

1 Introduction
One of the most interesting subjects for studying metal nanoclusters is to understand how their physical and chemical properties change as a function of size [1–15]. The size-selected metal nanoclusters are under investigation as the foundation for designing a new generation of catalysts [5,15–17]. By studying the properties of nanoclusters as a function of size, the researchers hope to provide better insight into factors governing transition-metal-centered chemistry. Also, it can provide important clues to the understanding of the mechanism of catalysis and other chemical properties [18,19]. More theoretical and experimental studies on the clusters improve the process of understanding their structures, electronic properties, and catalysis [1,4,5,7,11–14,17]. The geometry and electronic properties of nanoclusters have been successfully predicted by theoretical methods. In the literature, there are many reports that investigate the properties of nanoclusters. Among these studies, nickel nanoclusters are the subject of many experimental and theoretical studies because these nanoclusters are very efficient catalysts for the hydrogenation and dehydrogenation of olefin reactions [11,20].
Ethylene is an important intermediate in the petrochemical industry. It is predominantly produced by the thermal or catalytic cracking of hydrocarbons [21]. The small amounts of coproduced acetylene in the ethylene stream can poison catalysts of ethylene polymerization [21]. The reaction of catalytic-selective hydrogenation is the preferred method to reduce the amount of acetylene because of its relative ease. In this reaction, at first, a hydrogen molecule is chemisorbed on a catalyst surface, and in the next step, this surface hydrogen reacts with acetylene. In fact, the dissociative adsorption of the hydrogen molecule is a necessary step for the hydrogenation of acetylene [21].
By the use of the theoretical studies we can specify some of the elementary reaction steps on catalyst surfaces and provide ideas about the reaction pathways. The density functional theory (DFT) calculations have been used to examine the adsorption modes for atomic hydrogen, acetylene, vinyl, and ethylene on different metal surfaces [7,10,13,14,17,22–24]. For instance, Chen and Vlachos [25] investigated the hydrogenation of ethylene and dehydrogenation of ethane on Pt(111) and Pt(211) using DFT method. The DFT calculation of selective hydrogenation reactions over PdAg bimetallic catalysts was investigated by Gonzalez et al. [26]. Also, the thermodynamic properties of the acetylene and ethylene hydrogenation on some bimetallic PdAg alloy catalytic surfaces were theoretically studied by Sheth et al. [27].
The present study aims at reporting the results of hydrogenation of acetylene and ethylene on nickel nanoclusters by DFT calculations. The first step in the hydrogenation is the dissociative adsorption of hydrogen molecule, and therefore, we initially studied the quality of the adsorbed hydrogen on the nickel nanoclusters. Then, the adsorption of acetylene, ethylene, along with the vinyl and ethyl intermediates is discussed. Furthermore, we investigate the activity and selectivity of these surfaces.
2 Methodology
All the geometry optimization of ground states and transition states (TSs) were carried out through a high quantum level of DFT-D (Dispersion-corrected Density Functional Theory) approach implemented in the DMol3 program package [28,29]. In this work, the PBE (Perdew, Burke and Ernzerhof) functional [30] with Grimme's long-range dispersion correction [31] (PBE-G) was used. We selected fine grid for the numerical integration of the exchange–correlation functions and related matrix elements. The effective core potentials (ECPs) and double-numerical quality basis function with polarization function for H atoms (DNP) were selected for all atoms. We considered the high spin multiplicity of the Ni nanoclusters to find the lowest energy spin state (1, 3, 5, and 7 for odd multiplicity systems and 2, 4, 6, and 8 for even multiplicity systems). The spin state with the lowest electronic energy was used for the computation of geometry and electronic properties. No symmetry constraints are imposed for obtaining the equilibrium geometries of the structures for each possible spin multiplicity. The stationary geometries were characterized as minima or TSs by analytical frequency calculations at the same level as the geometry optimizations. To determine the activation energy for a specific reaction pathway, a TS was identified by the complete linear synchronous transit and the quadratic synchronous transit methods. Also, the TS structure was confirmed by the nudged elastic band method using TS confirmation calculations.
The adsorption energy (Eads) was calculated as follows:
| (1) |
| (2) |
3 Results and discussion
3.1 Acetylene and ethylene adsorption on the Nin (n = 2–10) nanoclusters
Acetylene adsorption: in our previous work [32], we investigated the geometric structures of Nin (n ≤ 10) nanoclusters in their lowest energy state. In this work, we used the most stable structure of the Nin (n = 2–10) nanoclusters as substrates and the adsorption energy of C2H2 on these nickel nanoclusters is calculated. All structures are optimized at the PBE-G/DNP-ECP level of theory. The optimized structure with the maximum adsorption energy is determined as the most stable structure. The most stable adsorption modes of C2H2 on the Nin (n = 2–10) nanoclusters, as well as the adsorption energies (Eads) and corresponding multiplicities (2S + 1) are reported in Table 1.
The most stable adsorption modes and corresponding multiplicities (2S + 1) for the adsorption of C2H2 on the Nin nanoclusters. The adsorption energies (Eads) are reported in kJ/mol.
According to Table 1, the C2H2 adsorption on these nickel nanoclusters is selective in π-coordination. This means that the two C atoms prefer to bond with the same Ni atom. In all structures, the CH bonds are distorted out of the acetylene plane (Table 1). We can explain this condition by the electron donation and electron back donation between the hybridized orbital of the Nin nanoclusters and the hybridized orbital of the acetylene molecule. The results show a good overlap between these orbitals and the increase in interactions between the nickel nanoclusters and the acetylene molecule as well. The range of the adsorption energy for the C2H2 adsorption is −146.27 to −177.34 kJ/mol. From comparison of the Nin nanoclusters, it can be said that the acetylene adsorption on the Ni5 and Ni4 nanoclusters is more effective than other nickel nanoclusters.
Ethylene adsorption: to investigate the adsorption of ethylene on the Nin (n = 2–10) nanoclusters, different structures were designed and optimized at the PBE-G/DNP-ECP level of theory. The optimized structure with the maximum adsorption energy is determined as the most stable structure.
The most stable adsorption modes of C2H4 on the Nin (n = 2–10) nanoclusters, as well as the adsorption energies (Eads) and corresponding multiplicities (2S + 1) are reported in Table 2. Accordingly, the C2H4 adsorption on these nickel nanoclusters is selective in π-coordination. The range of C2H4 adsorption energy is −126.23 to −153.89 kJ/mol. The ethylene adsorption on the Ni5 and Ni4 nanoclusters is more effective than other nickel nanoclusters.
The most stable adsorption modes and corresponding multiplicities (2S + 1) for the adsorption of C2H4 on the Nin nanoclusters (the adsorption energies (Eads) are reported in kJ/mol).
One of the catalytic drawbacks for acetylene hydrogenation is that acetylene is more easily adsorbed on the surface than the hydrogen molecule, and therefore, the catalytic active sites are occupied. Therefore, a catalyst can be effective when the adsorption energy of a hydrogen molecule on its surface is higher than that of an acetylene or ethylene.
The behavior of a material in bulk and nanoscale is very different and in most cases, nanoscale materials are much more active than the bulk. In our previous work [32], we investigated the adsorption energy for hydrogen on the Nin (n = 2–10) nanoclusters.
To find a catalyst with favorable conditions, the adsorption energies of hydrogen, acetylene, and ethylene on the Nin (n = 2–10) nanoclusters are compared. We plotted the absolute values of hydrogen, acetylene, and ethylene adsorption energies versus nanocluster size, n (Fig. 1). According to Fig. 1, the Nin (n = 2–10) nanoclusters show similar behavior in the adsorption of acetylene and ethylene. However, their behavior is different in adsorbing hydrogen molecule.
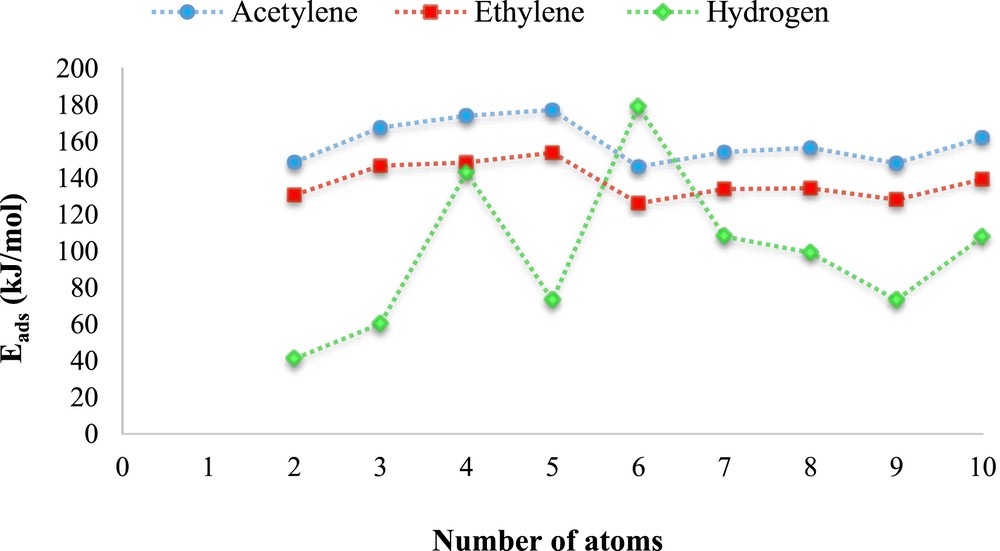
The absolute values of adsorption energies of hydrogen, acetylene, and ethylene on the Nin (n = 2–10) versus nanocluster size (n).
Except for the Ni6 nanocluster, the adsorption energy of the hydrogen molecule on the Nin (n = 2–10) nanoclusters is lower than that of acetylene and ethylene, whereas on the Ni6 nanocluster the adsorption energy of the hydrogen molecule is higher than that of acetylene and ethylene. Therefore, the Ni6 nanocluster was selected and its catalytic behavior for the acetylene and ethylene hydrogenation was studied.
3.2 Hydrogenation of acetylene on the Ni6 nanocluster
In general, the reaction paths for the hydrogenation of acetylene and ethylene are thought to follow the Horuiti–Polanyi [33] mode where a sequence of hydrogenated C2Hx species is formed by the sequential addition of single hydrogen atoms to the adsorbed hydrocarbon intermediate (Scheme 1). Initially, this reaction path was proposed by Bond and co-workers who studied the kinetics of acetylene hydrogenation over different transition metal surfaces [34,35]. Here, we use the reaction paths, shown in Scheme 1, for the study of acetylene hydrogenation.
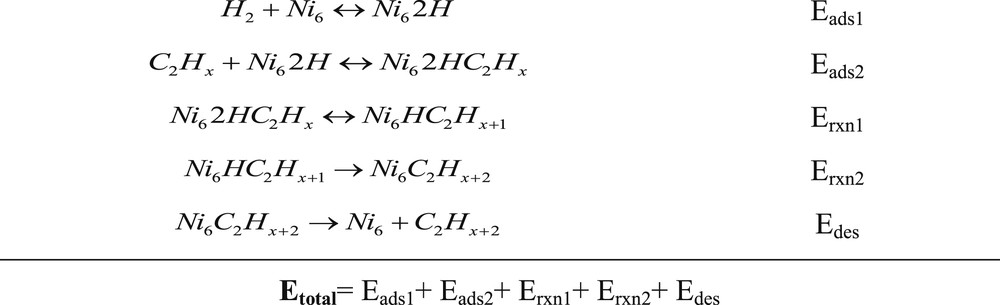
Reaction paths for the hydrogenation of C2Hx (x = 2, 4).
On the basis of our previous work [32], H2 was adsorbed dissociatively on the Ni6 nanocluster. The adsorption energy for the hydrogen atom on the Ni6 nanocluster surfaces (Εads1) was investigated. Fig. 2 shows the charge distribution over the Ni6 nanoclusters, visualized via the electrostatic potential (ESP) surface, V(r), map. This visualization can be used for finding the reactive sites of the molecules. The relative abundance and absence of electrons are, respectively, related to the sites with the lowest (red region) and highest (blue region) values of ESP energy. The maximum ESP energy value on the local surface of the Ni6 nanocluster is 87.17 kJ/mol (Fig. 2a). This result clearly explains the site-selectivity; the site with the value of 87.17 kJ/mol is the most favorable site because the ESP on its local surface is the most positive, and hence nucleophilic reagents tend to be attracted to this site.
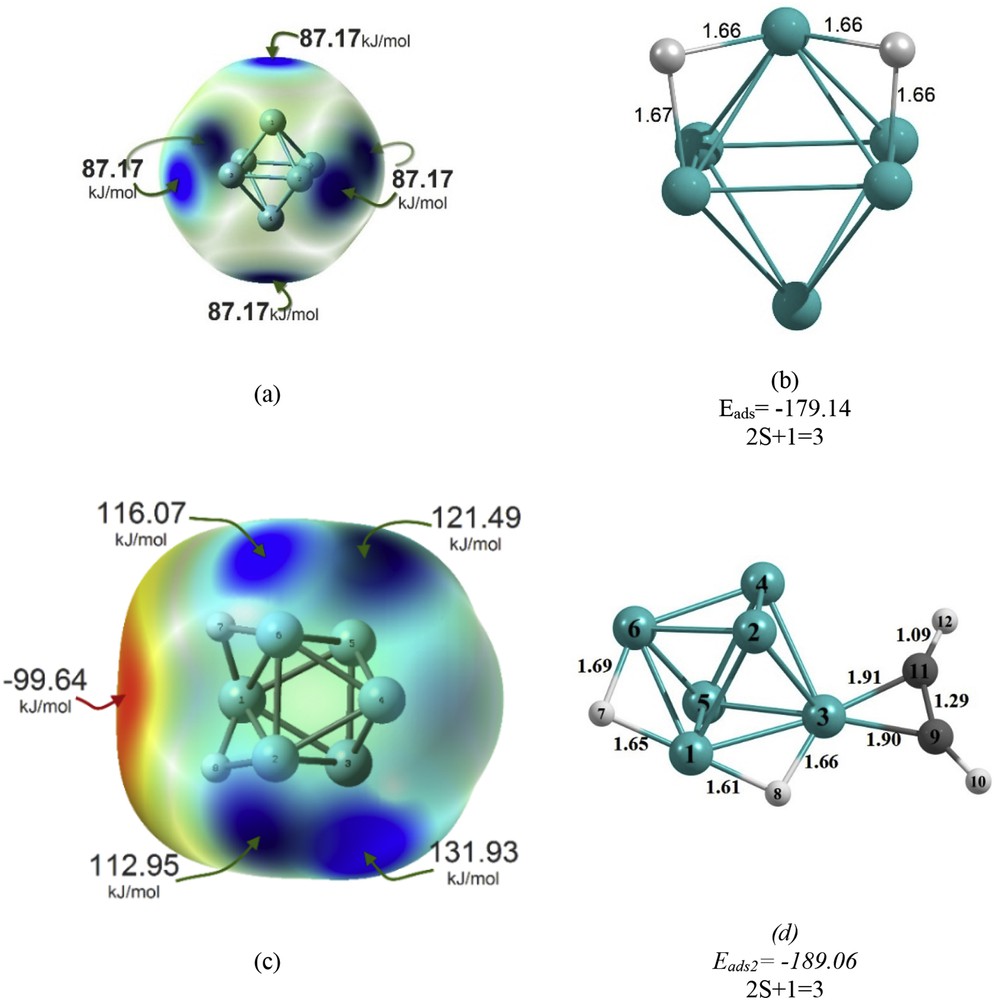
(a) The ESP of the Ni6 nanocluster, (b) the most stable structure of H2 on the Ni6 nanocluster (Ni62H), (c) the ESP of the Ni62H complex, and (d) the most stable structure of C2H2 on the Ni62H complex surface, as well as the adsorption energy (Eads), corresponding multiplicity (2S + 1), and CC and CH bond lengths and binding distances. The adsorption energy (Eads2) in kJ/mol and bond lengths and binding distances in Å are also reported.
To obtain the most stable structure of Ni6H2, according to Fig. 2a, H2 is located on the four active sites, individually. The optimized structure with the maximum adsorption energy is determined as the most stable structure. The most stable adsorption mode of H2 on the Ni6 nanocluster, as well as the adsorption energy (Eads1) and binding distances, is reported in Fig. 2b. Accordingly, the hydrogen molecule is dissociatively adsorbed on the Ni6 nanocluster. The adsorption energy for the H2 adsorption is −179.14 kJ/mol. In this complex, the minimum value of NiH bond length is 1.66 Å.
Fig. 2c also shows the ESP map over the Ni62H complex. Accordingly, the maximum ESP energy values on the local surface of the Ni62H complex are 131.93 and 121.49 kJ/mol, respectively. A minimum energy structural search is performed in studying the adsorption of C2H2 on the Ni62H complex for the most favored adsorption sites. The most stable adsorption mode of acetylene adsorption on the Ni62H complex surface, as well as the adsorption energy (Eads2) and CC and CH bond lengths and binding distances, is reported in Fig. 2d. The results show that between the two main reaction pathways (π and di-σ-bonded), π-bonded acetylene is determined as the most stable structure (Fig. 2d). The adsorption energy (Εads2) of most stable acetylene on the Ni62H complex is found to be −189.06 kJ/mol. The CC bond length of the Ni62HC2H2 shows an increase of 0.08 Å with respect to that of unadsorbed acetylene (1.21 Å). In the Ni62HC2H2 structure, the CH bonds (1.09 Å) are distorted out of the acetylene plane (Fig. 2d).
The reaction between surface hydrogen atom and acetylene forms a surface vinyl (C2H3) intermediate. To find the intermediate structure, the distance between the nearest hydrogen atom and the acetylene is scanned, and an initial guess for the intermediate structure is obtained. Therefore, the surface vinyl intermediate is optimized on the Ni6H complex to establish its most stable structure and corresponding reaction energy (Erxn1). According to Fig. 3a, the adsorbed C2H3 tilts along the surface and Ni atom is getting closer to one side, suggesting that one of the C atoms of vinyl has a stronger interaction with the surface. This is compatible with the theory of chemisorption presented by Hoffmann and co-workers [36,37]. According to this theory, the orbital overlap and Pauli repulsion are balanced in the favored adsorption site. In the surface vinyl intermediate, all the CH bond lengths are 1.10 Å and the CC and minimum NiC bond lengths are 1.40 and 1.91 Å, respectively. The NiC bond helps stabilize the electron deficiency of the CH group in the vinyl intermediate.

The most stable structure of (a) vinyl on the Ni6H complex (Ni6HC2H3), (b) TS structure for acetylene hydrogenation to vinyl on the Ni6H complex, (c) ethylene on the Ni6 nanocluster surface (Ni6C2H4), and (d) TS structure for vinyl hydrogenation to ethylene on the Ni6 nanocluster. The reaction energies (Erxn) in kJ/mol and bond lengths and binding distances in Å are also reported.
The reaction energy for Ni62HC2H2 → Ni6HC2H3 (Erxn1) is computed to be −57.39 kJ/mol. The TS structure for acetylene hydrogenation to vinyl is shown in Fig. 3b. The activation energy (Ea1) for the hydrogenation of acetylene to vinyl is +113.94 kJ/mol. In this TS, the bond lengths of NiH and CH are 2.44 and 1.69 Å, respectively.
The produced vinyl can react with another neighboring surface hydrogen atom to form a surface ethylene. Therefore, the surface ethylene intermediate is optimized on the Ni6 nanocluster to establish its most stable structure and corresponding reaction energy (Erxn2). According to Fig. 3c, ethylene adsorption on the nickel nanocluster is selective in π-coordination. In this structure, all the CH bond lengths are 1.09 Å and the CC and minimum NiC bond lengths are 1.41 and 2.02 Å, respectively. According to Scheme 1, we consider Ni6HC2H3 → Ni6C2H4 and the reaction energy (Erxn2) is computed to be −47.95 kJ/mol. The TS structure for the hydrogenation of vinyl to ethylene is shown in Fig. 3d. The activation energy for the hydrogenation of vinyl to ethylene is +150.12 kJ/mol. The NiH distance and CH bond length are 3.14 and 1.99 Å, respectively.
Also, the ethylene desorption energy (Edes) is computed according to Scheme 1. We consider Ni6C2H4 → Ni6 + C2H4 and the energies of the most stable structure of Ni6C2H4, Ni6, and C2H4 are calculated, and the desorption energy (Edes) is computed to be +126.13 kJ/mol.
Using Scheme 1, we calculate the total reaction energy for the acetylene hydrogenation to ethylene to be exothermic by −247.40 kJ/mol. Fig. 4 shows an overall energy diagram for the hydrogenation of acetylene to ethylene. The reaction energies for the hydrogenation of acetylene to vinyl and vinyl to ethylene are −57.39 and −47.95 kJ/mol, respectively. The activation energy of acetylene hydrogenation to vinyl is 36.17 kJ/mol higher than that of the hydrogenation of vinyl to ethylene. The Arrhenius equation gives the relationship between the activation energy and the rate constant at which a reaction proceeds [38]. From this equation, it can be said that the second step of acetylene hydrogenation is faster than the first step. The results of many experimental and theoretical studies on the Ni surface suggest that vinyl hydrogenation has smaller Ea than acetylene hydrogenation [39–42].

The overall energy diagram for the hydrogenation of acetylene to ethylene. All energies are in kJ/mol.
3.3 Hydrogenation of ethylene on the Ni6 nanocluster
To study the ethylene hydrogenation, according to Scheme 1, interaction between surface hydrogen atom and ethylene on the Ni6 nanocluster surface is investigated. At first, the adsorption of ethylene on the Ni62H complex surface is studied. To obtain the most stable structure of Ni62HC2H4, C2H4 is located individually on the active sites (Fig. 2c). The most stable adsorption mode of ethylene adsorption on the Ni62H complex surface, as well as the adsorption energy (Eads2) and CC and CH bond lengths and binding distances, is reported in Fig. 5a. The results show that π-bonded ethylene is determined as the most stable structure (Fig. 5a). The adsorption energy (Εads2) of ethylene on the Ni62H complex is found to be −168.63 kJ/mol. The CC bond length of the Ni62HC2H4 shows an increase of 0.08 Å with respect to that of unadsorbed ethylene. In the Ni62HC2H4 structure, the CH and minimum NiC bond lengths are 1.09 and 2.01 Å, respectively. The surface ethylene can react with neighboring surface hydrogen atom to form a surface ethyl (C2H5) intermediate. Therefore, the surface ethyl intermediate is optimized on the Ni6H complex surface. The results show that ethyl is adsorbed on the Ni6 nanocluster surface via di-σ-bonding involving two metal atoms (Fig. 5b). The surface ethyl tilts along the NiNi bond and ethyl is getting closer to one side. This means that one of the Ni atoms has a stronger interaction with the surface ethyl. The CC and NiC distances for surface ethyl are 1.53 and 2.06 Å, respectively. The NiC bond helps stabilize the electron deficiency of CH2 group in the ethyl intermediate.

The most stable structure of (a) ethylene on the Ni62H complex (Ni62HC2H4), (b) ethyl on the Ni6H complex surface (Ni6HC2H5), (c) TS structure for ethylene hydrogenation to ethyl on the Ni6H complex, (d) ethane on the Ni6 nanocluster surface (Ni6C2H6), and (e) TS structure for ethyl hydrogenation to ethane on the Ni6 nanocluster. The reaction energies (Erxn) in kJ/mol and bond lengths and binding distances in Å are also reported.
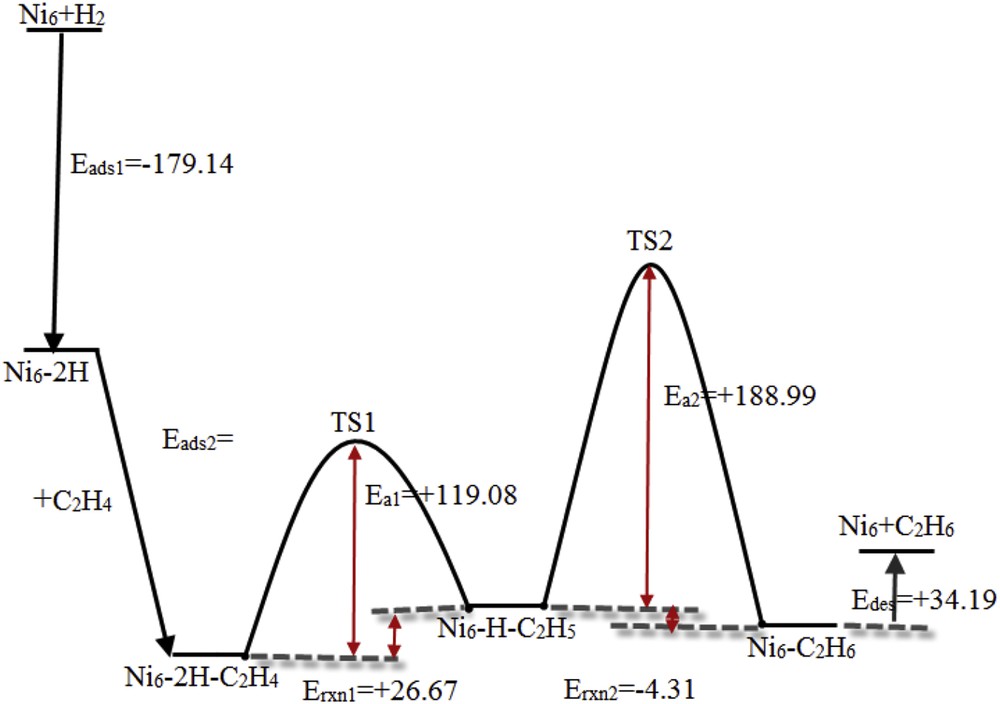
Following Scheme 1, the reaction energy for the hydrogenation of ethylene to ethyl (Erxn1) is calculated to be +26.67 kJ/mol. The activation energy of this step of hydrogenation is +119.08 kJ/mol. The CC bond length is 1.51 Å. The NiH distance increases to 2.13 Å and CH bond length decreases to 1.56 Å (Fig. 5c).
The reaction of surface ethyl intermediate with atomic surface hydrogen produces surface ethane (Fig. 5d). The result shows that the ethane adsorption on the Ni6 nanocluster is selective in di- σ-coordination. According to Scheme 1, the reaction energy for ethyl hydrogenation to ethane (Erxn2) is computed to be −4.31 kJ/mol. The TS structure is shown in Fig. 5e. The energy to activate ethyl to form surface ethane is calculated to be +188.99 kJ/mol. The activation energy for the production of ethane (second step) is higher than that for the hydrogenation of ethylene to ethyl. We consider Ni6C2H6 → Ni6 + C2H6, and the desorption energy (Edes) is computed to be +34.19 kJ/mol.
Using Scheme 1, we calculated the total reaction energy for the ethylene hydrogenation to ethane to be exothermic by −291.21 kJ/mol. Fig. 6 shows an overall energy diagram for the hydrogenation of ethylene to ethane on the Ni6 nanocluster surface. The reaction energies for the hydrogenation of ethylene to ethyl and ethyl to ethane are +26.67 and −4.31 kJ/mol, respectively. The values of Ea for the hydrogenation of ethylene to ethyl and hydrogenation of ethyl to ethane are +119.08 and +188.99 kJ/mol, respectively. Therefore, the second step of ethylene hydrogenation is faster than the first step.
The overall energy diagram for the hydrogenation of ethylene to ethane. All energies are in kJ/mol.
3.4 Activity and selectivity of the Ni6 nanocluster catalyst
In Section 3.2, it has been shown that the Ni6 nanocluster can be used as a catalyst for the hydrogenation of acetylene. In Table 3, it can be seen that the forward activation energy of hydrogenation of vinyl to ethylene (+150.12 kJ/mol) is smaller than the reverse activation energy of hydrogenation of acetylene to vinyl (+171.33 kJ/mol), and it seems that all of the produced vinyl intermediate can be converted to ethylene.
The activation energy and overall reaction energies for two steps of hydrogenation of acetylene and ethylene on the Ni6 nanocluster (all energies are in kJ/mol).
| Energy | Forward | Reverse | Forward | Reverse |
| Acetylene hydrogenation | ||||
| C2H2* + H* → C2H3* | C2H3* + H* → C2H4* | |||
| Ea | +113.94 | +171.33 | +150.12 | +198.07 |
| Erxn | −57.39 | +57.39 | −47.95 | +47.95 |
| Ethylene hydrogenation | ||||
| C2H4* + H* → C2H5* | C2H5* + H* → C2H6* | |||
| Ea | +119.08 | +92.40 | +188.99 | +193.30 |
| Erxn | +26.67 | −26.67 | −4.31 | +4.37 |
According to Table 3 and Fig. 6, the Ni6 nanocluster is not a useful catalyst for the hydrogenation of ethylene to ethane. The reverse activation energy of hydrogenation of ethylene to ethyl (+92.40 kJ/mol) is smaller than the forward activation energy of hydrogenation of ethyl to ethane (+188.99 kJ/mol). In the reverse hydrogenation of ethylene to ethyl, the produced CH bond in the ethyl intermediate can be broken and produce ethylene and hydrogen atom. Therefore, due to the faster rate of the reverse reaction, all of the produced ethyl intermediate can be converted to ethylene. According to the obtained results, the Ni6 nanocluster can selectively act in the hydrogenation of a mixture of acetylene and ethylene.
4 Conclusions
The hydrogenation of acetylene and ethylene on the nickel nanoclusters is investigated by the DFT using the Horuiti–Polanyi mode of the hydrogenation reaction. According to the performed calculations, among all the Nin (n = 2–10) nanoclusters, the Ni6 nanocluster can be used as catalysts in the reactions of hydrogenation of acetylene and ethylene. For the hydrogenation of acetylene, the surface hydrogen atom reacts with acetylene to form a surface vinyl intermediate and the produced vinyl reacts with another neighboring surface hydrogen atom to form a surface ethylene. This reaction on the Ni6 nanocluster is exothermic with the total energy of −247.40 kJ/mol. According to our results, the forward activation energy of hydrogenation of vinyl to ethylene is smaller than the reverse activation energy of hydrogenation of acetylene to vinyl, and it seems that all of the produced vinyl intermediate can be converted to ethylene. Also, the Ni6 nanocluster is not a useful catalyst for the hydrogenation of ethylene to ethane. The reverse activation energy of hydrogenation of ethylene to ethyl (+92.40 kJ/mol) is smaller than the forward activation energy of hydrogenation of ethyl to ethane (+188.99 kJ/mol). In the reverse hydrogenation of ethylene to ethyl, the produced CH bond in ethyl intermediate can be broken and produce ethylene and hydrogen atom. Therefore, due to the faster rate of the reverse reaction, all of the produced ethyl intermediate can be converted to ethylene. According to the obtained results, the Ni6 nanocluster can selectively act in the hydrogenation of a mixture of acetylene and ethylene.