

1 Introduction
Metal oxide nanostructures are intensively studied [1]. The behavior of nanostructured oxide is governed by both the oxidation process and the size reduction process [2]. The electronic processes of oxidation destroy the initially metallic bonds to create new kinds of bonds with specific properties, such as polarization, localization, and transportation of charge, which determine the behavior of the oxides to vary from their parent metals [3]. Oxygen interaction with atoms of metals relates to the technical processes of corrosion, bulk oxidation, heterogeneous catalysis, and so forth [4]. For examining nanostructures, many concepts developed in both molecular chemistry and solid-state physics have to be considered. It is possible to develop a “top-down” theory on the behavior of nanosystems starting from a solid and confining it to a limited size. Another chemical-like “bottom-up” approach is to start with a molecular system and expand its size [2].
Nanostructured materials are advantageous in offering huge surface to volume ratios, favorable transport properties, altered physical properties, and confinement effects resulting from the nanoscale dimensions, and have been extensively studied for several applications. The major domains of heterogeneous catalysis applied industrially concern:
- • oil refining, energy, and transport;
- • bulk chemicals;
- • polymers and materials and detergents and textiles;
- • fine chemicals, pharmaceutical and medical chemicals, and food and feed;
- • plant design/engineering and realization, catalyst design, and subsequent development of catalysts and of catalytic processes;
- • commercial production of catalysts in sufficient quantities;
- • monitoring and control of chemical reactions and plant operations; and
- • environmental issues.
Among these domains, this review describes recent developments of heterogeneous catalysis in ring opening of methylcyclopentane (MCP), selective partial oxidation reactions, and biodiesel on metal oxide catalysts.
2 Catalytic selective oxidation of hydrocarbons
This is an attractive field of reactions because the reaction produces valuable chemicals such as alcohols, aldehydes, acids, and so forth, and accordingly ca. 25% of the chemicals and intermediates are industrially produced by the catalytic oxidation processes [5,6]. Partial oxidation reactions have been largely studied since the 1960s both in the industry, particularly by researchers at SOHIO Co., and at the academia level, using mixed metal oxides in the form of the so-called multicomponent catalysts.
For instance, in the SOHIO process of propene oxidation to acrolein, the base catalyst was bismuth molybdate (α-phase Bi2MoO3), but Fe and comolybdates were added for better performances. This was assigned to epitaxial growth between both types of molybdates [7], which favors electron transfer through the solid catalysts as required in the redox mechanism, Mars and van Krevelen, and demonstrated by electrical conductivity measurements.
Recently, the selective catalytic oxidation of light alkanes has attracted much attention due to the shale gas revolution and the concern about petroleum [8,9]. Success in the reactions, however, has been limited so far despite tremendous efforts. This is due to the difficulty of the light alkane catalytic selective oxidations, which is derived from the chemical properties of light alkanes. Generally, CH bonds of hydrocarbons are the most affective parameters for its reactivity in catalytic oxidation, and in the case of light alkanes, severe reaction condition like high reaction temperature is usually required because of their low reactivity due to the strong CH bond. The severe reaction condition, however, often causes undesirable side reactions and consecutive reactions of the desired products, which makes it difficult to achieve high selectivity toward the desired products.
For a selective oxidation reaction, the main reaction mechanism is the Mars and van Krevelen mechanism, schematized below, which involves cations in variable oxidation states, such as Cr, Cu, Fe, Mo, V, and so forth.
| 2[CatO] + R-CH → 2[Cat] + R-CO + H2O |
| 2[Cat] + O2 (gas) → 2[CatO] |
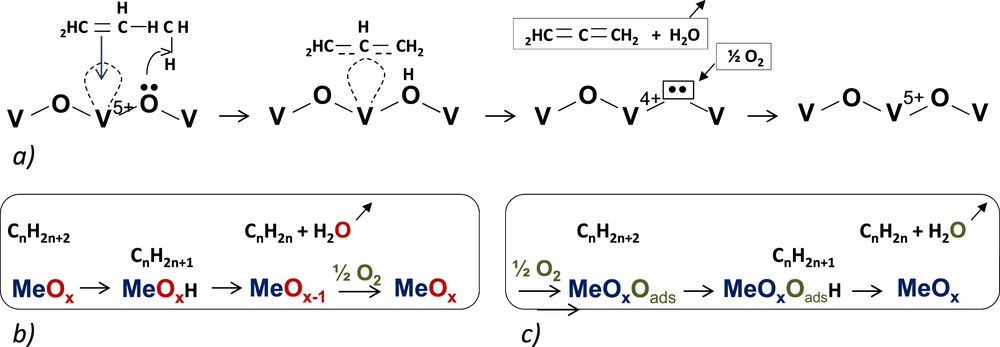
Simplified schemes of (a) propene adsorption OVO and formation of a π-allyl complex intermediate; (b) case of an alkane; and (c) effect of adsorbed oxygen.
Mixed metal oxides constitute the main catalyst families for selective partial oxidation of organic compounds and have been used industrially since very long (1960s) in the gas-phase partial oxidation of hydrocarbons to yield oxygenates (acids, alcohols, aminoacids, aldehydes, etc.) [10]. In most cases, the reaction mechanism is of redox-type so-called Mars and van Krevelen mechanism [11] described previously. Representative reactions are as follows: (1) oxidative dehydrogenationof short-chain alkanes (from C1 to C6) [12]; (2) propene partial oxidation/ammoxidation on bismuth molybdate/antimonate–based catalysts; (3) n-butane direct oxidation to maleic anhydride on VPO-type catalysts (vanadyl pyrophosphate (VO)2P2O7-based catalyst); and (4) propane direct oxidation/ammoxidation on mixed MoVTe(Sb)NbO catalysts [13]. The actual trends in partial oxidation processes are especially focusing on the use of biomass and light alkanes (C1 to C4), as a raw material.
As a general rule of selective partial oxidation reactions involving a redox mechanism, the concept of “7 pillars”, as proposed by Grasselli [14], has to be satisfied: (1) nature of lattice oxygen anions: nucleophilic (selective) rather than electrophilic, the later giving total oxidation. (2) Redox properties of the metal oxide (removal of lattice oxygen anions and its rapid reinsertion). (3) Host structure (permits redox mechanism to take place without structure collapse). (4) Phase cooperation in the multicomponent catalyst or supported catalyst (epitaxial growth and synergetic effects). (5) Multifunctionality (e.g., α-H abstraction and O/NH insertion). (6) Active site isolation (to avoid a too high lattice O surface mobility and thus overoxidation). (7) MO bond strength to be neither too weak (total oxidation) nor too strong (inactivity) (Sabatier principle).
Facile exchange of surface oxygens exists within domains (represented by arrows) but not between domains. Site isolation established between domains by picket fence of pyrophosphate groups posing oxygen diffusion barriers. Schematic representation of a surface structure of a type of polytype of (VO)2P2O7 is shown in Fig. 2. The arrows represent the facile pathways for surface oxygen mobility. This illustrates the site isolation principle by surface P2O7 entity groups, which constitute a barrier to oxygen diffusion. Taken from Fig. 13 of Ref. [14].
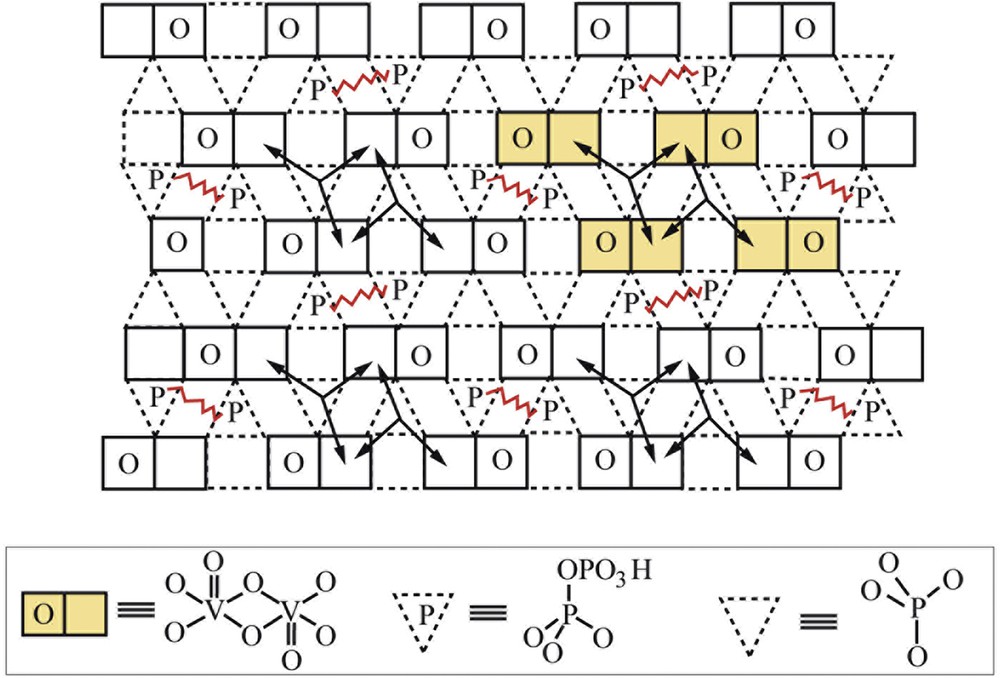
Site isolation by structure and composition: schematic of the (VO)2P2O7 surface structure.
Another example is presented in Fig. 3 for propane ammoxidation to acrylonitrile on MoVTeNbO mixed oxide.
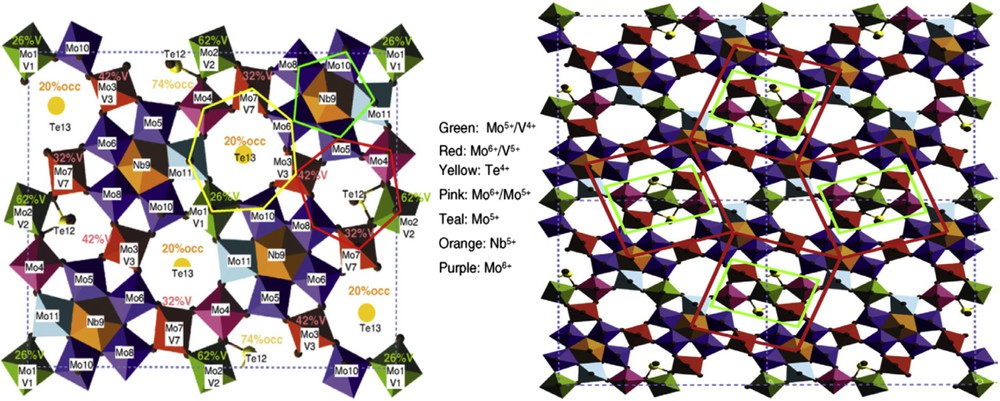
Bulk ab planes of M1 phase (right). Refined orthogonal structure had the space group Pba2, formula unit Mo7.8V1.2NbTe0.94O28.9 (M1), and lattice parameters, a = 2.1134 nm, b = 2.6658 nm, and c = 0.40146 nm with z = 4, from Ref. [15]. Active site: 2 × 2 unit cell structure model of M1 in [001] projection showing four isolated catalytically active centers (left). The active sites are isolated from each other by four Nb bipyramids that are surrounded by five MO6 octahedra. From Fig. 10 in Ref. [16].
Catalytic partial oxidation reactions are more efficient on mixed metal oxides based mainly on W, V, and Mo cations attached to other redox cations such as Sb, Nb, Te, Ta, as MO6 octahedra assembled, as shown in Fig. 4, and which are composed of several phases acting in synergy one with respect to the other(s) and leading to very complex chemical compositions as, for instance, in the case of the mixture of M1 and M2 phases in the MoVTeNbO catalyst used for propane ammoxidation/oxidation to acrylonitrile/acrylic acid. As the selectivity depends on the nature of surface oxygen species (O2−) associated with cation(s) Men+, with their bond energy MeO (of the lattice or with adsorbed O) and with their distribution on the surface, it is reasonable to conclude that oxidation reactions are demanding, that is, are structure sensitive. They are more demanding when the number of electrons involved in the reaction is high. For instance, if many catalysts are able to transform propane to propylene (reaction with 2e−) only three (CuO, BiMoO, and SnSbO) are able to transform propylene to acrolein (a 4e− reaction) and only one ((VO)2P2O7) for butane to maleic anhydride (a 14e− reaction) or (VTiO) for o-xylene oxidation to phthalic anhydride (a 12e− reaction).
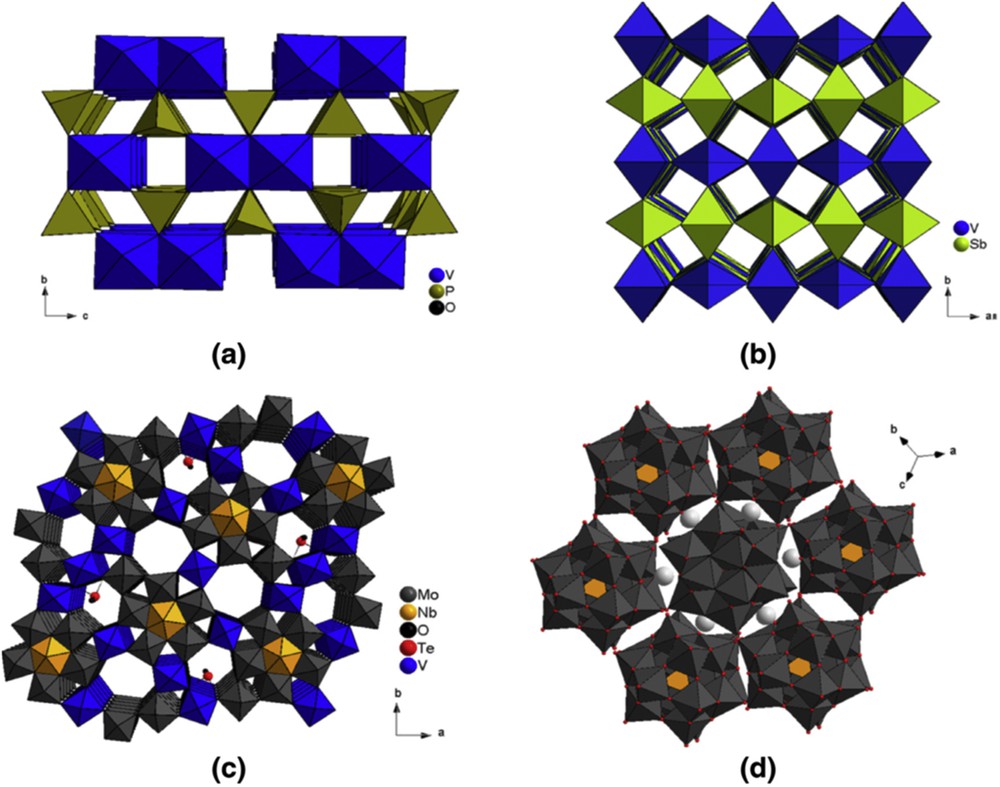
Structures (composed of MO6 octahedra) of the most studied catalysts in alkane partial oxidation: (a) vanadyl pyrophosphate (VPO), (b) VSbO rutile phase, (c) M1 phase, MoVTe(Sb)NbO, and (d) Keggin molybdophosphoric acid.
It has been proposed that the selectivity in olefins or oxygenated products depends on conversion extent (Fig. 5) and atomic arrangements around the active transition metal (Mo or V) as shown in Fig. 6.
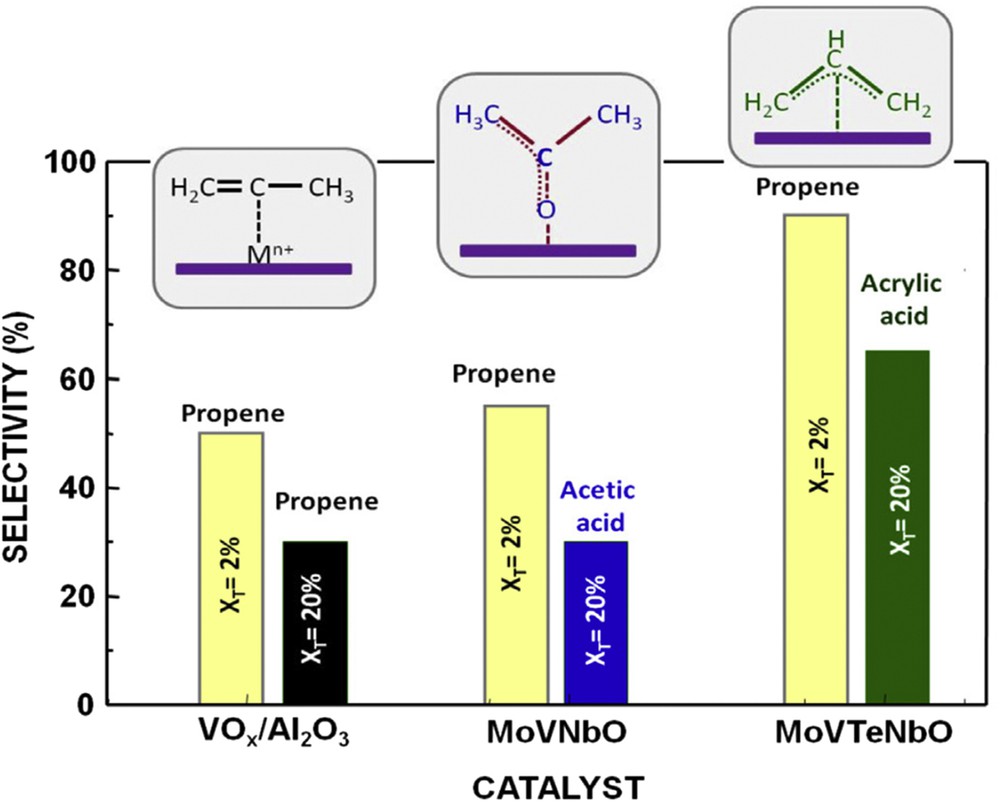
Selectivity to the main partial oxidation product achieved at low conversion (XT = 2%) and high conversion (XT = 20%) during propane oxidation at 400 °C on VOx/Al2O3; MoVNb mixed oxides calcined in air at 450 °C (amorphous); MoVTeNb mixed oxides heat-treated at 600 °C in N2 presenting M1 phase (MoVTeNbO). The main adsorbed species observed during the adsorption of propylene on each catalyst is shown. From Fig. 24.4 in Ref. [17].
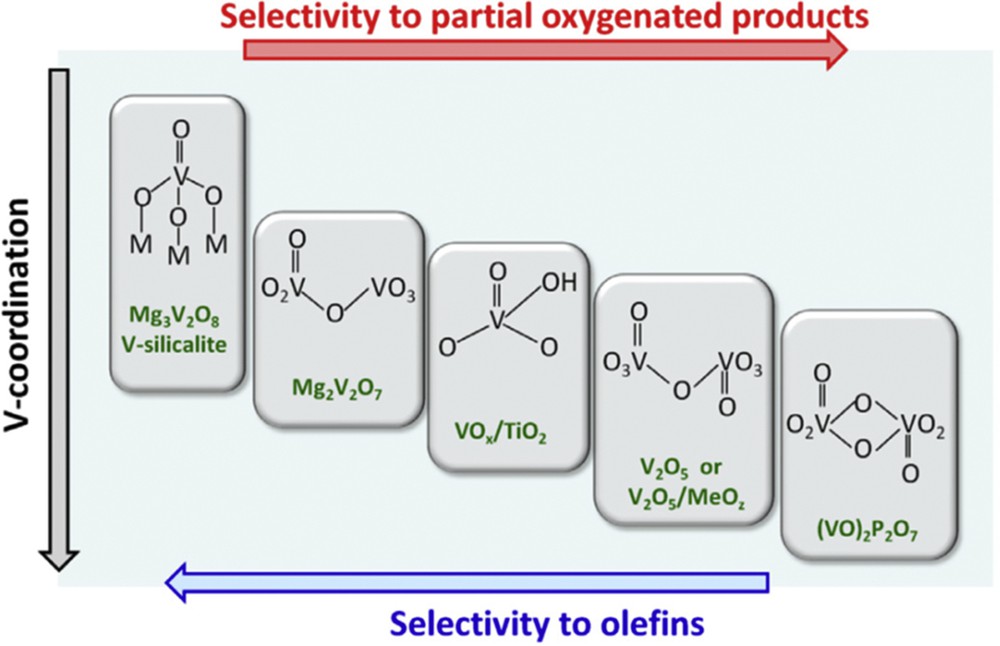
Importance of the V structure in V catalysts for H abstraction and O insertion. From Fig. 24.3 in Ref. [17].
3 Biodiesel and biocatalysis
In a post-petroleum era, catalysis will be central to overcoming the engineering and scientific barriers to economically feasible routes to alternative source of both energy and chemicals, notably bioderived and solar-mediated via artificial photosynthesis [18]. Although many alternative sources of renewable energy have the potential to meet future demands for stationary power generation, biomass offers the most readily implemented, low-cost solution to a drop-in transportation fuel for blending with/replacing conventional diesel [18,19] via the biorefinery concept. First-generation biofuels derived from edible plant materials received much criticism over the attendant competition between land usage for fuel crops versus traditional agricultural cultivation [18,20]. To be considered sustainable, second-generation bio-based fuels and chemicals are sought that use biomass sourced from nonedible components of crops, such as stems, leaves, and husks or cellulose from agricultural or forestry waste. Alternative nonfood crops such as switchgrass or Jatropha curcas [21], which require minimal cultivation and do not compete with traditional arable land or drive deforestation, are other potential candidate biofuel feedstocks. There is also growing interest in extracting bio-oils from aquatic biomass, which can yield 80–180 times the annual volume of oil per hectare than that obtained from plants [18,22]. Biodiesel is a clean burning and biodegradable fuel which, when derived from nonfood plant or algal oils or animal fats, is viewed as a viable alternative (or additive) to current petroleum-derived diesel [23]. Commercial biodiesel is currently synthesized via liquid base–catalyzed transesterification of C14eC20 triacylglyceride components of lipids with C1eC2 alcohols [18] into fatty acid methyl esters, which constitute biodiesel, alongside glycerol as a potentially valuable byproduct [18,24].
Biodiesel synthesis and use of glycerol are also important topics [19], although only one process has been industrialized for biodiesel production on ZnOAl2O3 catalysts. Note that the solid acids and bases applicable to biomass conversions should be water-tolerant catalysts [20]. Currently, acrolein is produced primarily by the gas-phase oxidation of propylene. However, as the use of propylene derivatives, particularly polymers, has recently experienced rapid growth and because this trend is likely to continue in the foreseeable future, the availability and cost of propylene is going to increase. One approach to substitute propylene relies on the use of new raw materials. For instance, catalysts and processes have been developed for the direct conversion of glycerol to acrolein [21]. However, such processes and catalysts face two major difficulties: (1) there is a more or less rapid deactivation, and (2) their selectivity is capped at an upper limit of less than 80%. There is thus a need for the development of novel routes for the production of acrolein. One approach may involve the conversion of allyl alcohol, which could be produced in an initial step by fermentation, through the use of processes such as the dehydration of 1,3-propanediol, or the dehydration/oxidation of glycerol [22].
Mesoporous oxides grafted with sulfonic acid group [18,25,26] surface coatings have been examined for biodiesel synthesis [27]. Phenyl and propylsulfonic acid SBA-15 catalysts are attractive materials in palmitic acid esterification [28]. Phenylsulfonic acid–functionalized silica are reportedly more active than their corresponding propyl analogues, in line with their respective acid strengths, but are more difficult to prepare. The KIT-6 mesoporous silica offers one solution to improve the in-pore accessibility of sulfonic acid sites. Superior molecular transport within the interconnected cubic structure of KIT-6 has been shown to facilitate biomolecule immobilization [18,29]. A family of pore-expanded propylsulfonic acid KIT-6 analogues, PrSO3H-KIT-6, prepared via MPTS grafting and subsequent oxidation, have been screened for FFA esterification with methanol under mild conditions. Such a conventionally prepared material exhibited 40% and 70% TOF enhancements for propanoic and hexanoic acid esterification, respectively, over an analogous PrSO3H–SBA-15 catalyst of comparable (5 nm) pore diameter, attributed to faster mesopore diffusion. However, pore accessibility remained rate limiting for esterification of the longer chain lauric and palmitic acids. Pore expansion of the KIT-6 mesopores up to 7 nm via hydrothermal aging doubled the resulting TOFs for lauric and palmitic acid esterification with respect to an unexpanded PrSO3H–SBA-15. It should be noted that the absolute conversions of FFAs over such tailored, inorganic solid acid catalysts remain significantly lower than those for commercial polymer alternatives, which possess superior acid site densities. Propylsulfonic acid–functionalized SBA-15 (SBA-15–PrSO3H) has also been evaluated for oleic acid esterification with methanol [18,30], showing good stability in boiling water, with the mesopore structure allowing facile diffusion of the acid to active sites. This catalyst exhibited similar activity to phenylethylsulfonic acid–functionalized silica gel and was superior to dry Amberlyst 15, reflecting the higher surface area and pore volume of the SBA-15–PrSO3H relative to the more strongly acidic phenylethyl mesoporous silica. The SBA-15–PrSO3H could be recycled by simple ethanol washing and drying at 80 °C, and maintained an esterification rate of 2.2 mmol min−1gcat−1. Simultaneous esterification and transesterification of vegetable oils with methanol has been performed with Ti-doped SBA-15 [18,31].
4 Ring-opening reactions
The worldwide depletion of oil reserves is expected to increase the future demand for diesel fuel. Concomitantly, environmental constraints necessitate the development of environmental friendly refining processes. With increasingly stringent environmental and economic regulations, the foremost challenge facing in all industries is to find greener processes with better atom efficiency [32–37]. In particular, the petrochemical industry has placed a strong research emphasis on the ring opening of naphthenic molecules over noble metal catalysts [38–43] to improve the cetane number of diesel fuels and to minimize harmful emissions. Diesel fuels can be improved by selective ring opening of naphthenic compounds after hydrogenation of aromatics compounds [44]. Taking into account the fact that each dearomatization reaction followed by a hydrogenation step leads to a cyclopentane, the understanding of MCP chemistry is very important. In this case, although MCP is not a component of diesel fuel, the ring opening of MCP must first be understood because it serves as a model molecule and provides the basis for understanding the behavior of all other molecules. The ring opening of MCP is one of the most-used model reactions for exploring the structural sensitivity of hydrocarbon conversion catalyzed by noble metals. Several works have been reported concerning the use of noble metals [38–43]. These catalysts have generated widespread interest because at low temperatures they exhibit the ability to promote the ring-opening reactions with atom efficiency and without unwanted side reactions such as cracking and enlargement. The distribution of these desired ring-opening products is governed by several factors, including the intrinsic nature of the metal [45], the platinum particle size [46–48], the interface length between the metal and support [49–51], the presence of carbonaceous residues [51–53], sulfiding [52], and the reaction conditions [50,51].
The ring-opening reaction of MCP can proceed by different mechanisms, which include associative [54] and dissociative [55] pathways. For the ring opening and cyclization of C5, a common adsorbed surface intermediate has been defined [46], which has sparked myriad discussions. Several structures have been proposed for this intermediate, including edgewise or flat geometries with associative or dissociative structures. The dissociative structure is a function of the degree of dehydrogenation of the surface intermediate. On Pt catalysts, a flat-lying intermediate with a low degree of dissociation has been suggested for selective ring opening [50,56]. This intermediate suggests that the rupture of the CeC bond in the proximity of the tertiary carbon atom was hindered [57]. A plausible explanation is based on the assumption that two metal atoms form the active ensemble. In this case, the chemisorption takes place by the rupture of the weakest CeH bond on the tertiary carbon atom, followed by CeC rupture on the neighboring metal atom. The presence of chemisorbed hydrogen is also necessary to prevent excessively deep dissociation. A flat-lying intermediate and an associative mechanism have also been suggested by Liberman [54], with hydrogen–metal ensembles attacking the chemisorbed ring without the dehydrogenation of the ensemble surface. Gault et al. [47,48] identified three different paths for the ring opening of MCP: dicarbene, π-adsorbed olefin, and metallocyclobutane intermediates. The dicarbene path occurs only at the unsubstituted secondary–secondary CeC bond [55]. In this case, the formation of 2-methylpentane (2-MP) and 3-methylpentane (3-MP) prevails in a nonstatistical distribution (selective mechanism), and these products were observed on Pt particles larger than 1.8 nm and with high coordination numbers [46]. This mechanism also prevails on Ir. The π-adsorbed olefin and metallocyclobutane paths occur at substituted CeC bonds, and the formation of 2-MP:3-MP:n-H occurs with a statistical distribution of 2:1:2 (nonselective mechanism). The π-adsorbed olefins require a flat adsorption of three neighboring carbon atoms, which interact with a single metal site on the catalyst surface. The formation of metallocyclobutane competes with the dicarbene path but exhibits a higher activation energy than that exhibited by the dicarbene mechanism. Gault et al. found that the relative importance of the selective and nonselective mechanisms was a function of the size of the Pt particles used. Other researchers, however, attempted to attribute the nonselective mechanism to the presence of adlineation sites on the metal–support boundary, whereas the selective mechanism was assigned to pure metallic sites [49,58]. The influence of particle size has been found to be different for reactions that involve Ir catalysts, which confirms that the nature of the ring-opening mechanism also depends on the catalytic metal [52]. Irrespective of the effects of particle size on Ir catalysts, the selective mechanism (dicarbene path) was found to prevail. Ir/SiO2 catalysts open the ring via a selective mechanism, whereas the Ir/Al2O3 catalyst opens the ring via a nonselective mechanism. In the latter case, the selectivity was found to be a support effect rather than a particle effect. However, the conversion of MCP has been studied on Pt/TiO2 [59–61]. A widespread view is that nonstoichiometric TiO2 supports provoke a surface migration of reduced TiOx species from the support to the metallic phase [62–64]. Thereafter, the catalytic behavior changes as a result of the decreased adsorption capacity of metals, the reduced catalytic activity and chemisorption capacity, and the changes in the selectivity of the MCP reaction [65]. Unique metal–support interactions between titanium and noble metals were observed for other reactions [66,67]. When Pt/TiO2 was reduced at 500 °C, the complete elimination of hydrogen and carbon monoxide chemisorption occurred; this result was associated with strong metal–support interactions (SMSIs). Otherwise, the extent of chemisorption elimination was strongly correlated with the reducibility of the support. In this case, the mechanism proposed is explained by increased d-orbital occupancy of the support, followed by covalent bond formation between support–metal ions and supported metal atoms. Moreover, theoretical studies [68] on surface clusters formed between Pt and the (TiO6)8− octahedron has indicated that covalent sharing between Pt and the support surface is indeed likely if one of the oxygen atoms is removed from the octahedron. The modification of chemisorptive and catalytic properties of dispersed metals by substrates, observed first by Schwab [69] and Solymosi [70], has found renewed interest since the discovery by Tauster et al. [62] of the so-called SMSI between Group-VIII noble metals and TiO2. Thereafter, the SMSI phenomenon was reported for other support–metal catalysts, including partially reducible oxides other than TiO2.
However, the conversion of MCP has been studied on PteIr/TiO2 bimetallic catalysts [71], because the coexistence of two metal sites is expected to modulate the peculiar behavior of such catalysts for catalytic reactions in a manner different from that of their two components. These catalysts exhibit high selectivity, activity, and stability as compared to their component pure metal particles. For the sake of argument, the bimetallic PteIr particles supported on γ-Al2O3 exhibited higher selectivity and a lower tendency for coke formation [71,72] as compared to a Pt catalyst. In the case of the catalysts that contain two metals, the specific behavior necessary for a desired catalytic reaction is tailored as a function of the distribution and topology of the two types of metal particles: (1) the two particles can be made bimetallic with a homogeneous composition by forming alloys; (2) the two metals may also exist as separate phases but in intimate contact via the surface segregation of one metal, in particular, the metal with the lower melting point, the smaller atomic radius, and the lower heat of vaporization is typically segregated on the surface [73–75]; and (3) metals may segregate into separate islands with each crystallite containing one of the respective metals.
The conversion of MCP on Pt/TiO2, Ir/TiO2, and PteIr/TiO2 catalysts, each prepared with low amounts of noble metals (0.5 wt %), was studied for the first time over the temperature range of 180–400 °C under hydrogen at atmospheric pressure [71]. The order of reactivity as a function of the temperature and total conversion rates was Ir/TiO2 at 180 °C > PteIr/TiO2 at 220 °C > Pt/TiO2 at 260 °C. All catalysts exhibited the ability to open the ring of MCP with atom efficiency. For the catalysts, a change in the reaction temperature provokes an alteration in the selectivity to ring opening of MCP in favor of selectivity to cracking reactions. The cracking products were formed in large amounts at high temperatures, with C1 compounds as the major products. Ir/TiO2 operates via the selective mechanism at low temperatures and via the nonselective mechanism at 220 °C due to the SMSI phenomenon. Pt/TiO2 operates via the nonselective mechanism, whereas the PteIr catalyst operates via the selective mechanism and shows Ir-like character. The synergy between PteIr as bimetallic particles was assessed by total conversion of MCP. The MCP ring-opening results indicated that the reaction takes place on Ir sites, which suggests that the bimetallic catalyst contains separate entities of the two metals. REP were absent on the Ir/TiO2 and PteIr/TiO2 catalysts and present on Pt/TiO2 only at high temperatures. Taken together, the results [71] revealed that the Ir/TiO2 catalyst was more active and was the most promising catalyst for the ring opening of MCP at low temperatures.
Despite the effectiveness of supported noble metals, which efficiently catalyze the conversion of MCP with atom efficiency, the high cost and limited availability of precious metals restrict their wide application. A practical solution is to find a clean and efficient heterogeneous catalyst to replace noble metal catalysts. It is cheaper to use base metal oxide catalysts because of the environmental and economic advantages, good catalytic performance, and the competitiveness with the noble metals. In this context, mesoporous materials appear to be good candidates for the conversion of hydrocarbons because of a large internal surface area, high thermal stability, and surface properties that create new opportunities for heterogeneous catalysis [76,77]. However, it is noteworthy that few reports are available on the catalytic properties of mesoporous materials for the conversion of MCP. Although the discovery of mesoporous materials has brought about the beginning of a new age in the field of material synthesis, these materials are known to be of limited use as catalysts because the catalytic functionalities are missing. These mesoporous materials can be functionalized by depositing heteroatoms onto the silica framework [78–80]. A promising approach for the creation of active sites is that of substituting heteroatoms for Si by one-pot synthesis, which creates active sites that enhance the activities of mesoporous solids and allow catalysis [78–80]. The non-noble metal oxides of molybdenum, tungsten, and iron appear to be efficient and environmental alternatives to the expensive noble metals. The results showed [80] that the efficiency of the catalyst is dependent on the density of active sites. Mo, Fe, and MoeFe catalysts have attracted significant interest because of their high efficiency as catalysts in environmentally important processes [81–87]. The efficiency of these catalysts is governed by the size of their oxide nanoparticles, which can be controlled by the dispersion on the support. A high dispersion leads to an increased number of catalytically active sites; however, the presence of strong particle–support interactions often reduces their activity [81–87]. In contrast, weak interactions with the support favor oxide particle agglomeration at elevated temperatures. Therefore, the strategy for optimizing the size of supported oxide nanoparticles to create uniform and well-dispersed supported transition metal oxides (Mo, Fe, MoeFe) for the conversion of MCP is to use the mesoporous support. The goal is to understand that the basic surface chemistry of these oxides in a reductive environment greatly lags behind that of noble metals. Iron is of high interest due to its wide range of applications in catalysis [83,88–90]. However, most studies of redox mesoporous catalysts published thus far dealt with oxidative environments. Despite the successful development of catalytically active Fe-containing mesoporous materials, little is known about their behavior in reductive environment and the influence of isomorphous substitution on their properties. It is known [91] that the isomorphous substitution of Fe3+ in the silicate framework leads to acid catalysts by creating SiOFe bonds and being the genesis of Brønsted acidity. To obtain a gradation of the acidity of the Fe–mesoporous samples, a challenging controlled-synthesis method was used to substitute iron for silicon atoms to obtain several Si/Fe ratios. Fe–TUD-1 catalysts with Si/Fe ratios of 85, 65, and 45 were prepared [92] via hydrothermal one-pot synthesis. The ability of these catalysts was tested by examining the conversion of the reaction of MCP with hydrogen at atmospheric pressure and temperatures between 200 and 400 °C. The active sites, tetrahedrally coordinated Fe and isolated atomic Fe sites, were responsible for the endocyclic CC bond rupture between substituted secondary–tertiary carbon atoms, whereas the small clusters serve as active sites for the successive CC bond rupture. The Fe content in the calcined samples is almost the same as the Fe content in the wet gel. This correlation indicates that most of the Fe was incorporated into the mesoporous TUD-1 by hydrothermal one-pot synthesis at 180 °C.
Isolated tetrahedral Fe3+ entities appear to be selective sites for n-H, whereas the small aggregates serve as active sites for the C1 cracking products, as previously proposed for Fe/KIT-6 prepared by a solid–solid method [85,92]. For completeness, the reactivity of isolated tetrahedrally/octahedrally coordinated Fe cations and small aggregates in the Fe–TUD-1 catalysts were evaluated in the conversion of MCP. The Fe–TUD-1 catalysts with different Si/Fe ratios were investigated for the first time [92]. The effect of the temperature on the activity was investigated in the temperature range of 200–400 °C for Si/Fe ratios between 85 and 45. Before the catalytic tests, all samples were reduced for 4 h in hydrogen at 500 °C [92]. All catalysts displayed an activity in the conversion of MCP [92]. Increasing the iron content of TUD-1 significantly enhanced the MCP conversion, suggesting that iron can function as the active sites in this reaction. For all catalysts, the activity increased with temperature. If evaluated at the same reaction temperature, the order of conversion (catalytic performance) of the samples was as follows: Fe–TUD-1, Si/Fe = 45 > Fe–TUD-1, Si/Fe = 65 > Fe–TUD-1, Si/Fe = 85. This order is in good agreement with the sequence of the density of active sites of these catalysts. n-H was the only species among the ring-opening products that was formed irrespective of the reaction temperature or the Fe loading [92]. The ring-opening reaction was dominant in the samples with Si/Fe ratios of 85 and 65, irrespective of the reaction temperature. These results suggest that the highly isolated, tetrahedrally coordinated Fe entities in the samples with Si/Fe ratios of 85 and 65 were responsible for the rupture of endocyclic CC bonds in MCP between the substituted secondary–tertiary carbon atoms [92]. The results were very interesting because a different behavior was observed when compared with noble metal catalysts. For example, Pt/Al2O3 catalysts can open the MCP ring by breaking the substituted or the unsubstituted CC bonds in MCP [55,57] to form 2-MP, 3-MP, and n-H. Ir/Al2O3 catalysts exhibit the tendency to break the endocyclic CC bonds in MCP between unsubstituted secondary–secondary carbon atoms, which only form 2-MP and 3-MP [57,88,93]. However, the ability of Ni/Al2O3 catalysts for the conversion of MCP must be noted [94]. An interesting work [95] reported the selective formation of n-H on noble metal catalysts, but the experimental conditions were different. The conditions in that work were 10–50 torr under H2. Logically, the 1–6 cyclization is favored under these conditions, as already reported [96]. For high Fe loading on the sample with a Si/Fe ratio of 45, the ring-opening reaction with the selective formation of n-H prevailed at the low temperature of 200 °C. Only the highly isolated, tetrahedrally coordinated Fe entities are favorable sites for the endocyclic rupture of CC bonds at the substituted position. For the other reaction temperature, the cracking C1 product was dominant. Taking into account the UV–vis results, isolated tetrahedrally/octahedrally coordinated Fe species and small Fe clusters seem to coexist in this sample [92].
This result is consistent with the suggestion that the tetrahedrally coordinated and atomically isolated sites are responsible for the rupture of endocyclic CC bonds between substituted secondary–tertiary carbon atoms. It must be noted that the ring-opening reaction is dominant but not exclusive, suggesting that only a part of the Fe ions are involved in the ring-opening reaction and that the mononuclear species of Fe formed in these cases can be responsible for the successive rupture of CC bonds (which will be discussed in Section 3). A decrease in the n-H selectivity with the temperature is a common feature observed in all samples [92]. However, these results were significantly better than what was observed for W mesoporous catalysts [80], for which no products derived from ring-opening reactions were observed under the same conditions or for Fe/KIT-6, Mo/KIT-6, and FeMo/KIT-6 prepared by solid–solid methods in which the cracking products prevailed [85]. This reaction of ring opening of MCP, also called “methylcyclopentane hydrogenolysis”, was extensively studied in the 1970s and was exclusively performed on noble metals, mainly on alumina-supported platinum catalysts with various mean metallic particle sizes. As a consequence of such studies, it was noted that in the skeletal rearrangements of hydrocarbons, two types of isomerization mechanisms occur: the bond shift and the cyclic mechanism. To distinguish between them, the 13C tracer technique was used—Scheme 1 [55]. It was noticed that the cyclic mechanism was strongly favored on small Pt aggregates with diameters less than 2 nm and that three types of hydrogenolysis take place: a nonselective one, occurring on highly dispersed catalysts and corresponding to an equal chance of breaking any CC bond of the ring; a selective one, allowing only the rupture of bisecondary CC bonds; and a “partially selective” mechanism competing with the selective one on catalysts of low dispersion. For the results, which have only n-H formed, we have to assume the reaction of a metallocyclobutane intermediate with the exocyclic methyl, as already suggested [97]. After the metallocyclobutane exocyclic intermediate is created, the CC bond rupture of the ring leads to the formation of a carbene–olefin, which gives a π-adsorbed vinyl group via a 1,2 hydride shift, and n-hexane is formed.
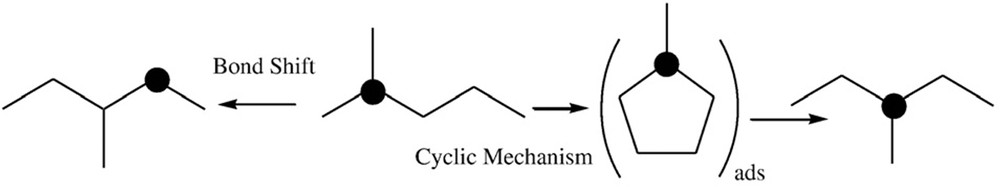
Ref. [92].
In addition, using deuterium exchange reactions on Fe films, it was observed that the vinylic hydrogen is very mobile and is very easily exchanged [98]. Such observations on Fe catalysts reinforce the proposed mechanism, which may take place on the Fe–TUD-1—Scheme 2. Such a mechanism can explain the predominant n-H formation, the aromatization, and the extensive cracking reactions when the conversion or the temperature is increased.
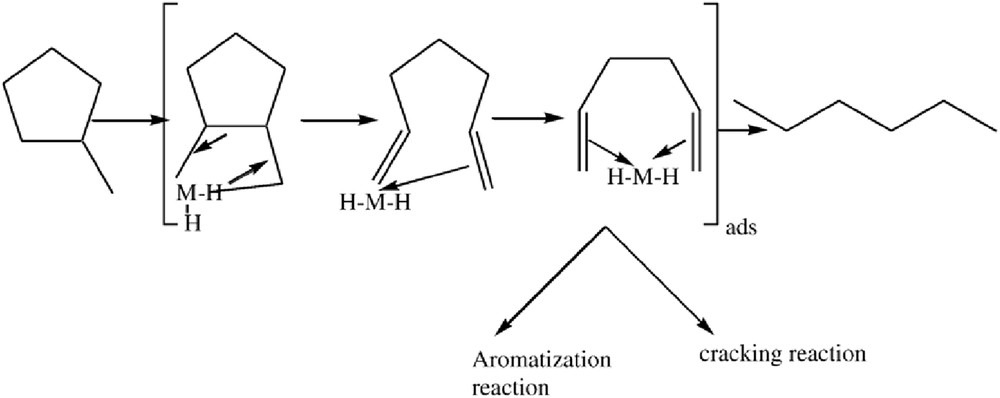
Ref. [92].
Regardless of the Fe loading in the TUD-1 and the reaction temperature, the only major product formed was determined to be C1, indicating that a deep cracking process occurred with the Fe–TUD-1 catalysts. The high selectivity toward C1 is attributed, most likely, to the hindered desorption of products that are formed, followed by the successive CC bond rupture on the Fe active sites present on the TUD-1. Compared with the samples with Si/Fe ratios of 85 and 65, impressive values of selectivity toward cracking reactions were observed for the sample with a Si/Fe ratio of 45 in the temperature range of 250–400 °C, which suggests that octahedral Fe entities/small clusters and high temperatures increase the C1 selectivity. The color of this sample with a Si/Fe ratio of 45 was gray, whereas the color of the samples with Si/Fe ratios of 65 and 85 remained white after the catalytic tests [92]. It seems that these small clusters are susceptible to easily forming the iron carbide, taking into account the fact that in the FeOFe clusters, the binding energy of OFe is small as compared with that of CFe. When the Fe is isolated and tetrahedrally coordinated in the SiOFe framework, the binding energy is strong as compared with CFe and the Fe carbide cannot be formed, which is supported in this case by the white color after catalytic tests [92]. Noticeably, for the sample with a Si/Fe ratio of 45 (the point of the framework partial damage) and a high temperature, the convertibility between Fe clusters and Fe carbide was observed. These latter moieties are responsible for the cracking reaction [92]. Benzene was only detected for the sample with a Si/Fe ratio of 45. Enlargement of the MCP ring was only observed at temperatures of 400 °C. This result can be attributed to thermodynamic phenomena because aromatization is favored at high temperatures [92]. The ring-enlargement product formation was hindered at low temperatures and at low Fe contents, this result represents an advantage of Fe catalysts due to their ability to hinder aromatization. This also holds true for the samples with high Fe loadings but at low temperatures [92].
5 Conclusions
Over the past year, there have been a number of exciting developments in the synthesis and characterization of metal oxide nanostructures for selective/partial oxidation reactions, ring opening of MCP, and biocatalysis. Of increasing importance have been the metal oxides and porous oxides probing the conversion and selectivities of catalytic reaction. The MCP reaction has been successfully studied of other on different metal oxide catalysts of much cheaper main component. Efficiency and stability was shown to be inferior to those of noble metals but quite reasonable for further possible applications.
The general aspects of heterogeneous selective oxidation reactions are as follows: (1) Flexibility of the oxide surface under catalytic (redox) conditions (living surface) avoiding any structure collapse during the redox process. (2) Collective properties: electron and oxygen anion transfer (electrical and cationic conductivities), changes in adsorption properties. (3) Influence of the coadsorbate on the bond between the adsorbate and the surface. (4) Role of the catalyst surface adlayer composition/structure (amorphous vs crystallized) on activity and selectivity as very often met. (5) Oxygen species involved in the reaction: lattice oxygen anions, weakly adsorbed oxygen species. (6) Mechanism and anisotropy of oxygen migration. (7) Importance of surface species, which do not play a direct role in the reaction mechanism, for example, H2O, carbon deposit (fouling), as spectators.
Advances in the development and applications of heterogeneous catalysts are driven by several factors:
- • A need to focus on economics. Considerable R&D should be focused on technology that uses ethane and natural gas as feedstock, as well as methanol and derivatives. In this regard, the search for energy improvement will continue, as it has in the past, even with low energy and oil prices.
- • A need for greenhouse gas reduction. Greenhouse gases can be reduced by several methods. Enhanced oil recovery using CO2 is the least expensive way to go, but with the drop in the price of crude oil, we are seeing less of this. Of course, this could change if the price of crude oil goes back up. Carbon capture and sequestration is the next alternative and considerable work is being done in this area.
- • The conversion of CO2 to chemicals. Catalytically transforming carbon dioxide to chemicals is challenging because of thermodynamic limitations—more energy may be required to produce these chemicals than from other feedstock, and if this causes more CO2 to be released into the atmosphere, this does not serve the purpose. However, there is elegance to pursue this route and we expect there will be considerable research in this area. Research projects are underway at several universities and industry-sponsored institutions. Among the most interesting are those that are developing catalyst systems and use sunlight as a source for at least a part of the energy requirements.
- • The use of biomass as feedstock. The catalytic conversion of biomass has gained tremendous attention in recent years as this uses a renewable material and also biomass as it grows, absorbs CO2 from the atmosphere. There is a tremendous amount of R&D in this area, and we would expect that many chemicals will ultimately be produced from bio-based technology. In this context, it will be important to focus on using nonedible biomass. Furthermore, the research is likely to shift toward using catalytic processes, rather than enzymatic processes, because reaction rates with enzymes are typically quite slow.
- • With the success achieved by Siluria with their oxidative coupling technology and the ability to produce ethylene from methane at a very low cost (as long as natural gas remains cheap), there will continue to be strong emphasis to conduct increased R&D in this area.
- • Meanwhile, because so many chemicals are currently produced from syngas, particularly in China, there will also be continued interest and increasing amounts of R&D in this area.
- • Low alkane production from shale has modified the oil needs and prices in the past 10 years but may not last very long.
Acknowledgments
I.F. gratefully acknowledges Centre National de la recherche Scientifique France for financial support.