

1 Introduction
Vanadium–metal oxide catalysts are very interesting materials widely used in the selective oxidation of light alkanes also known as oxidative dehydrogenation (ODH) process [1]. In recent years, numerous studies were devoted to the effect of the metal oxide (Al2O3, Nb2O5, SiO2, TiO2, ZrO2, or MgOSiO2) [2–6] on the physicochemical properties and catalytic behavior of these materials. It has been found through the various characterization techniques that the nature and the environment of the vanadium species on the catalyst surface strongly depend on the structure and acid–base character of the metal oxide. This suggests that there is a synergy effect developed between the vanadium and the metal oxide, generating different acid–base and redox surface properties, which may have a direct impact on the orientation of the catalytic reaction. The magnesium oxide (MgO) is one of the most used because its combination with vanadium gives rise to VMgO mixed oxides, which have proved their high reactivity in the ODH reactions [7–9]. Three mixed phases, Mg3V2O8, α-Mg2V2O7, and MgV2O6 can be formed in these catalysts depending on the vanadium content, the preparation method and the calcination temperature [10–12]. The structural and textural properties of the VMgO mixed oxides strongly affect their catalytic properties. However, contradictory results have been reported about the active phase responsible for the formation of alkenes. Most of the studies report that magnesium orthovanadate (Mg3(VO4)2) is the active phase and that the isolated tetrahedral species (VO4) present in this phase are responsible for the activation of the CH bond of the alkane [13,14]. Recently, other researchers found that the catalytic performances of Mg3V2O8 were improved when other phases such as MgO and α-Mg2V2O7 were present; this was assigned to the interactions between the different phases [15]. The formation of V2O5 crystallites with VO double bonds mainly favors the complete oxidation and the production of oxygenated compounds [5,16]. Therefore, the most suitable VMgO catalysts for the olefins' production are those containing isolated tetrahedral species (VO4) and without V2O5 crystallites, because it has been concluded after a broad study of these literature results that the vanadium content could determine the type of metal oxide species present on the surface. Besides, the preparation procedure can influence the morphology of the catalysts giving phases that differ in the activities, according to some work in the literature [5].
The aim of the present work is to study the physicochemical properties of two series of VMgO mixed oxides obtained using impregnation (IMP) and sol–gel (SG) methods as a function of the preparation method, the vanadium loading, and the calcination temperature, to establish a correlation between these different parameters. This work reports also the evaluation and the comparison of the catalytic properties of the elaborated VMgO samples in the selective oxidation of n-butane according to the structural, textural, and surface properties.
2 Materials and methods
2.1 Preparation of the materials
VMgO catalysts with two different vanadium loadings, 30 and 60 wt % of V2O5 (with respectively molar ratio MgO/V2O5 = 10.5 and 3), were prepared using SG synthesis and wet IMP route.
2.1.1 Sol-gel method
Ammonium metavanadate (NH4VO3, Aldrich) was dissolved in water at 80 °C and an equimolar amount of citric acid (p.a., Merck) was added to this solution. During this step, citric acid acted as a weak reducer turning the vanadium solution initially yellow/orange (V5+) to dark blue (V4+) [17,18]. Similarly, an equimolar solution of magnesium nitrate (Mg(NO3)2·6H2O, Aldrich) and citric acid was prepared by addition of citric acid to the magnesium nitrate solution heated at 80 °C. The vanadium/citric acid solution was added dropwise at 80 °C and under vigorous stirring to the magnesium/citric acid solution. No precipitation occurs. The solution was kept at 80 °C until a highly viscous gel was formed by water evaporation. The green color is ascribable to the simultaneous presence of V5+ and V4+ [11,12]. The gel (dark blue) was dried at 140 °C for 15 h. Heating NH4NO3 being a dangerous manipulation, the necessary precautions were taken during the preparation process (only a small amount of the catalyst is prepared in a single operation). Low densities foams, of various shades of green depending on the vanadium content, were formed after this step.
2.1.2 Impregnation method (IMP)
An appropriate amount of MgO (Fluka, 98%) was added to a basic aqueous solution of NH4VO3 (pH = 11, T = 70 °C). The mixture was stirred for 1 h. The resulting suspension was evaporated to dryness at 85 °C, followed by further drying at 110 °C for 2 h.
The dried powders (SG and IMP) were crushed and calcined at 550 or 650 °C for 6 h (5 °C/min) to obtain the final catalysts denoted as x-SG-T and x-IMP-T, where x (30% or 60%) is the percentage weight of V2O5, SG (or IMP) stands for the preparation procedure, and T (550 or 650 °C) is the temperature of calcination. For comparison, pure V2O5 was obtained from the calcination of NH4VO3 at 600 °C for 4 h (5 °C/min).
2.2 Characterization techniques
X-ray diffraction (XRD) analysis was carried out using a theta–theta D8 Advance (Bruker) powder diffractometer with Cu Kα radiation (0.154 nm) operated at 30 kV and 30 mA, and equipped with a 1 D LynxEye detector. The LynxEye detector was set to a 3° opening and the scanning range was 10°–80° by steps of 0.01°.
Fourier transform infrared spectra were recorded at room temperature using a SHIMADZU 8400S spectrometer. The experiments were performed on thin wafers obtained by pressing a mixture of the catalyst with KBr (ratio, 1:100).
The elemental composition of the catalysts was determined by X-ray fluorescence (XRF) spectroscopy using a XEPOS spectrometer (X-rays Energy Pro Operating Software, AMETEK instrument). Quantitative values are obtained using calibration curves from standard samples for each element.
The Brunauer–Emmett–Teller (BET) surface areas were determined by measuring nitrogen physisorption isotherms at 77 K using a BelSorp Max apparatus. Before measurement, samples were outgassed under vacuum at 300 °C for 6 h.
The acid properties of the calcined samples were measured by the NH3–temperature-programmed desorption (NH3-TPD) analysis using a Micromeritics AutoChem 2910 apparatus. The sample (200 mg) was pretreated at 350 °C under a flow of argon (20 mL/min) for 30 min, then cooled to 80 °C, and subjected to a flow of 5% NH3 in helium (20 mL/min) for 30 min. The excess of NH3 was removed under a flow of helium (20 mL/min) for 30 min. Ammonia desorption was carried out by heating the sample from 80 to 850 °C (7 °C/min), under He flow (20 mL/min). The amount of adsorbed ammonia (NH3) and the temperatures at which desorption takes place have been recorded using a thermal conductivity detector (TCD) coupled to a mass spectrometer (GSD320) to discriminate between desorbed NH3 and desorbed water, as a dehydration or dehydroxylation of the sample during the TPD experiment cannot be excluded.
Samples were characterized by scanning electron microscopy (SEM) using a Hitachi (SU-70) SEM FEG ultra-high resolution microscope at 3.4 mm working distance with an accelerating voltage of 1 kV. Sample preparation consisted simply in depositing the powder on a carbon tape (no metallization).
Transmission electron microscopy (TEM) and energy dispersive X-ray spectroscopy (EDS) investigations were performed using a JEOL 2010 microscope operating at 200 kV with a LaB6 filament and equipped with an Orius CCD camera (Gatan) and a Princeton Gamma-Tech (PGT) dectector (EDS spot as low as 20 nm diameter). Samples were characterized either as powders or as microtomed slices (the powdered samples were embedded in epoxy resin and cut into thin slices (ca. 50 nm) with an ultramicrotome tool equipped with a diamond knife). The Mg/V ratios have been calculated based on the relative intensity of the EDS signals associated with V and Mg K lines. Sample 60-SG-550, which has been shown by XRD to be made of an almost pure Mg3V2O8 phase and showed a very homogeneous distribution of V and Mg, has been used to calibrate the data (calibration has been set so as to obtain a Mg/V ratio of 1.5 for this sample).
X-ray photoelectron spectroscopy (XPS) spectra were carried out on an Οmicron X-ray photoelectron spectrometer. XPS measurements were performed in ultra-high vacuum conditions (1 × 10−8 Pa) at room temperature with an ESCA+ (electron spectroscopy for chemical analysis) spectrometer using Al Kα (1486.6 eV) as exciting energy. The characteristic X-ray irradiation source works at 280 W (20 mA, 14 kV) with a 1 mm2 spot size. After data collection, the binding energies were calibrated with respect to the binding energy (BE) of C1s, set at 284.7 eV. Spectra processing was carried out using the Casa XPS software package.
2.3 Catalytic test
The catalytic activity of the prepared materials was tested in the ODH of n-butane using oxygen of air as an oxidant. The catalytic test was carried out at atmospheric pressure in a conventional fixed bed setup consisting of a mixing chamber, a glass reactor (internal diameter of 15 mm), and an on-line gas chromatograph (Agilent 7890A equipped with TCD (thermal conductivity) and FID (flame ionization) detectors). Hydrocarbons were analyzed with the FID whereas the products of deep oxidation were characterized with the TCD. The sample (200 mg) was tested at a reaction temperature of 500 °C, measured using a thermocouple placed in a thermowell centered in the catalyst bed and with a feed ratio air/n-butane = 5. The total flow rate used was 30 mL/min. Blank measurements, without catalyst, were performed to check the reactor inertia and the influence of the reaction temperature. No butane conversion was recorded up to 450 °C. From this temperature, a very low butane conversion was observed (<1%), which reached about 3% at 500 °C with high production of cracking products (methane, ethane, and propane). Catalytic properties were evaluated in terms of conversion of n-butane and selectivity of product (i), and were calculated as follows:
3 Results and discussion
3.1 Spectroscopic characterizations
The XRD patterns obtained for the two series of IMP- and SG-calcined catalysts are presented in Fig. 1. For 30-SG-550 and 30-IMP-550 samples (with low V2O5 loading), MgO periclase (ICDD 45-0946) is the main phase observed at 2θ = 49.2° and 62.3°, independently of the preparation method. However, the size of the MgO crystallites depends strongly on the preparation procedure and very small MgO domains are obtained using the SG procedure. Moreover, other weak and broad signals are also observed, whose positions (2θ = 29.5° and 35.2°) roughly match with little-crystallized Mg3(VO4)2 phase (ICDD 73-0207). In the 60-SG-550 and 60-IMP-550 catalysts (high V2O5 loading), three phases are observed for the sample prepared by IMP: MgO, Mg3V2O8, and α-Mg2V2O7 (ICDD 31-0816), whereas for the sample prepared by the SG method, the diffractogram shows the formation of the almost pure phase Mg3V2O8 (in agreement with V/Mg molar ratio in this sample). For each sample, the same crystallographic phases are observed after calcination at 650 °C, and the only modification upon high temperature calcination is a narrowing of the XRD peaks (this result could be because of an increase in the size of the crystalline domains).
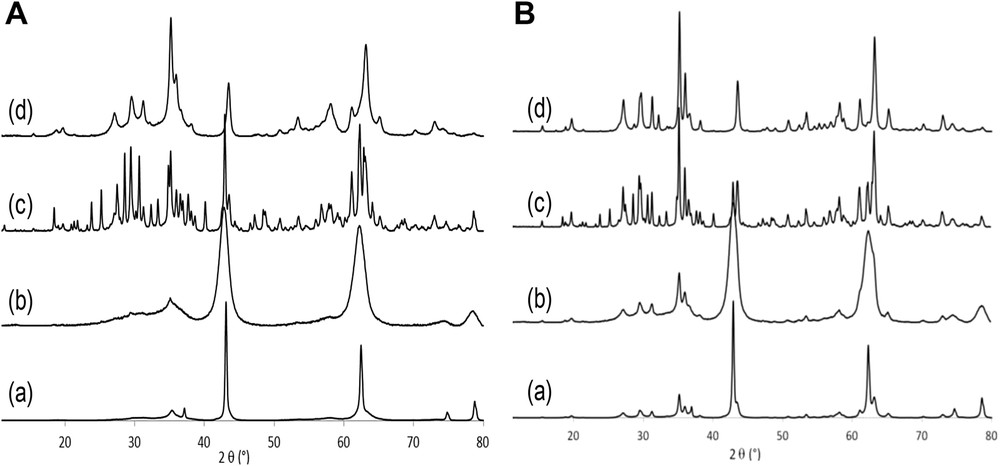
X-ray diffractograms of (a) 30-IMP-T; (b) 30-SG-T; (c) 60-IMP-T; and (d) 60-SG-T samples. (A) At T = 550 °C and (B) at T = 650 °C for 6 h.
These XRD results suggest that the crystalline structure of the samples depends on the preparation method and the vanadium content, whereas calcination temperature essentially controls the size of the crystalline domains. Moreover, whatever the calcination temperature, the loading, and the preparation method, crystalline V2O5 is never observed.
To evaluate quantitatively the weight fraction of each crystalline phase and the size of the crystalline domains, a refinement of the patterns has been performed. The obtained results are regrouped in Table 1. It can be concluded from the calculated data that the intimacy of the magnesium and vanadium precursors in the material before calcination clearly plays a major role in the final crystalline composition after calcination: for the samples prepared by the SG method (i.e., the samples for which a close proximity of the magnesium and vanadium precursors in the starting material is expected) the Mg3V2O8 phase is formed under milder conditions compared to the samples prepared by the IMP method. Moreover, the size of Mg3V2O8 crystalline domains is significantly smaller for the samples prepared by SG. Whatever the preparation procedure, calcination at 650 °C induces a significant increase in the size of the crystallographic domains.
Structural parameters of IMP and SG samples.
| Preparation method | Catalyst | Phase composition | % Phase (XRD)a | Average crystallite size (nm)b |
| Impregnation (IMP) | 30-IMP-550 | MgO Mg3V2O8 | 78 22 | 38 6 |
| 30-IMP-650 | MgO Mg3V2O8 | 72 28 | 48 25 | |
| 60-IMP-550 | MgO Mg3V2O8 α-Mg2V2O7 | 9 48 44 | – 28 57 | |
| 60-IMP-650 | MgO Mg3V2O8 α-Mg2V2O7 | 7 77 16 | – 46 56 | |
| Sol-gel (SG) | 30-SG-550 | MgO Mg3V2O8 | 95 5 | 6 4 |
| 30-SG-650 | MgO Mg3V2O8 | 51 49 | 10 18 | |
| 60-SG-550 | MgO Mg3V2O8 | 12 88 | 19 15 | |
| 60-SG-650 | Mg3V2O8 α-Mg2V2O7 | >99 <1 | 34 |
a Percentage phase calculated from the refinement of XRD patterns using Quanto software.
b Crystallite size calculated using the Debye–Scherrer formula.
The infrared spectra (600–1200 cm−1 region) of the IMP and SG samples are shown in Fig. 2. Similar spectra were obtained for the two calcinations temperatures (550 and 650 °C). The attributions of the Fourier transform infrared bands were effectuated according to Busca et al. [19] results. For the 60-IMP-550, the bands at 966, 821, and 665 cm−1 indicate the presence of α-Mg2V2O7, whereas the bands at 918 and 686 cm−1 are characteristic of the presence of Mg3V2O8. The identification of these two phases in this sample is in agreement with the XRD results. More surprisingly the spectrum of 60-SG-550 also shows a weak shoulder at 966 cm−1, which could indicate that this sample also contains a very low fraction of α-Mg2V2O7 that could not be detected by XRD. Moreover, the larger peak width of Mg3V2O8 bands (e.g., the band at 918 cm−1) could be because of the small size of the Mg3V2O8 particles in this sample. In the case of 30-IMP-550 sample, the bands are merged in three broad and ill-defined bands that impede a clear identification of the phases present in this sample; the shoulder at 966 cm−1 could nevertheless indicate the presence of a small fraction of α-Mg2V2O7 as found by XRD. For the 30-SG-550 sample, the large and broad peak is characteristic of the presence of the little-crystallized Mg3V2O8 phase in this sample. It is important to emphasize here that, for all the four samples, the absence of the V2O5 is clearly established by the absence of the band at 1020 cm−1 characteristic of the VO stretching [10,16], which is consistent with the XRD results.
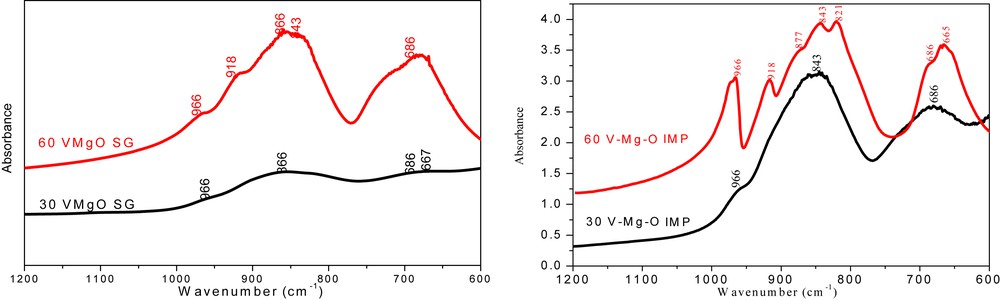
Infrared spectra of the VMgO mixed oxides SG (left) and IMP (right) after calcination at 550 °C for 6 h.
3.2 Composition, textural, and acid properties
The vanadium percentage and BET surface areas of the calcined samples (before catalytic test) are listed in Table 2. The measured vanadium loading (obtained by XRF analysis) is in good agreement with the expected values (fixed in the preparation step). For the IMP samples, the BET surface areas (SBET) of the samples are higher than that of the starting MgO (SBET = 7.8 m2/g). Whatever the vanadium loading and calcination temperature, the SG method leads to a higher specific surface area in comparison with the IMP process (SBET30-SG > SBET30-IMP, and SBET60-SG > SBET60-IMP). For the two series of materials (IMP and SG), the specific surface area decreased with increasing the vanadium content and the calcination temperature. From the N2-sorption isotherms (Fig. 3), it is important to note that the 30-SG-550 sample presents a fairly large porosity compared to the other samples.
Vanadium loadings (measured by XRF) and BET results.
| Preparation method | Sample | % V2O5 | SBET (m2/g) |
| – | Commercial MgO | 00 | 7.8 |
| Calcination of NH4VO3 | V2O5 | 100 | 1.3 |
| Impregnation (IMP) | 30-IMP-550 | 30.8 | 45.5 |
| 30-IMP-650 | 31.8 | 24.5 | |
| 60-IMP-550 | 63.8 | 24 | |
| 60-IMP-650 | 65.2 | 5.4 | |
| Sol–gel (SG) | 30-SG-550 | 33.8 | 116 |
| 30-SG-650 | 36 | 73.4 | |
| 60-SG-550 | 62.2 | 34 | |
| 60-SG-650 | 60.7 | 14 |

N2-sorption isotherms of IMP and SG samples calcined at 550 °C.
Table 3 presents the acidity results of VMgO mixed oxides calcined at 550 °C, obtained via the NH3-TPD analysis. These results indicate that the four samples only have a very small amount of acid sites between 8 and 16 μmol NH3/g. Based on the low temperature of NH3 desorption on these samples (ca. 250 °C), this acidity is rather weak. These results are very different from those reported previously by Elkhalifa and Friedrich [20] who concluded from the same technique, the presence of a very large amount (more than 1.5 mmol g−1) of strong acid sites (desorption peaks at 450 and 650 °C) in MgO and VMgO mixed oxides. Their results are probably erroneous as such a high density of strong acid sites is unlikely (especially for MgO whose basic character is well documented). One likely reason for their observation of two desorption signals at high temperature is the desorption of other species (such as H2O or CO2) that cannot be distinguished from NH3 using a TCD. Furthermore, the preparation method seems to influence the acidic properties of our VMgO catalysts, as the two SG samples exhibited a twice weaker density of acid sites compared to the IMP ones.
Acidity of VMgO samples calcined at 550 °C.
| Sample | Total acidity (μmol NH3/g) | Approximate desorption temperature (°C) |
| 30-IMP-550 | 15.6 | 250 |
| 60-IMP-550 | 16.3 | 240 |
| 30-SG-550 | 8.4 | 250 |
| 60-SG-550 | 8.8 | 260 |
3.3 Surface characterizations
The SG and IMP samples after calcination at 550 °C have been further characterized using SEM and TEM techniques. Analysis of the SEM images of the four samples (Fig. 3) reveals that relatively large grains (at least 5 μm) are formed for all compositions and preparation procedures. However, the structure of the particles that compose these grains differs significantly: for the IMP samples the grains seems to be made of an assembly of crumbled sheets, especially for the lowest loading (30-IMP-550, Fig. 4a), whereas the grains the SG samples prepared appear at first sight to have a smooth surface. A closer inspection of these samples (see insets in Fig. 4b and d) reveals the presence of smaller particles.

SEM images of (a) 30-IMP-550; (b) 30-SG-550; (c) 60-IMP-550; and (d) 60-SG-550.
TEM measurements coupled with EDS analysis have also been performed (see Fig. 5).

TEM images of (a) 30-IMP-550; (b) 30-SG-550; (c) 60-IMP-550; (d) 60-SG-550. The circled areas represent the zones on which an EDS analysis has been performed.
The “crumpled-sheet” structure of the particles of sample 30-IPM-550 is confirmed by the TEM analysis (Fig. 5a). Surprisingly, this sample appears very homogeneous based on TEM analysis, although XRD has revealed the presence of two crystalline phases (MgO and Mg3V2O8 in 3:1 ratio). The Mg/V ratio measured from EDS on different zones gives a value of 2.4 ± 0.1, which corresponds to a very homogeneous distribution of Mg and V, but intriguingly this value is well below the value that is expected from the chemical composition (5.3). To further investigate this point, sample 30-IMP-550 has been characterized as ultramicrotomed slices (see Fig. 6a) to get an insight inside the catalysts grains. This measurement confirms, as expected from the XRD analysis, the presence of two phases: a vanadium rich phase with a Mg/V = 2.3 ± 0.1, which shows the same crumpled-sheet structure observed with regular TEM, and a MgO rich phase (Mg/V > 80) in the form of dense MgO nodules. This phase could probably not be observed by regular TEM (and EDS) as it is located at the center of the catalyst grains. The Mg/V ratio of 2.3 measured on the crumpled sheet, although slightly higher than the value expected for a pure Mg3V2O8 phase (Mg/V = 1.5), is consistent with the presence of this phase. TEM and EDS analysis of sample 60-IMP-550 confirms the complexity of this sample. It reveals the presence of MgO-rich domains (Mg/V > 18) and vanadium-rich domains (Mg/V ≈ 1) that could correspond to the α-Mg2V2O7 phase observed by XRD. The Mg3V2O8 phase, whose presence has been ascertained by XRD, was not clearly identified here, but it is not surprising as TEM can only be used on a very limited part of this sample because of the large size of the grains. Coming back to the SEM analysis and by analogy with sample 30-IMP-550, the Mg3V2O8 phase could be associated with the crumpled-sheet domains of the grains (see inset in Fig. 5c).

TEM of microtomed slices of (a) 30-IMP-550 and (b) 30-SG-550. The circled areas represent the zones on which an EDS analysis has been performed.
TEM analysis of sample 30-SG-550 (Fig. 5b) confirms that the grains are made of small particles (see inset). The sample is rather homogeneous with a Mg/V ratio of 5 ± 0.1. Nevertheless, according to the XRD analysis (and chemical composition of this sample) one would expect some heterogeneity in this sample that contains both MgO and Mg3V2O8 phases. To explain this point, this sample has also been analyzed as microtomed slices. TEM images of microtomed slices coupled with EDS analysis confirm (Fig. 6b) the presence of two phases: a Mg-rich phase (Mg/V = 5.5 ± 2), likely the MgO phase detected by XRD, and a V-rich phase (Mg/V = 1.2 ± 0.2), likely the Mg3V2O8 phase. Both phases appear to be composed of aggregates of smaller particles. For the 60-SG-550, the TEM image (Fig. 5d) confirms that the micrometer-sized grains of this sample (as seen on the SEM image of Fig. 4d) are made of smaller particles with a fairly homogeneous size of about 50–100 nm (see inset). This is significantly larger than the particle size of 15 nm determined based on the XRD analysis, indicating a probable contribution of strain to the broadening of the XRD peaks. The EDS analysis of this sample shows a high homogeneity (1.5 ± 0.1), in agreement with the XRD analysis of this sample (about 88% of Mg3V2O8).
The surface composition of the IMP and SG mixed oxides calcined at 550 °C (before catalytic test) was investigated by XPS measurements. The BE corresponding to V2p3/2, Mg1s, and O1s and the atomic ratios Mg/V are reported in Table 4.
XPS quantification results of IMP and SG mixed oxides calcined at 550 °C.
| Sample | Binding energy (eV) | Mg/V ratio | |||
| V2p3/2 | O1s | ||||
| OI | OII | OIII | |||
| 30-IMP-550 | V4+: 515.69 (9.9%) V5+: 517.08 (90.1%) | 529.92 (51.13%) | 531.56 (41.54%) | 533.00 (7.33%) | 6.3 |
| 30-SG-550 | V5+: 517.27 (100%) | 529.88 (17.3%) | 531.79 (60.47%) | 533.36 (22.24%) | 20.7 |
| 60-IMP-550 | V5+:517.08 (100%) | 530.13 (40.57%) | 531.71 (42.35%) | 533.09 (17.08%) | 4.0 |
| 60-SG-550 | V5+: 517.27 (100%) | 529.87 (59.32%) | 530.95 (31.14%) | 532.29 (9.54%) | 3.2 |
XPS results showed only one V2p3/2 peak (at 517.08–517.27 eV) in all calcined samples except in the case of 30-IMP-550 in which two peaks were observed at 517.08 (90.1%) and 515.69 eV (9.9%). The values of the main peak (at higher BE) indicate that vanadium is present at the surface in the V5+ oxidation state [21,22]. The second component of V2p3/2 (with low BE) observed only on the spectrum of 30-IMP-550 is associated with the presence of a small amount of reduced V4+ species, which is possibly because of the influence of the ultra-high vacuum during the XPS analysis of this sample [15]. The photopeak O1s could be decomposed into three components with maxima at 530.00 ± 0.15, 531.4 ± 0.4, and 532.8 ± 0.5 eV, associated with three distinct oxygen surface species [23,24]. The first peak (OI) with the lowest BE was assigned to lattice O2− according to the literature data [17,25,26]. The second and third peaks (labeled OII and OIII) were associated with the adsorbed oxygenated species: the second peak was assigned to adsorbed CO2 or carbonate, whereas the third one was associated with adsorbed O− and O2− species [27,28]. As the second peak (OII) is shifted to about +1.4 eV with reference to the first one, another possible assignment for this peak could be surface hydroxyls. Indeed Coustet and Jupille [29] studied the XPS signature of surface hydroxyls of aluminum and magnesium oxide and observed a peak shift of +1.6 eV (with regards to the O1s peak of the lattice oxygen). In the literature [22,30,31], the selective oxidation is classically associated with nucleophilic oxygen O2−, whereas electrophilic oxygen (O− and O2−) species are associated with deep oxidation, but other authors [32] suggest that OIII (O− and O2−) species can participate in selective oxidation. The relative contributions of the OI, OII, and OIII peaks are not the same for our catalysts. Indeed, the surface area of each oxygen peak varies according to the vanadium loading and the preparation method. The highest contribution of lattice oxygen (OI) is obtained for the 60-SG-550 sample; for this method the proportion of surface lattice oxygen increased with increasing vanadium composition from 30% to 60%. Inversely, with the IMP method, the proportion of lattice oxygen decreases with the vanadium loading. Compared to the three other catalysts, the 30-SG-550 contains a much lower fraction of lattice oxygen. This is likely because of the much higher surface area (and hence higher surface to volume ratio) of this solid compared to the three other samples (see Table 3).
For all samples, the quantitative XPS analysis revealed a noticeable enrichment of the catalytic surface with magnesium. Indeed the Mg/V ratio measured by XPS is significantly higher than the bulk Mg/V ratios, which can be calculated based on the chemical composition (Mg/V = 5.2 for the 30-SG and 30-IMP samples and Mg/V = 1.5 for the 60-SG and 60-IMP samples). This may suggest that an important part of vanadium is integrated in the bulk vanadate crystallites, which leaded to a diminution of the total amount of vanadium at the surface. Among the four samples, 30-SG-550 sample exhibits, by far, the highest magnesium surface enrichment (Mg/V = 20.7, compared to a bulk Mg/V = 5.2), this ratio is much larger than that obtained for the 30-IMP-550 solid (Mg/V = 6.3, i.e., a value close to the bulk Mg/V ratio). This large difference could be related to the high surface area of 30-SG-550 solid (116 m2/g compared with 45.5 m2/g for the 30-IMP-550 sample). Indeed, if one assumes that the MgO phase is the main contributor of this large surface area, an exacerbated density of surface Mg (compared with surface V) is expected for this sample.
3.4 Catalytic properties
The results of reactivity of pure V2O5 and the VMgO mixed oxides during the partial oxidation of butane (T = 500 °C, air/n-butane = 5, and total flow rate = 30 mL/min) are presented in Fig. 7 and Table 5. The catalytic performances are compared based on the preparation method, the vanadium content, and the calcination temperature. For all catalytic experiments, the obtained products were butenes (1-butene, cis-2-butene, trans-2-butene, and isobutene), cracking products (methane, ethane, and propane), COx (CO + CO2), isobutane, and butadiene (in trace).
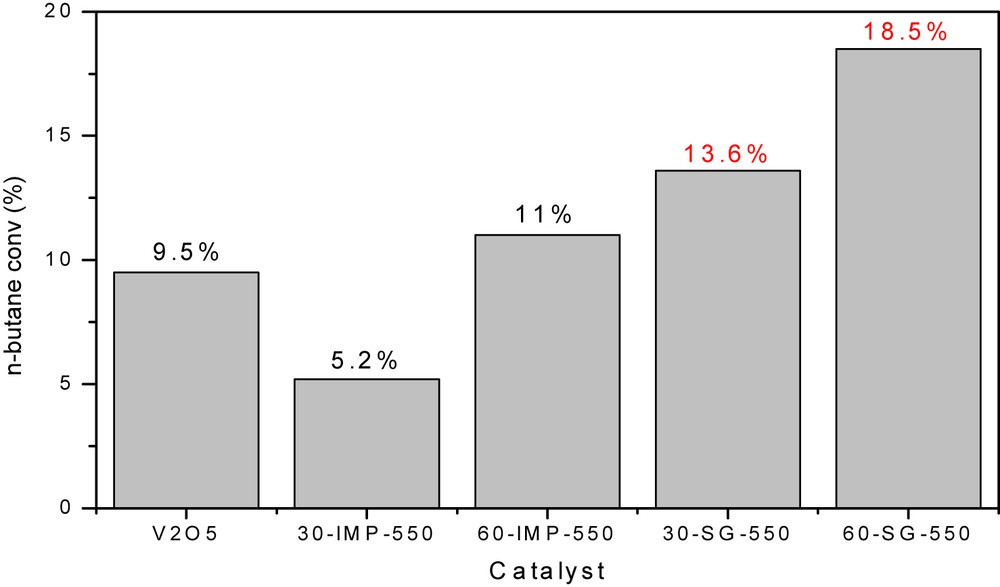
Evolution of the n-butane conversion as a function of the vanadium loading and the preparation procedure.
Influence of the preparation method, vanadium loading, and calcination temperature on the catalytic performances of the VMgO catalysts: Tr = 500 °C, air/n-butane = 5, and total flow rate = 30 mL/min.
| Catalyst | C4H10 Conversion (%) | Selectivities (%) | ||||
| Cracking | COx | Butenes | Isobutane | Butadiene | ||
| V2O5 | 9.5 | 14.5 | 71.5 | 12.2 | 1.8 | 0.0 |
| 30-IMP-550 | 5.2 | 36.7 | 11.5 | 45.6 | 6.2 | 0.0 |
| 30-IMP-650 | 5.1 | 40.2 | 10.4 | 45.4 | 4 | 0.0 |
| 30-SG-550 | 13.6 | 7.9 | 15.1 | 72.6 | 4.3 | 0.1 |
| 30-SG-650 | 8.4 | 13.1 | 13.9 | 68.8 | 4.2 | 0.0 |
| 60-IMP-550 | 11.0 | 35.1 | 5.2 | 49.9 | 8.5 | 1.3 |
| 60-IMP-650 | 14.9 | 47 | 2.2 | 43.2 | 6.8 | 0.8 |
| 60-SG-550 | 18.5 | 33.6 | 3.1 | 60.3 | 3 | 0.0 |
| 60-SG-650 | 9.0 | 36.9 | 17.9 | 42.7 | 2.5 | 00 |
Vanadium pentoxide is, despite its very low specific surface area (SBET = 1.3 m2/g), active in the partial oxidation of butane (up to 9% of butane conversion). In our experimental conditions, V2O5 oxide showed a high selectivity for COx products (up to 71%) (Table 5), which can be related to the VO double bond of V2O5 crystallites, as suggested in the literature [11,16,34]. Indeed, it was reported in these works that the presence of VO double bond favors the deep oxidation.
For the VMgO mixed oxides (see Fig. 7), the obtained results clearly confirmed that the SG method leads to better catalytic activity compared to the IMP one. Indeed, the conversion of butane decreases in the following order: 60-SG-T > 30-SG-T > 60-IMP-T > 30-IMP-T. These results revealed that there is an obvious correlation between the catalytic activity, the preparation procedure, and the vanadium content. In fact, the good catalytic activity of SG samples can be attributed to their high specific surface area, their small crystallite size, and the homogeneity of particles distribution compared to the IMP samples. We note also that, whatever the method of preparation used, the activity in butane conversion of VMgO catalysts increases with increasing the vanadium content (from 30% to 60%), which can be assigned to the increase in the proportion of Mg3(VO4)2 active phase as shown by the XRD quantitative results (see Table 1). In fact, most of studies reported that the CH bond of the adsorbed alkane is activated by the surface oxygen of the (VO4) species [36–38]. According to Blasco et al. [39], these active species are present in the bulk of orthovanadate phase (Mg3(VO4)2) or present on the catalyst surface as distorted (VO4) tetrahedral environments.
In terms of selectivity to butene isomers (Fig. 8), the production of the 1-butene is the most important whereas that of the isobutene is the lowest, regardless of the preparation method and the vanadium loading. The selectivity order of the four isomers is as follows: 1-butene > trans-2-butene > cis-2-butene > isobutene. However, it can be noted that for the IMP method, the selectivities to 1-butene and trans-2-butene are close, whereas for the SG procedure, the production of 1-butene is about twice that of trans-2-butene. Comparing the total selectivity to butenes (Table 5), the SG-550 catalysts showed the best selectivities estimated at 72.6% and 60.3% for 30-SG-550 and 60-SG-550, respectively, against 45.6 and 49.9% for 30-IMP-550 and 60-IMP-550 samples. The highest selectivity of the SG solid can be attributed primarily to their higher surface area (see Table 3), smaller particles size (see TEM analysis), and their adequate porosity compared to their homologue IMP samples. Moreover, the acid–base character of the catalytic surface generated by the nature of surface oxygen species also contributes in the orientation of the selectivity. According to NH3-TPD measurements (see Table 3), it has been found that the SG solids have a lower surface acidity compared to the IMP solids. This very weak acidity favors the rapid desorption of olefins from the surface, which reduce the probability of deep oxidation reactions [40,41]. However, the highest selectivity of 30-SG-550 toward butenes (72.6%) may seem in contradiction with the small proportion of lattice oxygen O2− in this sample (estimated by XPS, see Table 4) compared with the 60-SG-550. As already mentioned, nucleophilic O2− species are usually considered as the active centers for the activation of CH bond and formation of olefins, whereas the electrophilic species OIII (O− and O2−) are usually associated with deep oxidation. This smaller proportion of O2− must rather be regarded as an artifact linked to the high surface area and the high proportion of MgO phase in this sample (as testified with the high fractions of adsorbed oxygen species OII and OIII). This high selectivity to butenes obtained with the 30-SG-550 sample can then be explained by the fact that under reaction conditions (in the presence of O2 gas) and at relatively high temperatures, the electrophilic oxygen species present on the catalytic surface are removed and there is a regeneration of lattice oxygen O2− species [42,43], according to a redox mechanism (O2(ads) is adsorbed oxygen on the catalytic surface):
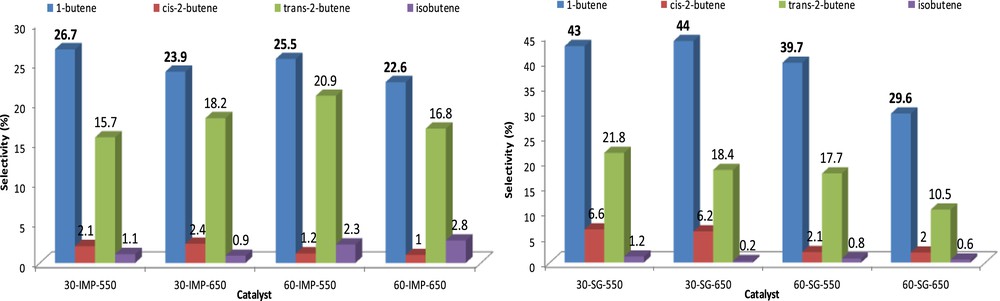
Evolution of each isomer selectivity as a function of the vanadium content, the calcination temperature, and the preparation method: Tr=500 °C, air/n-butane = 5, and total flow rate = 30 mL/min.
The influence of the calcination temperature on catalytic properties of our VMgO mixed oxides was also examined using the same reaction conditions (Table 5 and Fig. 8). In the case of IMP solids, the catalytic activity remains low and weakly affected by the calcination temperature. The same result was observed for the selectivity to butenes, we note, for example, that for the 60-IMP-T catalyst, a higher calcination temperature leads to a slight decrease in the butenes selectivity (from 49.9% to 43.2%) in favor of the selectivity of cracking products (35.1%–47%). A different behavior is observed for the SG solids for which an increase in the calcination temperature (from 550 to 650 °C) leads to a decrease in the butane conversion especially for 60-SG-T catalyst (from 18.5% to 9%) and to a significant change in product distribution with a sharp increase in the selectivity to COx products (from 3.1% to 17.9%) at the expense of butenes selectivity (from 60.3% to 42.7%). This degradation of catalytic performances upon increasing the calcination temperature is probably partly due to the decrease in the specific surface area and the agglomeration of particles, but it could also be related to the absence of MgO and/or α-Mg2(V2O7) (the Mg3(VO4)2 phase represents more than 99%). As reported in the literature [9], the simultaneous presence of these phases improves the catalytic properties of VMgO mixed oxides, whereas the presence of a pure Mg3V2O8 phase decreases the selectivity of oxidehydrogenation products. This has been interpreted by a synergy effect between the coexisting phases. Moreover, the increase in the calcination temperature does not change the selectivity order of the different isomers, so the 1-butene remains the main isomer formed over our samples.
According to these obtained results, it may be concluded that the high catalytic activity and selectivity toward butenes can be enhanced by choosing the preparation method that leads to low crystallite size and high specific surface area, the nature of crystalline phases present in the catalyst and the nature of oxygen surface species, which can be controlled by the operating reaction conditions. These results are in good agreement with the literature data that reported the key role of these parameters in the orientation of the selective oxidation process [36,37].
4 Conclusions
In the present work, VMgO mixed oxides with different vanadium loadings were prepared using wet IMP and SG methods. Structural and textural properties of these catalysts were found very sensitive to the vanadium loading, the preparation method, and the calcination temperature. The SG solids differ from IMP solids by the higher concentration of Mg3V2O8 orthovanadate in the catalyst, smaller size of the particles, greater specific surface area, and a better homogeneity with uniform particles distribution. These samples all have very weak acidic properties especially the SG samples, which favor the desorption of butenes from the catalytic surface. The best catalytic performances in the selective oxidation of butane were obtained over the SG catalysts. These good performances of the SG samples (higher n-butane conversion and selectivity to butenes) were in good agreement with their low crystallite size, high specific surface area, good homogeneity, and lower surface acidity compared to the IMP samples. The electrophilic oxygen species adsorbed on the catalytic surface (O− and O2−) can also be responsible for the oxidehydrogenation process and this in the presence of O2 gas atmosphere. The increase in the calcination temperature strongly influenced the catalytic properties of the VMgO mixed oxides with high vanadium loading (60%), which can be assigned to a change in the phase composition of the catalyst at Tc = 650 °C. It was confirmed that the presence of almost a pure Mg3V2O8 phase (absence of MgO and α-Mg2V2O7) is not beneficial for the selective oxidation of butane to butenes.