, 1H); 13C NMR (100.5 MHz, CDCl3) δ: 43.8 (−), 120.4 (CH), 127.6 (CH), 127.8 (2C, CH), 127.9 (2C, CH), 128.7 (2C, CH), 128.8 (2C, CH), 129.7 (CH), 134.7 (Cquat), 138.1 (Cquat), 141.4 (CH), 165.8 (Cquat, CO); MS (EI, 70 eV) m/e (%) = 237 (M+).
4.2.4 N-Benzyl 3-chlorocinnamide (5b)
Colorless needles; mp 112 °C [Lit. mp 108–109 °C [30]); νmax (KBr/cm−1) 3295, 3058, 2927, 1652, 1616, 1538, 1424, 1331, 1228, 1079, 1035, 969, 858, 787, 696, 664; δH (400 MHz, CDCl3) 4.57 (2H, d, 3J = 5.6 Hz), 5.97 (1H, b, NH), 6.40 (1H, d, 3J = 15.6 Hz), 7.27–7.37 (8H, m), 7.48 (1H, s), 7.61 (1H, d, 3J = 15.6 Hz); δC (100.5 MHz, CDCl3) 43.9, 121.7, 126.2, 127.4, 127.7, 127.9, 128.8, 129.6, 130.1, 134.8, 136.6, 137.9, 140.0, 165.2 (CO).
4.2.5 N-Octyl cinnamide (5c) [31]
Colorless solid; IR (KBr) νmax: 3285, 3063, 2954, 2926, 2853, 1668, 1653, 1548, 1476, 1460, 1333, 1224, 993, 869, 768, 721 cm−1; 1H NMR (CDCl3, 400 MHz) δ: 0.86 (t, 3J = 6.8 Hz, 3H), 1.26–1.30 (m, 10H), 1.54 (m, 2H), 3.36 (dt, 3J = 6.0 Hz, 3J = 6.0 Hz, 2H), 6.38 (d, 3J = 15.6 Hz, 1H), 7.33–7.35 (m, 3H), 7.47–7.49 (m, 2H), 7.61 (d, 3J = 15.6 Hz, 1H); 13C NMR (CDCl3, 100.5 MHz) δ: 14.1 (CH3), 22.6 (−), 27.0 (−), 29.2 (−), 29.3 (−), 29.7 (−), 31.8 (−), 39.8 (−), 120.7 (CH), 127.7 (2C, CH), 128.8 (2C, CH), 129.6 (CH), 134.8 (Cquat), 140.8 (CH), 165.9 (Cquat, CO); MS (EI, 70 eV) m/e (%) = 233 (M+).
4.2.6 N-Piperidinyl cinnamide (5d)
Colorless solid; mp 123 °C (mp 122 °C [32]); νmax (KBr/cm−1) 3078, 3027, 2933, 2849, 1645, 1588, 1458, 1442, 1281, 1249, 1223, 1018, 992, 764; δH (400 MHz, CDCl3) 1.57–1.70 (6H, m), 3.61 (4H, br s), 6.89 (2H, d, 3J = 15.2 Hz), 7.30–7.50 (3H, m), 7.48–7.52 (2H, m), 7.62 (2H, d, 3J = 15.2 Hz); δC (100.5 MHz, CDCl3) 24.6 (CH2), 25.8 (br s, CH2), 26.6 (br s, CH2), 43.3 (br s, CH2), 47.1 (br s, CH2), 117.7 (CH), 127.7 (2C, CH), 128.7 (2C, CH), 129.4 (CH), 135.5 (Cquat), 142.1 (CH), 165.3 (Cquat, CO).
4.2.7 N-Pyrrolidinyl cinnamide (5e)
Pale yellow solid; mp 99 °C (mp 93–94 °C [33]); νmax (KBr/cm−1) 3060, 2968, 2873, 1652, 1600, 1452, 1433, 986, 865, 761, 706, 681, 568, 547, 487; δH (400 MHz, CDCl3) 1.84–2.01 (4H, m), 3.56–3.63 (4H, m), 6.71 (1H, d, 3J = 15.6 Hz), 7.30–7.37 (3H, m), 7.50–7.53 (2H, m), 7.68 (1H, d, 3J = 15.6 Hz); δC (100.5 MHz, CDCl3) 24.3 (CH2), 26.1 (CH2), 46.1 (CH2), 46.6 (CH2), 118.8 (CH), 127.8 (2C, CH), 128.7 (2C, CH), 129.5 (CH), 135.3 (Cquat), 141.7 (CH), 164.7 (Cquat, CO).
4.2.8 N-Benzyl 4-methoxycinnamide (5g) [34]
Colorless needles; mp 145–146 °C; νmax (KBr/cm−1) 3282 (s, NH), 3030, 2929, 2834, 1644, 1605, 1528, 1252, 1217, 1174, 1028, 972, 825, 752, 699; δH (400 MHz, CDCl3) 3.81 (3H, s, OCH3), 4.54 (2H, d, 3J = 5.6 Hz), 6.11 (1H, m), 6.86 (2H, d, 3J = 8.4 Hz), 7.26–7.34 (5H, m), 7.42 (2H, d, 3J = 8.5 Hz), 7.62 (1H, d, 3J = 15.6 Hz); δC (100.5 MHz, CDCl3) 43.8, 55.3 (OCH3), 114.2 (2C), 118.0, 127.4, 127.5, 127.9 (2C), 128.7 (2C), 129.4 (2C), 138.2, 141.0, 160.9, 166.2 (CO).
4.3 General procedure for the preparation of carboxylic acid anhydrides
4.3.1 Cinnamic anhydride (9a)
To a solution of triphenylphosphine (910 mg, 3.47 mmol) in dry dichloromethane (10 mL) bromotrichloromethane (710 mg, 3.58 mmol) was added, and the resulting mixture was stirred at a rate for 30 min to give a reddish-brownish solution. Thereafter, cinnamic acid (4a, 451 mg, 3.05 mmol) was added, and the resulting mixture was held at reflux for 5 h. Thereafter, cinnamic acid (451 mg, 3.05 mmol) and subsequently triethylamine (310 mg, 3.05 mmol) were added, and the reaction mixture was held at reflux for 8 h. Then, the cooled solution was concentrated in vacuo, and the concentrate was subjected to rapid column chromatography on a silica gel [CH2Cl2-hexane 2:1] to give cinnamic anhydride (9a, 605 mg, 71%) as colorless crystals; mp 139–140 °C (mp 138 °C [35]); IR (KBr) νmax: 3060, 3027, 1767, 1701, 1632, 1450, 1305, 1270, 1228, 1201, 1074, 960, 858, 761, 690, 676, 551 cm−1; 1H NMR (CDCl3, 400 MHz) δ: 6.53 (d, 3J = 16.0 Hz, 2H), 7.43–7.45 (m, 6H), 7.57–7.59 (m, 4H), 7.86 (d, 3J = 16.0 Hz, 2H); 13C NMR (CDCl3, 100.5 MHz) δ: 116.7 (2C, CH), 128.6 (4C, CH), 129.1 (4C, CH), 131.3 (2C, CH), 133.7 (2C, Cquat), 148.7 (2C, CH), 162.5 (2C, Cquat, CO).
4.3.2 Phthalic anhydride (8a)
Colorless solid; mp 130 °C (Lit. 130–131 °C [36]); IR (KBr) νmax: 1851, 1762, 1598, 1469, 1258, 1108, 907, 713, 533 cm−1; 1H NMR (400 MHz, CDCl3) δ: 7.90–7.92 (m, 2H), 8.02–8.04 (m, 2H); 13C NMR (CDCl3, 100.5 MHz) 125.7 (2C, CH), 131.3 (2C, Cquat), 136.0 (2C, CH), 162.8 (Cquat, CO); MS (EI, 70 eV) m/e (%) = 148 (M+, 20), 104 (100).
4.3.3 Anthranyl-9-acrylic anhydride (8b)
As an orange solid; mp 216–218 °C (Lit. 216–218 °C [37]); IR (KBr) νmax: 3052, 1760, 1706, 1123, 935, 913, 744, 702 cm−1; 1H NMR (CDCl3, 400 MHz) δ 6.63 (d, 3J = 16.0 Hz, 2H), 7.50–7.59 (m, 8H), 8.04 (dm, 3J = 8.0 Hz, 4H), 8.29 (dm, 3J = 7.6 Hz, 4H), 8.51 (s, 2H), 8.93 (d, 3J = 16.0 Hz, 2H); 13C NMR (CDCl3, 100.5 MHz) δ: 124.9 (4C, CH), 125.4 (4C, Cquat), 125.5 (4C, CH), 126.9 (4C, CH), 128.1 (2C, Cquat), 129.0 (4C, CH), 129.3 (2C, CH), 129.5 (4C, Cquat), 131.2 (2C, CH), 146.4 (2C, CH), 162.0 (2C, Cquat, CO). Anal. Calcd for C34H22O3 (478.54): C, 85.34%; H, 4.63%. Found: C, 85.25%; H, 4.73%.
4.3.4 Benzoic anhydride (8c)
Slowly crystallizing, colorless solid; mp 41 °C (Lit. 42 °C [21a]); IR (neat) νmax: 3065, 3010, 1786, 1724, 1599, 1491, 1452, 1315, 1213, 1173, 1040, 1018, 995, 873, 778, 701, 615 cm−1; 1H NMR (CDCl3, 400 MHz) δ: 7.50–7.54 (m, 4H), 7.65–7.69 (m, 2H), 8.14–8.17 (m, 4H); 13C NMR (CDCl3, 100.5 MHz) 128.8 (2C), 128.9 (4C), 130.6 (4C), 134.6 (2C), 162.4 (2C, Cquat, CO); MS (EI, 70 eV) m/e (%) = 226 (M+, 5), 105 (100).
4.3.5 4-Bromobenzoic anhydride (8d)
Colorless solid; mp 221–224 °C [Lit. mp 217–218 °C [38a], 225 °C [38b]; IR (KBr) νmax: 1786, 1721, 1586, 1480, 1396, 1226, 1173, 1067, 1004, 824, 826, 793, 738, 676, 625 cm−1; 1H NMR (CDCl3, 400 MHz) 7.68 (d, 3J = 8.8 Hz, 2H), 8.00 (d, 3J = 8.8 Hz, 2H); 13C NMR (CDCl3, 100.5 MHz) 127.5 (2C, Cquat), 130.2 (2C, Cquat), 131.9 (4C, CH), 132.4 (4C, CH), 161.4 (2C, Cquat, CO); MS (EI, 70 eV) m/e (%) = 340 (M+), 338 (M+), 185, 183.
4.3.6 3-Chlorocinnamic anhydride (9b)
Silvery shiny needles, mp 117–118 °C; IR (KBr) νmax: 3079, 1758, 1709, 1629, 1134, 940, 784, 665 cm−1; 1H NMR (CDCl3, 400 MHz) δ: 6.52 (d, 3J = 16.0 Hz, 2H), 7.35–7.46 (m, 6H), 7.57 (s, 2H), 7.78 (d, 3J = 16.0 Hz, 2H); 13C NMR (CDCl3, 100.5 MHz) δ: 118.0 (2C, CH), 126.8 (2C, CH), 128.2 (2C, CH), 130.4 (2C, CH), 131.2 (2C, CH), 135.2 (2C, Cquat), 135.4 (2C, Cquat), 147.1 (2C, CH), 161.9 (2C, Cquat, CO). Anal. Calcd for C18H12Cl2O3 (347.19) C, 62.27%; H, 3.48%. Found: C, 62.43%; H, 3.57%.
4.3.7 Bis(2,5-dimethoxycinnamic) anhydride (9c)
Pale yellow solid; mp 103–105 °C; IR (KBr) νmax: 3078, 3008, 2947, 2912, 1837, 1714, 1704, 1625, 1579, 1498, 1458, 1428, 1322, 1290, 1255, 1206, 1086, 1049, 1018, 985, 947, 866, 805, 719, 669 cm−1; 1H NMR (CDCl3, 400 MHz) δ: 3.80 (s, 6H, 2 OCH3), 3.86 (s, 6H, 2 OCH3), 6.60 (d, 3J = 16.0 Hz, 2H), 6.87 (d, 3J = 8.8 Hz, 2H), 6.96 (dd, 3J = 8.8 Hz, 4J = 2.8 Hz, 2H), 7.06 (d, 4J = 2.8 Hz, 2H), 8.12 (d, 3J = 16.0 Hz, 2H); 13C NMR (CDCl3, 100.5 MHz) 55.8 (2C, 2 OCH3), 56.1 (2C, 2 OCH3), 112.5 (2C, CH), 113.6 (2C, CH), 117.5 (2C, CH), 118.3 (2C, CH), 123.2 (2C, Cquat), 143.8 (2C, CH), 153.3 (2C, Cquat), 153.5 (2C, Cquat), 163.2 (2C, Cquat, CO). Anal. Calcd for C22H22O7 (398.41): C, 66.32%; H, 5.57%. Found: C, 66.74%; H, 5.62%.
4.3.8 Bis(3,4-dimethoxycinnamic) anhydride (9d)
Pale yellow solid; mp 177–179 °C; IR (KBr) νmax: 3119, 3072, 2995, 2993, 2960, 2931, 2835, 1756, 1716, 1627, 1598, 1581, 1513, 1461, 1425, 1270, 1163, 1444, 1084, 1023, 1001, 869, 815 cm−1; 1H NMR (CDCl3, 200 MHz) δ: 3.95 (s, 12H, 4 OCH3), 6.41 (d, 3J = 15.6 Hz, 2H), 6.91 (d, 3J = 8.4 Hz, 2H), 7.10 (s, 2H), 7.19 (d, 3J = 8.4 Hz, 2H), 7.81 (d, 3J = 15.6 Hz, 2H); 13C NMR (CDCl3, 50.3 MHz) δ: 55.9 (2C, 2 OCH3), 56.0 (2C, 2 OCH3), 109.9 (2C, CH), 111.1 (2C, CH), 114.4 (2C, Cquat), 123.6 (2C, CH), 126.8 (2C, CH), 148.5 (2C, CH), 149.3 (2C, Cquat), 152.0 (2C, Cquat), 162.9 (2C, Cquat, CO). Anal. Calcd for C22H22O7 (398.41): C, 66.32%; H, 5.57%. Found: C, 66.02%; H, 5.62%.
4.4 General procedure for the preparation of esters
4.4.1 Ethyl 4-nitrobenzoate (11a)
To a solution of triphenylphosphine (910 mg, 3.47 mmol) in dry dichloromethane (20 mL) bromotrichloromethane (710 mg, 3.58 mmol) was added, and the resulting mixture was stirred at a rate for 30 min to give a reddish-brownish solution. 4-Nitrobenzoic acid (509 mg, 3.04 mmol) was added and the solution was stirred at reflux for 1 h. Thereafter, dry ethanol (0.50 mL, 400 mg, 8.7 mmol) was added dropwise via a syringe. The resulting mixture was stirred at reflux for 12 h. Thereafter, it was cooled and subjected directly to column chromatography on a silica gel (ether/CHCl3/hexane 1:1:3) to give 11a (515 mg, 87%) as colorless crystals; mp 56–59 °C; IR (KBr) νmax: 3120, 3057, 2990, 1716, 1607, 1526, 1351, 1279, 1103, 871, 841, 787, 714, 505 cm−1; 1H NMR (CDCl3, 400 MHz) δ: 1.41 (t, 3J = 7.2 Hz, 3H), 4.42 (q, 3J = 7.2 Hz, 2H, OCH2), 8.19 (d, 3J = 9.2 Hz, 2H), 8.26 (d, 3J = 9.2 Hz, 2H); 13C NMR (CDCl3, 100.5 MHz) δ: 14.2 (CH3), 62.0 (OCH2), 123.5 (2C, CH), 130.6 (2C, CH), 135.8 (Cquat), 150.4 (Cquat), 164.7 (Cquat, CO). Anal. Calcd for C9H9NO4 (195.17): C, 55.39%; H, 4.65%; N, 7.18%. Found: C, 55.53%; H, 4.69%; N, 7.10%.
4.4.2 Isopropyl 4-nitrobenzoate (11b)
Colorless crystals; mp 110–112 °C; IR (neat) νmax: 3112, 3081, 3076, 2987, 2939, 2880, 1713, 1525, 1282, 1100, 1011, 918, 875, 841, 788, 718 cm−1; 1H NMR (CDCl3, 400 MHz) δ: 1.38 (d, 3J = 6.4 Hz, 6H, 2 CH3), 5.27 (hept., 3J = 6.4 Hz, 1H), 8.18 (d, 3J = 9.2 Hz, 2H), 8.25 (d, 3J = 9.2 Hz, 2H); 13C NMR (CDCl3, 100.5 MHz) δ: 21.8 (2C, 2 CH3), 69.7 (OCH), 123.4 (2C, CH), 130.6 (2C, CH), 136.2 (Cquat), 150.4 (Cquat), 164.2 (Cquat, CO). Anal. Calcd for C10H11NO4 (209.20): C, 57.41%; H, 5.30%; N, 6.70%. Found: C, 57.33%; H, 5.45%; N, 6.68%.
4.4.3 Benzyl 3-chlorobenzoate (11c) [39]
Colorless oil; νmax (neat/cm−1) 3067, 3034, 2954, 2888, 1723, 1575, 1278, 1254, 1126, 1073, 747, 696; δH (400 MHz, CDCl3) 5.36 (2H, s, OCH2), 7.35–7.45 (6H, m), 7.52 (1H, d, 3J = 8.0 Hz), 7.95 (1H, d, 3J = 8.0 Hz), 8.04 (1H, s); δC (100.5 MHz, CDCl3) 67.1 (OCH2), 127.8 (CH), 128.3 (2C, CH), 128.4 (2C, CH), 128.7 (CH), 129.7 (CH), 129.8 (CH), 131.8 (Cquat), 133.1 (CH), 134.5 (Cquat), 135.6 (Cquat), 165.2 (Cquat, CO).
4.4.4 Heptyl 2-chlorobenzoate (11d) [40]
Colorless oil; νmax (neat/cm−1) 2956, 2929, 2857, 1732, 1593, 1469, 1436, 1292, 1250, 1118, 1051, 747; δH (400 MHz, CDCl3) 0.88 (3H, t, 3J = 7.0 Hz, CH3), 1.26–1.58 (8H, m), 1.72–1.77 (2H, m), 4.32 (2H, t, 3J = 6.4 Hz, OCH2), 7.28–7.45 (3H, m), 7.79 (1H, dd, 3J = 7.6 Hz, 4J = 2.0 Hz); δC (100.5 MHz, CDCl3) 14.1 (CH3), 22.6 (CH2), 26.0 (CH2), 28.6 (CH2), 28.9 (CH2), 31.7 (CH2), 65.7 (OCH2), 126.5 (CH), 130.5 (Cquat), 130.9 (CH), 131.3 (CH), 132.3 (CH), 133.6 (Cquat), 165.9 (Cquat, CO).
4.4.5 3-O-Methylestra-1,3,5(10)-trien-17β-yl 4-(E)-methoxycinnamate (11e)
As a colorless solid, mp 126–128 °C; IR (KBr/cm−1) νmax: 2919, 2866, 2836, 1701, 1636, 1604, 1575, 1512, 1497, 1323, 1257, 1205, 1172, 1033, 984, 967, 828, 812; 1H NMR (400 MHz, CDCl3) δ: 0.90 (3H, s, CH3), 1.25–2.31 (13H, m), 2.85–2.87 (2H, m), 3.77 (3H, s, OCH3), 3.83 (3H, s, OCH3), 4.82 (1H, dd, 3J = 9.2 Hz, 3J = 8.0 Hz), 6.32 (1H, d, 3J = 16.0 Hz), 6.63 (1H, d, 4J = 2.8 Hz), 6.70 (1H, dd, 3J = 8.8 Hz, 4J = 2.8 Hz), 6.90 (2H, d, 3J = 8.8 Hz), 7.20 (1H, d, 3J = 8.4 Hz), 7.48 (2H, d, 3J = 8.8 Hz), 7.62 (1H, d, 3J = 16.0 Hz); 13C NMR (100.5 MHz, CDCl3) δ: 12.2, 23.3, 26.2, 27.2, 27.7, 29.8, 37.0, 38.6, 43.2, 43.8, 49.8, 55.2 (OCH3), 55.4 (OCH3), 82.6 (OCH), 111.5 (CH), 113.8 (CH), 114.3 (2C, CH), 116.1 (CH), 126.4 (CH), 127.2 (Cquat), 129.7 (2C, CH), 132.5 (Cquat), 137.9 (Cquat), 144.0 (CH), 157.4 (Cquat), 161.3 (Cquat), 167.4 (Cquat, CO). Anal. Calcd for C29H34O4. (446.58): C, 78.00%; H, 7.67%. Found: C, 78.14%; H, 7.44%.
4.4.6 3-O-Methylestra-1,3,5(10)-trien-17β-yl 3,4-(E)-dimethoxycinnamate (11f)
As a colorless solid, mp 162–165 °C; νmax (KBr/cm−1) 3010, 2953, 2863, 2843, 2805, 2698, 1612, 1596, 1510, 1445, 1417, 1294, 1257, 1155, 1139, 1024, 848, 812, 782, 616, 570; δH (400 MHz, CDCl3) 0.90 (3H, s, CH3), 2.84–2.88 (2H, m), 1.25–2.31 (13H, m), 3.77 (3H, s, OCH3), 3.91 (3H, s, OCH3), 3.92 (3H, s, OCH3), 4.83 (1H, dd, 3J = 8.0, 3J = 7.6 Hz, OCH), 6.32 (1H, d, 3J = 16.0 Hz), 6.63 (1H, d, 4J = 2.8 Hz), 6.70 (1H, dd, 3J = 8.4 Hz, 4J = 2.8 Hz), 6.86 (1H, d, 3J = 8.4 Hz), 7.05 (1H, d, 4J = 2.0 Hz), 7.10 (1H, dd, 3J = 8.4 Hz, 4J = 2.0 Hz), 7.20 (1H, d, 3J = 8.4 Hz), 7.61 (1H, d, 3J = 16.0 Hz); δC (100.5 MHz, CDCl3) 12.2, 23.3, 26.2, 27.2, 27.7, 29.8, 37.0, 38.6, 43.2, 43.8, 49.8, 55.2 (OCH3), 55.9 (OCH3), 56.0 (OCH3), 82.7 (OCH), 109.5, 111.0, 111.5, 113.8, 116.2, 122.6, 126.4, 127.5, 132.5, 137.9, 144.3, 149.2 (Cquat), 151.0 (Cquat), 157.4 (Cquat), 167.3 (Cquat, CO). Found: C, 75.76%; H, 7.32%. Calcd. for C30H36O5. (476.60): C, 75.60%; H, 7.61%.
4.5 General procedure for the preparation of O-acyloximes
4.5.1 3-Nitroacetophenone oxime 4-methoxycinnamate (13e)
To a solution of triphenylphosphine (910 mg, 3.47 mmol) in dry dichloromethane (10 mL) bromotrichloromethane (710 mg, 3.58 mmol) was added, and the resulting mixture was stirred at a rate for 30 min to give a reddish-brownish solution. Then, 4-methoxycinnamic acid (4d, 542 mg, 3.04 mmol) was added, and the mixture was stirred at reflux for 45 min. Thereafter, 3-nitroacetophenone oxime (12c, 545 mg, 3.03 mmol) was added, and the mixture was stirred at reflux for another 12 h. Column chromatography on a silica gel (CH2Cl2) gave (E)-1-bromo-2-(4-methoxyphenyl)ethene (6, 77 mg, 12%) [41] as a colorless solid; mp 52–56 °C (Lit. mp 58–59 °C [42]); νmax (KBr/cm−1) 2958, 2837, 1607, 1512, 1256, 1028, 950, 927, 836, 777, 526; δH (400 MHz, CDCl3) 6.60 (1H, d, 3J = 14.0 Hz), 6.84 (2H, d, 3J = 8.4 Hz), 7.03 (1H, d, 3J = 14.0 Hz), 7.22 (2H, d, 3J = 8.4 Hz); δC (100.5 MHz, CDCl3) 55.3 (OCH3), 104.0 (CH), 114.7 (2C, CH), 127.3 (2C, CH), 128.7 (Cquat), 136.5 (CH), 159.6 (Cquat) and 13e (826 mg, 80%) as a pale yellow solid; mp 130–133 °C; IR (KBr) νmax: 1754, 1624, 1602, 1573, 1524, 1513, 1351, 1309, 1286, 1249, 1178, 1102, 1026, 981, 921, 825, 735 cm−1; 1H NMR (400 MHz, CDCl3) δ: 2.50 (s, 3H, CH3), 3.84 (s, 3H, OCH3), 6.46 (d, 3J = 16.0 Hz, 1H), 6.92 (d, 3J = 8.8 Hz, 2H), 7.53 (d, 3J = 8.8 Hz, 2H), 7.60 (dd, 3J = 8.0 Hz, 3J = 8.0 Hz, 1H), 7.82 (d, 3J = 16.0 Hz, 1H), 8.20 (dm, 3J = 8.0 Hz, 1H), 8.29 (dm, 3J = 8.0 Hz, 1H), 8.56 (dd, 4J = 2.0 Hz, 4J = 1.6 Hz, 1H); 13C NMR (100.5 MHz, CDCl3) δ: 14.4 (CH3), 55.4 (OCH3), 112.4 (CH), 114.4 (2C, CH), 122.1 (CH), 125.1 (CH), 126.9 (Cquat), 129.7 (CH), 130.1 (2C, CH), 132.9 (CH), 136.8 (Cquat), 146.5 (CH), 148.4 (Cquat), 160.5 (Cquat), 161.8 (Cquat), 164.6 (Cquat). Anal. Calcd for C18H16N2O5 (340.33): C, 63.63; H, 4.85; N, 8.19%. Found: C, 63.89; H, 4.87; N, 7.96%.
4.5.2 3-O-Methyl estra-1-3,5(10)-trien-17-one-17-oxime N-3,4-(E)-dimethoxycinnamate (13a)
As a colorless solid, mp 152–154 °C (isopropanol/diethyl ether); IR (KBr/cm−1) νmax 3061, 2930, 2864, 2837, 1734, 1627, 1597, 1513, 1465, 1420, 1307, 1266, 1236, 1162, 1124, 1021, 979, 863, 816 cm−1; 1H NMR (CDCl3, 400 MHz, CDCl3) δ: 0.83–2.91 (m, 15H), 1.05 (s, 3H, CH3), 2.91 (s, 3H, OCH3), 3.77 (s, 3H, OCH3), 3.92 (s, 3H, OCH3), 6.37 (d, 3J = 16.0 Hz, 1H), 6.63 (d, 4J = 2.8 Hz, 1H), 6.71 (dd, 3J = 8.8 Hz, 4J = 2.8 Hz, 1H), 6.87 (d, 3J = 8.4 Hz, 1H), 7.06 (d, 4J = 2.0 Hz), 7.13 (dd, 3J = 8.4 Hz, 4J = 2.0 Hz, 1H), 7.21 (d, 3J = 8.8 Hz, 1H), 7.71 (d, 3J = 16.0 Hz, 1H); 13C NMR (CDCl3, 100.5 MHz) δ: 17.1, 22.9, 26.1, 27.2, 27.3, 29.6, 33.7, 38.2, 43.8, 45.5, 52.8, 55.2 (OCH3), 55.9 (OCH3), 56.0 (OCH3), 109.7, 111.0, 111.5, 113.8, 113.9, 122.7, 126.4, 127.4, 132.1, 137.6, 145.4, 149.2, 151.2, 157.5, 165.1, 178.7 (Cquat, CO). Found: C, 73.69%; H, 7.37%; N, 2.73%. Calcd. for C30H35NO5 (489.60): C, 73.59%; H, 7.21%; N, 2.86%.
4.5.3 Fluoren-9-one oxime benzoate (13b)
As a yellow solid, mp 180–183 °C; IR (KBr) νmax: 1743, 1597, 1449, 1319, 1248, 1178, 1091, 1081, 1064, 1026, 975, 889, 785, 731, 711, 644 cm−1; 1H NMR (CDCl3, 400 MHz) δ: 7.30–7.69 (m, 9H), 8.01 (d, 3J = 7.6 Hz, 1H), 8.21 (d, 3J = 7.2 Hz, 2H), 8.29 (d, 3J = 7.6 Hz, 1H); 13C NMR (CDCl3, 100.5 MHz) δ: 120.1 (CH), 120.4 (CH), 123.6 (CH), 128.5 (CH), 128.6 (CH), 128.8 (2C, CH), 128.9 (Cquat), 129.9 (3C, CH), 130.1 (Cquat), 131.7 (CH), 132.6 (CH), 133.6 (CH), 134.5 (Cquat), 141.1 (Cquat), 142.8 (Cquat), 159.1 (Cquat), 164.2 (Cquat). Anal. Calcd for C20H13NO2. (299.32) 0.2H2O: C, 79.30%; H, 4.46%, N 4.62%. Found: C, 79.26%; H, 4.34%; N, 4.64%.
4.5.4 Fluoren-9-one oxime cinnamate (13c)
As a pale yellow solid; mp 126–128 °C; IR (KBr) νmax: 3029, 1746, 1634, 1449, 1308, 1116, 951, 788, 761, 733, 646 cm−1; 1H NMR (CDCl3, 400 MHz) δ: 6.74 (d, 3J = 16.0 Hz, 1H), 7.28–7.66 (m, 11H), 7.97 (md, 1H), 7.98 (d, 3J = 16.0 Hz, 1H), 8.31 (md, 1H); 13C NMR (CDCl3, 100.5 MHz) δ: 114.9 (CH), 120.1 (CH), 120.3 (CH), 123.5 (CH), 128.4 (3C, CH), 128.5 (CH), 129.0 (2C, CH), 130.1 (CH), 130.2 (Cquat), 130.9 (CH), 131.7 (CH), 132.6 (CH), 134.1 (Cquat), 134.5 (Cquat), 141.2 (Cquat), 142.6 (Cquat), 147.3 (CH), 158.4 (Cquat), 164.6 (Cquat, CO). Anal. Calcd for C22H15NO2 (325.36): C, 81.21%; H, 4.65%; N, 4.30%. Found: C, 81.49%; H, 4.70%; N, 4.36%.
4.5.5 3-Nitroacetophenone oxime benzoate (13d)
As a colorless solid, mp 139–141 °C; IR (KBr) νmax: 1746, 1530, 1349, 1246, 1057, 1021, 931, 776, 738, 707, 676, 670 cm−1; 1H NMR (400 MHz, CDCl3) δ: 2.59 (s, 3H, CH3), 7.49–7.53 (m, 2H), 7.61–7.63 (m, 2H), 8.13 (d, 3J = 7.6 Hz, 2H), 8.26 (d, 3J = 7.6 Hz, 1H), 8.32 (dd, 3J = 7.6 Hz, 4J = 1.2 Hz, 1H), 8.61 (s, 1H); 13C NMR (100.5 MHz, CDCl3) δ: 14.6 (CH3), 122.1 (CH), 125.3 (CH), 128.7 (2C, CH), 129.7 (3C, 2 CH, Cquat), 129.7 (CH), 132.9 (CH), 133.6 (CH), 136.6 (Cquat), 148.4 (Cquat), 161.3 (Cquat), 163.4 (Cquat). Anal. Calcd for C15H12N2O4. (284.27): C, 63.38%; H, 4.25%, N 9.85%. Found: C, 63.36%; H, 4.27%; N, 9.88%.
4.5.6 6-Methoxy-1-tetralone oxime 3,4-dimethoxycinnamate (13f)
As a pale pink solid; mp 148–150 °C; IR (KBr) νmax: 3002, 2975, 2837, 2833, 1724, 1630, 1616, 1600, 1514, 1459, 1423, 1353, 1258, 1160, 1124, 1071, 1028, 988, 938, 877, 816, 767, 598 cm−1; 1H NMR (CDCl3, 200 MHz) δ: 1.89 (m, 2H), 2.75 (dd, 3J = 6.0 Hz, 3J = 6.0 Hz, 2H), 2.92 (dd, 3J = 6.0 Hz, 3J = 6.0 Hz, 2H), 3.81 (s, 3H, OCH3), 3.92 (s, 6H, 2 OCH3), 6.45 (d, 1H, 3J = 16.0 Hz), 6.66 (br s, 1H), 6.76–7.17 (m, 3H), 7.08 (br s, 1H), 7.74 (d, 1H, 3J = 16.0 Hz), 8.16 (d, 1H, 3J = 8.6 Hz); 13C NMR (50.3 MHz, CDCl3) δ: 21.4 (CH2), 25.6 (CH2), 29.9 (CH2), 55.3 (OCH3), 55.9 (OCH3), 56.0 (OCH3), 109.7 (CH), 111.0 (CH), 113.0 (2C, CH), 113.6 (Cquat), 121.7 (Cquat), 122.8 (CH), 127.4 (Cquat), 127.6 (CH), 142.8 (CH), 145.6 (CH), 149.2 (Cquat), 151.3 (Cquat), 161.5 (Cquat), 165.0 (Cquat, CO). Anal. Calcd for C22H23NO5. (381.42): C, 69.28%; H, 6.08%, N 3.67%. Found: C, 69.41%; H, 5.95%; N, 3.73%.
4.5.7 2,5-Dimethoxyacetophenone oxime 4-nitrobenzoate (13g)
As yellow needles; mp 141–145 °C; IR (KBr) νmax: 3109, 2992, 2969, 2937, 2836, 1741, 1606, 1527, 1499, 1468, 1410, 1349, 1269, 1224, 1088, 1067, 1040, 1011, 857, 713 cm−1; 1H NMR (CDCl3, 400 MHz) 2.48 (s, 3H, CH3), 3.79 (s, 3H, OCH3), 3.81 (s, 3H, OCH3), 7.00 (s, 1H), 7.88 (d, 3J = 8.8 Hz, 1H), 8.18 (d, 3J = 8.8 Hz, 1H), 8.29 (d, 3J = 9.2 Hz, 2H), 8.34 (d, 3J = 9.2 Hz, 2H); 13C NMR (CDCl3, 100.5 MHz) δ: 17.8 (CH3), 56.0 (OCH3), 56.1 (OCH3), 112.4 (CH), 115.3 (CH), 116.9 (CH), 123.7 (2C, CH), 125.2 (Cquat), 130.7 (2C, CH), 134.8 (Cquat), 150.7 (Cquat), 151.7, (Cquat), 162.0 (Cquat), 164.7 (Cquat), 166.8 (Cquat). Anal. Calcd for C17H16N2O6. (422.43): C, 59.30%; H, 4.68%, N 8.14%. Found: C, 59.62%; H, 4.81%; N, 8.18%.
- - Preparation of the single crystal of 13g:
Single crystals of compound 13g were obtained by very slow evaporation (5 days) in a mixture of CH2Cl2/diethyl ether/hexane at room temperature.
- - X-ray crystallography of 13g:
A single crystal of the title compound with a dimension of 0.75 × 0.33 × 0.10 mm was chosen for X-ray diffraction study. The data were collected on an Oxford Diffraction SuperNova, diffractometer with an Atlas detector. A Mo-micro-focus sealed-tube X-ray-source was used. The data were collected at 100 K.
Acknowledgements
Ms. M. Al Azani thanks the UAEU for a PhD scholarship.