

1 Introduction
Nanoscience and nanotechnology are the latest revolutionary developments in science and engineering, which are rapidly moving forward. To measure the new and improved properties of nanomaterials requires a close connection among the physical, chemical, biological, and engineering sciences. One of the most widely used nanomaterials is fullerene. A fullerene is a molecule of carbon in the form of a hollow sphere, an ellipsoid, tube, or other shape. Harry Kroto, Richard Smalley, and Robert Curl discovered fullerenes in 1985 and won the 1996 Nobel Prize in chemistry [1,2]. Much experimental and theoretical work has been done to discover and provide fullerenes, in particular C60. A fullerene is a deceptive molecule; it is a nonlinear optical (NLO) nanocluster and has a large number of conjugated double bonds. These characteristics suggest large nonlinear hyperpolarizabilities. Many studies on the NLO properties of fullerenes are either experimental or theoretical [3–12]. Because of the widespread use of optical materials in information storage, optical communication, and computing and optical switching devices, the design of high-performance NLO materials is an important area of physical chemistry research [13–28]. The presence of CH bonds usually has a harmful effect on NLO responses; therefore, pure carbon systems such as fullerenes are good candidates for exploitation of NLO properties. Because additional electrons play an important role in increasing the first hyperpolarizability (β0), species that contain extra electrons can show a significant NLO response [29–38]. Recently, M3O (M = Li, Na, and K) has been used to increase the NLO response [39]. The C20 molecule, with a dodecahedral cage structure, is the smallest and truly unique member of the fullerene family [40]. It is thought that the smallest fullerene C20 with positive electron affinity is a new organic nanoelectronic material. Its structure is very simple and highly symmetrical, consisting of 12 five-membered rings only [41]. In this work, we present a density functional theory (DFT) study on the exohedral interaction between lithium atoms and fullerene C20.
2 Methods
The nanostructure of fullerene C20 and Lin@C20 (n = 1–6) structures was completely optimized at the theoretical B3LYP/6-31+G(d) level [42,43]. All calculations were performed using the unrestricted spin method. Geometry optimization also was performed using the CAM-B3LYP [44] and WB97XD [45] methods in conjunction with the 6-31G(d), 6-31+G(d), and 6-311+G(d) basis sets for all complexes to examine the extent of difference in geometries optimized using long-range corrected calculation. B3LYP is a reliable method for the study of nanostructures [46–49]. Frequency calculations of all optimized structures were done at the B3LYP/6-31+G(d) level. The density of state was calculated in GaussSum.3.3.9 [50]. The HOMO–LUMO (highest occupied molecular orbital–lowest unoccupied molecular orbital) gap energy (Eg) of structures was determined from the difference between ƐH (HOMO energy) and ƐL (LUMO energy). Polarizability (α) and β0 as linear optical and NLO properties, respectively, were examined at the theoretical CAM-B3LYP/6-31+G(d) level. The β0 is a factor in the NLO response coefficient and can be calculated using the following equations:
Because the S2 values for the nanoclusters with one unpaired electron were in the range of 0.751–0.785, the computational methods were found to be reliable and spin contamination negligible. All calculations were done in Gaussian 09 package [51]. Natural bond orbital (NBO) analysis [52] was also performed.
3 Results and discussion
Fig. 1 shows the optimized structures of all nanostructures studied.
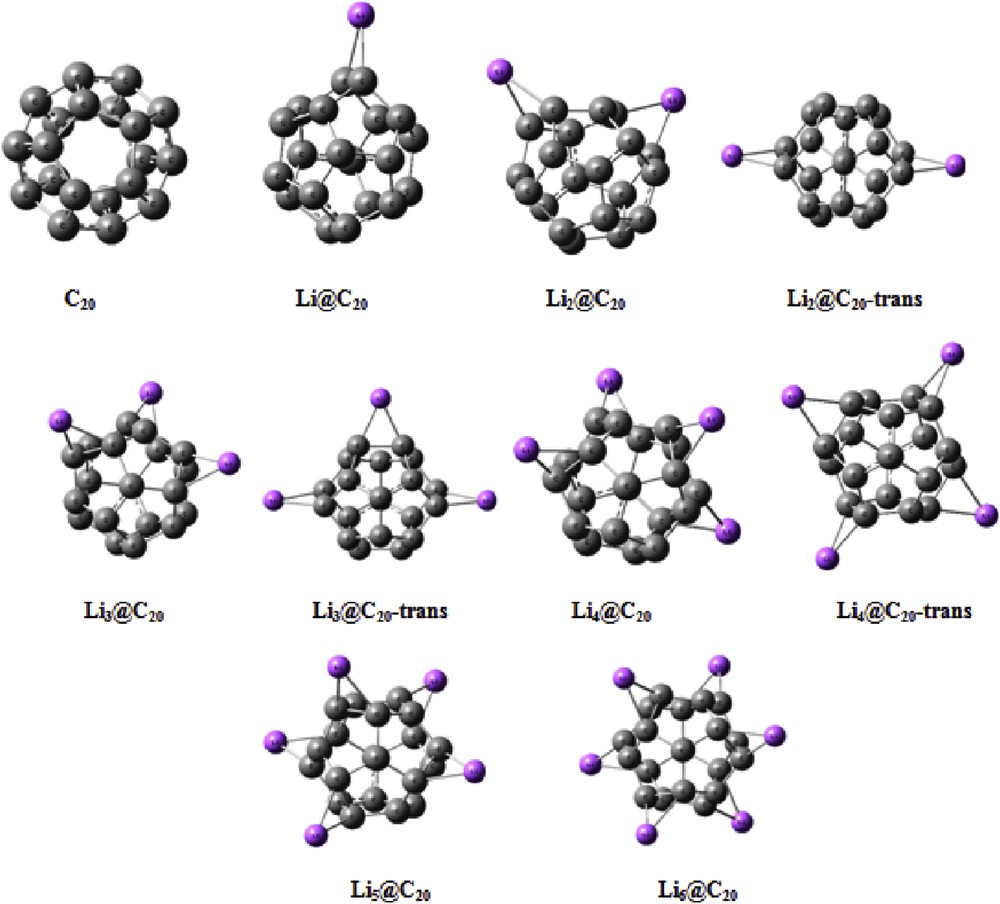
Optimized structures of C20 and Lin@C20 nanoclusters.
After optimization and frequency calculations, the energetic properties of all nanoclusters were calculated, for example, the binding energy (Eint) was determined from the following equation:
Binding energy, change in enthalpy, free energy, and entropy of Lin@C20 formation.
| Structure | Eint (kcal/mol) | ΔH (kcal/mol) | ΔG (kcal/mol) | ΔS (kcal/mol) |
| Li@C20 | −48.6200 | −49.1642 | −40.5604 | −0.0288 |
| Li2@C20 | −91.3402 | −91.2412 | −73.6572 | −0.0589 |
| Li2@C20-trans | −99.1734 | −98.5856 | −81.0481 | −0.0588 |
| Li3@C20 | −124.1404 | −124.5445 | −99.7328 | −0.0832 |
| Li3@C20-trans | −133.7791 | −133.7569 | −108.4452 | −0.0849 |
| Li4@C20 | −172.3620 | −171.7263 | −137.7813 | −0.1138 |
| Li4@C20-trans | −175.8398 | −175.1099 | −141.2144 | −0.1138 |
| Li5@C20 | −193.1880 | −193.7764 | −152.9408 | −0.1369 |
| Li6@C20 | −203.2263 | −204.3173 | −155.9811 | −0.1621 |
As seen in Table 1, the addition of lithium atoms to the surface of C20 is highly exothermic. The binding energy, enthalpy, and free energy of the Lin@C20 formation increased as the number of lithium atoms increased. The enthalpy change per lithium atom was calculated as the fraction
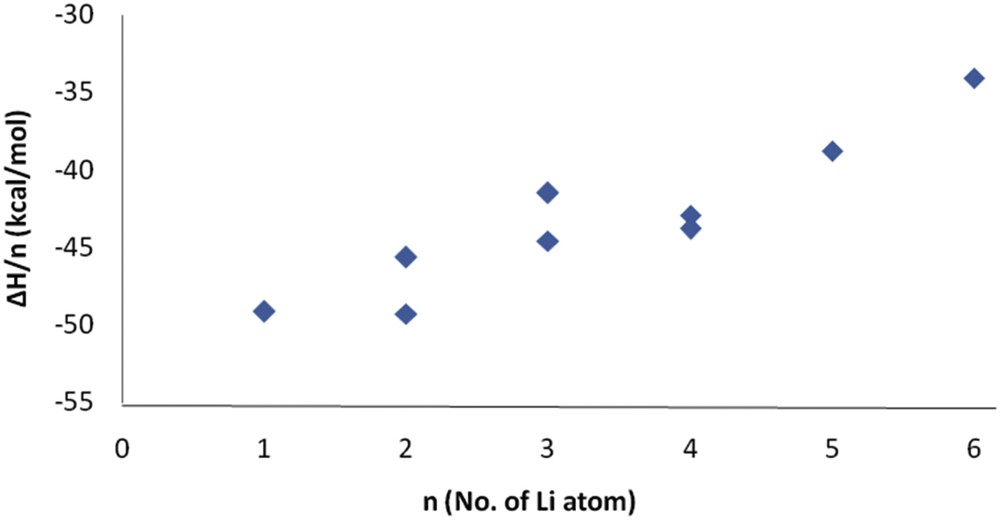
Changes in
Fig. 2 shows that when the number of Li atoms increased, the enthalpy of the reaction per lithium atom decreased. Although this value for the sixth lithium atom is negative, the reaction remains exothermic. To confirm the results of the adsorption energy of Li onto the C20 surface, the interaction energies were calculated using the WB97XD and CAM-B3LYP methods and 6-31G(d), 6-31+G(d), and 6-311+G(g) basis sets. The results are presented in Table 2. As seen, the interaction energies for all structures showed a similar trend for different methods and basis sets.
Eint (kcal/mol) values of nanostructures calculated with different methods and basis sets.
| Compound | Method | |||||
| Cam-B3LYP/6-31G(d) | Cam-B3LYP/6-31+G(d) | Cam-B3LYP/6-311+G(d) | WB97XD/6-31G(d) | WB97XD/6-31+G(d) | WB97XD/6-311+G(d) | |
| Li@C20 | −51.17 | −52.24 | −52.87 | −51.10 | −52.35 | −53.06 |
| Li2@C20 | −95.74 | −97.77 | −99.57 | −96.39 | −98.77 | −100.59 |
| Li2@C20-trans | −104.95 | −106.60 | −108.27 | −105.67 | −107.87 | −109.56 |
| Li3@C20 | −133.03 | −135.96 | −138.42 | −133.68 | −137.29 | −139.80 |
| Li3@C20-trans | −143.55 | −145.93 | −148.35 | −144.05 | −147.22 | −149.70 |
| Li4@C20 | −186.37 | −188.84 | −192.30 | −186.85 | −190.48 | −193.99 |
| Li4@C20-trans | −190.36 | −192.36 | −195.66 | −190.80 | −194.16 | −197.49 |
| Li5@C20 | −199.56 | −206.55 | −203.08 | −195.84 | −207.39 | −211.03 |
| Li6@C20 | −208.17 | −206.69 | −210.82 | −202.83 | −206.49 | −208.68 |
The HOMO (ƐH) and LUMO (ƐL) orbital energies were obtained from the density of state spectrum of each nanocluster. The ƐH, ƐL, and Eg are reported in Table 3. The HOMO and LUMO energies and the gap energy for fullerene C20 were −5.5, −3.62, and 1.88 eV, respectively. Other chemical parameters, which relate to the stability and reactivity of nanoclusters, such as the chemical potential (μ), global electrophilicity (ω), ionization potential (I), electron affinity (A), softness (γ), global hardness (η), and electronegativity (χ), were calculated and are reported in Table 3. The behavior of gap energy as a function of the number of lithium atoms (n) is shown in Fig. 3.
Chemical properties of the C20 and Lin@C20 nanostructures.
| Structure | ƐH (eV) | ƐL (eV) | Eg (eV) | %ΔEg | I (eV) | A (eV) | μ (eV) | η (eV) | γ (eV−1) | ω (eV) | χ (eV) |
| C20 | −5.5 | −3.62 | 1.88 | – | 5.5 | 3.62 | −4.56 | 0.94 | 1.0638 | 11.0604 | 4.56 |
| Li@C20 | −4.8 | −2.95 | 1.85 | −1.57 | 4.8 | 2.95 | −3.875 | 0.925 | 1.0811 | 8.1165 | 3.875 |
| Li2@C20 | −4.51 | −2.52 | 1.99 | 5.85 | 4.51 | 2.52 | −3.515 | 0.995 | 1.0050 | 6.2086 | 3.515 |
| Li2@C20-trans | −4.3 | −2.23 | 2.07 | 10.10 | 4.3 | 2.23 | −3.265 | 1.035 | 0.9662 | 5.1498 | 3.265 |
| Li3@C20 | −3.98 | −1.6 | 2.38 | 26.59 | 3.98 | 1.6 | −2.79 | 1.19 | 0.8403 | 3.2706 | 2.79 |
| Li3@C20-trans | −3.56 | −1.44 | 2.12 | 12.76 | 3.56 | 1.44 | −2.5 | 1.06 | 0.9434 | 2.9481 | 2.5 |
| Li4@C20 | −3.49 | −1.27 | 2.22 | 18.08 | 3.49 | 1.27 | −2.38 | 1.11 | 0.9009 | 2.5515 | 2.38 |
| Li4@C20-trans | −3.27 | −1.11 | 2.16 | 14.89 | 3.27 | 1.11 | −2.19 | 1.08 | 0.9259 | 2.2204 | 2.19 |
| Li5@C20 | −2.13 | −1.57 | 0.56 | −70.21 | 2.13 | 1.57 | −1.85 | 0.28 | 3.5714 | 6.1116 | 1.85 |
| Li6@C20 | −1.93 | −1.59 | 0.34 | −81.91 | 1.93 | 1.59 | −1.76 | 0.17 | 5.8823 | 9.1106 | 1.76 |
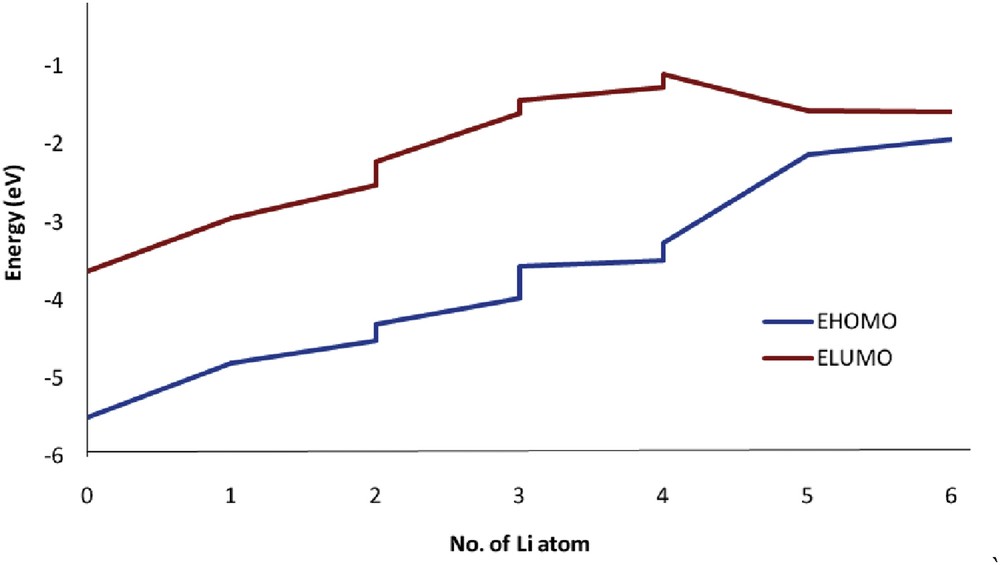
HOMO and LUMO energies as a function of the number of Li atoms adsorbed onto C20 surface.
Fig. 3 shows that adding Li atoms to the fullerene surface increased the HOMO and LUMO energies. The gap energy values increased up to the Li4@C20 structure, but the Li5@C20 and Li6@C20 structures show a sharp decrease in the gap energy and the greatest deviation in the gap energy deviation (70.21% and 81.91%, respectively).
Chemical hardness relates to the stability and reactivity of a chemical system. It measures the resistance of a compound to a change in the electron density or electron transfer distribution. Chemical hardness is directly proportional to the energy gap between HOMO and LUMO. An increase in hardness is a sign of a more stable and less reactive combination. In a chemical process, electrophilicity measures the capacity of a species to accept electrons; therefore, the stability of the structure after the receipt of the additional electronic charge by the system is shown [53]. A good reactive nucleophile molecule is characterized by a smaller amount of μ, ω, and, conversely, high electrophilicity with high values of μ and ω. Fig. 4 shows the changes in μ and ω relative to the number of Li atoms added to the C20 surface.
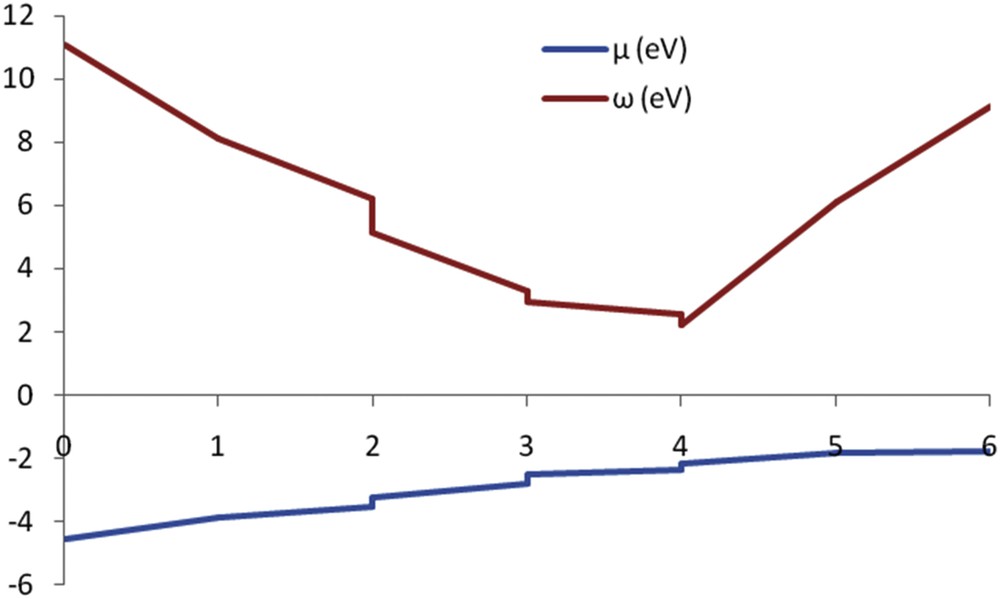
Changes in μ and ω relative to the number of Li atoms added to the C20 surface.
Fig. 4 shows the minimum global electrophilicity was attributed to four Li atoms adsorbed onto the C20 fullerene surface. Dipole moment (μ), α, and β0 as optical properties for all nanostructures were calculated and listed in Table 4. A high value for β0 is a prerequisite for the good behavior of NLO materials.
Total dipole moment, polarizability, and first hyperpolarizability of Lin@C20 nanostructures.
| Structure | μ (a.u.) | α (a.u.) | β0 (a.u.) |
| C20 | 0.0000 | 188.17 | 0.40 |
| Li@C20 | 2.8120 | 211.80 | 1565.87 |
| Li2@C20 | 4.1110 | 227.08 | 2840.01 |
| Li2@C20-trans | 0.0000 | 238.33 | 0.11 |
| Li3@C20 | 5.0202 | 253.81 | 4347.71 |
| Li3@C20-trans | 2.5999 | 257.87 | 3395.08 |
| Li4@C20 | 3.8429 | 274.07 | 3850.67 |
| Li4@C20-trans | 0.00016 | 285.19 | 0.30 |
| Li5@C20 | 1.9739 | 2088.69 | 4,034,196.74 |
| Li6@C20 | 0.00028 | 2804.79 | 302,865.14 |
It is evident that polarizability and hyperpolarizability depend on various factors. The degree of symmetry and extent of charge available in the molecules are two important factors. If the degree of symmetry increases, polarizability and hyperpolarizability must decrease. If the number of available electrons in the molecules increases, the probability of distortion as the result of external field increases and hyperpolarizability must increase. The s valence electron of the Li atom is pushed out and turns into a diffuse excess electron for the whole system and the extent of the existing electron increases as the result of Li atom adsorption onto its surface [54]. The higher polarizability and β0 for Li5@C20 and Li6@C20 nanostructures (Table 4) could be the result of higher electron transfer from the Li atoms to a cluster. However, the lower value of β0 for Li6@C20 can be explained by its higher degree of symmetry. NBO analysis was carried out in terms of the natural electron configuration and charge transfer. The value of the charge transfer (CT) from the Li atoms to the C20 fullerene is shown in Table 5.
Electron transfer values of Li atoms to C20 fullerene.
| Structure | CT value | Structure | CT value | Structure | CT value |
| Li@C20 | 0.85743 | Li3@C20 | 2.53233 | Li4@C20-trans | 3.30459 |
| Li2@C20 | 1.69844 | Li3@C20-trans | 2.52752 | Li5@C20 | 3.43132 |
| Li2@C20-trans | 1.69458 | Li4@C20 | 3.3199 | Li6@C20 | 3.62532 |
As shown in Table 5, increasing the number of Li atoms increased the CT values. As shown previously, the enthalpy of the Li atom adsorption improved as the CT increased. Fig. 5 plots the enthalpies of Li adsorption onto the C20 surface as a function of the CT value.
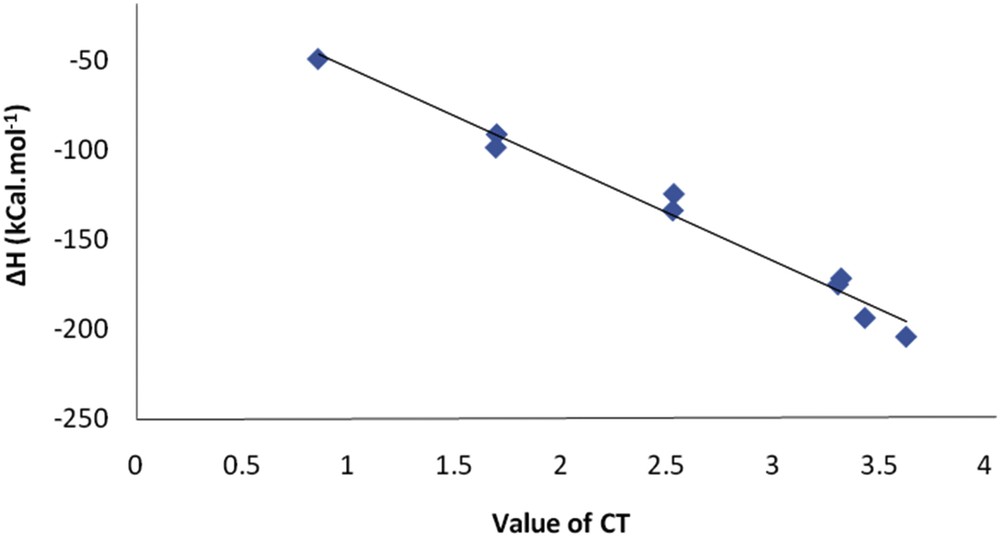
Change in enthalpy of Li adsorption as a function of CT value.
Fig. 5 reveals that increasing the electron transfer from the Li atoms to the C20 fullerene increases the exothermic reaction. As expected, the excess electrons transferred from the Li atoms to the C20 changed its electronic properties. As mentioned above, the NLO of the system altered in the presence of the Li atoms. The natural electron configurations of the Li atoms in the structures are summarized in Table 6. The results indicates that, by the adsorption of the Li atoms onto the surface of the C20, the p-character of the Li atoms in the Li⋯C interactions of the nanoclusters increased remarkably, particular in Li5@C20 and Li6@C20 nanostructures.
Natural electron configuration for the Li atom in nanoclusters.
| Compound | Spin multiplicity | Atom | Natural electron configuration |
| Li@C20 | 2 | Li | [He]2s(0.04)2p(0.10) |
| Li2@C20 | 1 | Li | [He]2s(0.03)2p(0.10)3p(0.01) |
| Li | [He]2s(0.04)2p(0.11)3p(0.01) | ||
| Li2@C20-trans | 1 | Li | [He]2s(0.06)2p(0.07)3p(0.01) |
| Li | [He]2s(0.06)2p(0.07)3p(0.01) | ||
| Li3@C20 | 2 | Li | [He]2s(0.04)2p(0.12) |
| Li | [He]2s(0.05)2p(0.12) | ||
| Li | [He]2s(0.04)2p(0.09) | ||
| Li3@C20-trans | 2 | Li | [He]2s(0.05)2p(0.11) |
| Li | [He]2s(0.06)2p(0.11) | ||
| Li | [He]2s(0.05)2p(0.11) | ||
| Li4@C20 | 1 | Li | [He]2s(0.05)2p(0.12) |
| Li | [He]2s(0.04)2p(0.13) | ||
| Li | [He]2s(0.05)2p(0.13) | ||
| Li | [He]2s(0.05)2p(0.12) | ||
| Li4@C20-trans | 1 | Li | [He]2s(0.08)2p(0.10) |
| Li | [He]2s(0.04)2p(0.12) | ||
| Li | [He]2s(0.08)2p(0.10) | ||
| Li | [He]2s(0.04)2p(0.12) | ||
| Li5@C20 | 2 | Li | [He]2s(0.13)2p(0.11) |
| Li | [He]2s(0.16)2p(0.13) | ||
| Li | [He]2s(0.12)2p(0.11) | ||
| Li | [He]2s(0.07)2p(0.12) | ||
| Li | [He]2s(0.46)2p(0.15) | ||
| Li6@C20 | 2 | Li | [He]2s(0.26)2p(0.14) |
| Li | [He]2s(0.26)2p(0.14) | ||
| Li | [He]2s(0.26)2p(0.14) | ||
| Li | [He]2s(0.26)2p(0.14) | ||
| Li | [He]2s(0.26)2p(0.14) | ||
| Li | [He]2s(0.26)2p(0.14) |
4 Conclusions
The present study calculated the structural, electronic, and optical properties of C20 and Lin@C20 (n = 1–6) nanoclusters. It was shown that the interaction of Li atoms onto the C20 surface is highly exothermic and the values of enthalpy per Li atom decreased slightly when the number of Li atoms increased. In addition, it can be seen that the adsorption of one to four Li atoms onto the C20 surface increased its gap energy. The addition of five or six Li atoms onto its surface decreased the value of Eg. NBO analysis of the nanoclusters showed that an increase in the number of Li atoms increased the value of electron transfer. It was shown that the enthalpy of the adsorption reaction increased as the value of the charge transfer reaction increased. The effect of the Li atoms on the NLO properties of C20 fullerene was explored. It was shown that the interaction of five or six Li atoms with C20 fullerene greatly increased its first hyperpolarizability, making the value of β0 for Li5@C20 very interesting.