

1 Introduction
Transition metal–based catalysis of CN bond formation through cross-coupling reactions is a powerful method for preparing important compounds that are highly demanded in pharmaceutical, material, and chemical industries [1–4]. For this reason the development of convenient and effective catalyst synthesis methods has attracted much attention. Some studies have reported successful CN coupling of nucleophilic aromatic groups with aryl halides by using catalysts based on palladium [2,4], nickel [3], and copper [5]. Lower cost of Cu-based catalysts has motivated their large-scale industrial applications. Therefore, synthetic chemists have attempted to focus on Cu-based catalysts and introduce milder methods for the synthesis of Cu-containing catalysts. One of the simplest and cheapest solutions of producing N-aryl nitrogen heterocycles is using the Ullmann-type coupling of aryl halides with nitrogen heterocycles [6–8]. Cu-based catalysts can catalyze Ullmann reactions, meantime their efficiencies can be promoted if their copper sources, ligands, bases, and other additives be selected, correctly.
Recently, several mild and efficient methods have been adopted for N-arylation of heterocyclic amines, for example, Refs. [9–11]. Although these reported methods of Cu-catalyzed N-arylation of amines are highly efficient, they rely on homogeneous catalysis and restrict separation of the catalysts from the reaction mixture. This issue can be overcome by developing recyclable and efficient heterogeneous catalysts through immobilization of catalytically active species, for example, organometallic complexes, on a solid support [12]. In this respect, many heterogeneous Cu-based catalytic systems have been proposed to perform cross-coupling reactions. Some notable examples include Merrifield resin–supported phenanthroline copper(I) complex [13], Cu immobilized on organic–inorganic hybrid materials [14], Cu immobilized on MCM-41 [15], Cu ferrite nanoparticles (NPs) [16], [Cu30I16(mtpmt)12(μ10-S4)] [17], Al2O3–supported Cu(II) catalyst [18], glycerol ingrained Cu [19], CuO nanocatalyst [20], and polymer-supported Cu(II) catalyst [21].
This study aims to prepare a silica-supported Cu catalyst. The major concern is that making silica-based hybrid materials with a high portion of organic functional groups is difficult because of the heterogenous distribution of active sites on silica particles. To achieve a homogeneous distribution of organic functional groups on silica-hybrid materials, several approaches, such as surface patterning, surface functionalization with dendrimers, and molecular/ion imprinting, have been reported [22–29]. As an alternative approach, our research group reported the design and preparation of an SBA-15/CCPy/Pd(II) nanocatalyst by grafting melamine-bearing pyridine groups on SBA-15 and depositing Pd NPs for stable catalysis of Suzuki–Miyaura reactions and N-arylation of indoles [[30], 4h]. Because the results of our previous works on synthetic application of nanocatalysts [31] were encouraging, herein we report a novel heterogeneous catalytic system based on Cu(OAc)2 immobilization on melamine-bearing pyridine-modified SiO2 particles (Scheme 1). This novel catalyst is air stable, can be reused, and shows excellent catalytic performance for CN coupling in Ullmann reactions.
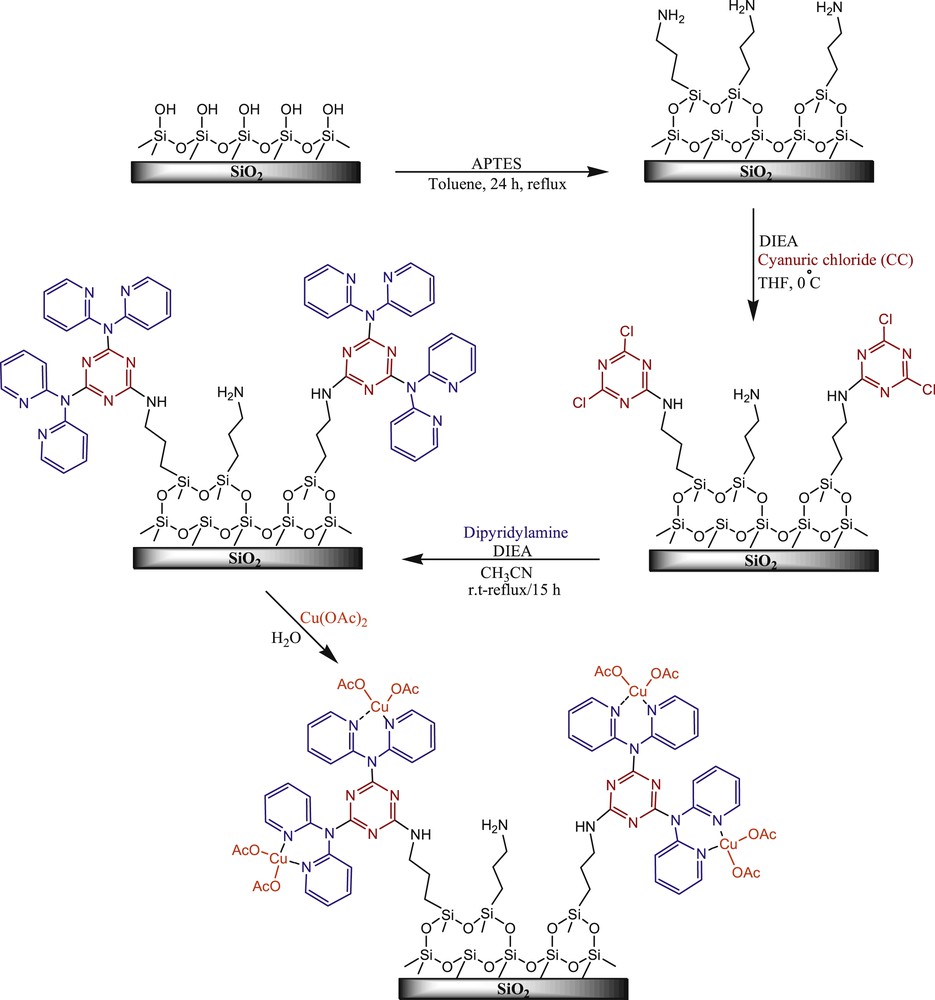
Schematic diagram of SiO2/CCPy/Cu(OAc)2 fabrications.
2 Experimental section
2.1 Preparation of SiO2/CCPy
Thirty milliliters of anhydrous toluene, 1.0 g of SiO2, and 0.36 g (3.0 mmol) of 3-aminoropropyl trimethoxysilane were added to a 100-mL round-bottom flask. The solution was refluxed under an inert atmosphere for 24 h, filtered, washed subsequently with toluene, dichloromethane, and methanol, and dried under reduced pressure at 80 °C for 10 h. In another 100-mL round-bottom flask, a solution of 1 g of aminopropyl-functionalized SiO2 in 35 mL THF was prepared and 0.5 mL of diisopropylethylamine was added to it. Then, 0.46 g (3 mmol) of cyanuric chloride (CC) was added to the functionalized SiO2 solution, at 0 °C. After 2 h, the solution was decanted and washed two times with 25 mL of THF. Then, 25 mL of acetonitrile and 1 mL of diisopropylethylamine were added to the residual solid. Next, 7 mmol dipyridylamine was added, the mixture was stirred for 2 h at room temperature and the resultant solution was refluxed for 12 h. After completion of the reaction, the solid product was filtered, washed with deionized water and then acetone, and dried at 80 °C for 12 h. In this way, dipyridylamine clung on triazine-functionalized SiO2 (SiO2/CCPy) was obtained.
2.2 Heterogenization of Cu(OAc)2 on the surface of SiO2/CCPy
Cu(OAc)2 (1.5 mmol) was dissolved in 50 mL of H2O and stirred for 0.5 h. Then, 1.0 g of the prepared SiO2/CCPy solid was added and stirred at room temperature for 24 h. After completion of the reaction, the blue solid product was filtered, washed with water, acetone, and ethanol, and dried under vacuum at 80 °C for 24 h to give SiO2/CCPy/Cu(OAc)2. Elemental analysis of the SiO2/CCPy/Cu(OAc)2 product by inductively coupled plasma-atomic emission spectrometry (ICP-AES) determined 0.32 mmol/g Cu loading.
2.3 N-Arylation of amines in the presence of SiO2/CCPy/Cu(OAc)2
In a 25-mL round-bottom flask, the synthesized catalyst (25 mg; 0.7 mol %), aryl halides (1 mmol), CH3CN (3 mL), amines (1 mmol), and Et3N (2 mmol) were mixed. Then, the mixture was stirred for the desired time at 50 °C. The reaction progress was observed by thin-layer chromatography (TLC). After completion of the reaction, the reaction mixture was cooled to room temperature and the catalyst was recovered by centrifugation and washed with ethanol and ethyl acetate. The combined organic layer was dried over anhydrous sodium sulfate and evaporated in a rotary evaporator under reduced pressure. The resultant material was purified by silica gel–based preparative TLC to separate the desired product.
2.4 Procedure for reusing the catalyst
At any specific reaction time, 5 mL of ethyl acetate was added to the reaction mixture and stirred for 5 min. After this time, the catalyst was separated by centrifugation. In the next step, the recovered solid was washed using ethanol and dried under vacuum. Then, the recovered catalyst was used in another catalytic run.
2.5 Large-scale N-arylation of indole in the presence of SiO2/CCPy/Cu(OAc)2
In a 250-mL round-bottom flask, the catalyst (0.5 g; 0.7 mol %), iodobenzene (20 mmol), CH3CN (60 mL), indole (20 mmol), and Et3N (40 mmol) were mixed. Then, the mixture was stirred for the desired time at 50 °C. The reaction was observed by TLC. After completion of the reaction, the reaction mixture was cooled to room temperature and the catalyst was separated by simple filtration and washed with ethanol and ethyl acetate. The combined organic layer was dried over anhydrous sodium sulfate and evaporated in a rotary evaporator under reduced pressure. The resultant material was purified by silica gel–based preparative TLC to obtain N-phenylindole with 95% yield.
3 Results and discussion
3.1 Catalyst preparation and characterization
The proposed catalyst was prepared by following the steps that are described in Scheme 1. In this synthesis process, first, SiO2 was chosen as the support material. Then, silica was reacted with 3-aminopropyltriethoxysilane to produce functionalized silica. In the next step, CC was immobilized on the surface of the functionalized SiO2 particles covalently while the temperature was under control. Then, the other two chloride functional groups of CC were replaced with 2 equiv of dipyridylamine through the formation of CN bonds between the triazine ring and the amine moieties. In the final step, the SiO2/CCPy/Cu(OAc)2 nanocatalyst was synthesized by reacting Cu(OAc)2 with SiO2/CCPy in water. According to ICP measurements, Cu loading of the prepared catalyst was about 0.32 mmol/g of the catalyst.
Fourier transform infrared (FTIR) spectroscopy was used to assure about incorporation of pyridine groups and melamine-based dendrimers in the silica framework. Fig. 1 shows the FTIR spectra of SiO2/CCPy and SiO2/CCPy/Cu(OAc)2. The typical 453, 794, and 1086 cm−1 bands of SiOSi are present in both samples and are attributed to the condensed silica network [32]. Also, the strong absorption band at about 3422 cm−1 (curves a and b) corresponds to the –OH stretching vibrations of the silanol groups [33]. The stretching frequencies of 2880 and 2950 cm−1 are associated with the asymmetric and symmetric CH stretching in the propyl chain, respectively. The new bands that have appeared at 1455 and 1561 cm−1 (curve a) belong to the aromatic triazine rings in the SiO2/CCPy sample and confirm formation of melamine-based dendrimers [27,34]. Also, availability of pyridine groups was verified by the 3050 cm−1 (CH aromatic stretching) and 1657 cm−1 (CN stretching) bands. These results approved successful functionalization of SiO2 by aminopropyl and melamine-bearing pyridine groups. The peak located at 1657 cm−1, in curve a, refers to metal–ligand coordination [35]. If this peak of curve a be compared with the most relevant peak in curve b, then it will be noticed that this peak has shifted to a lower frequency (from 1657 to 1648 cm−1). Therefore, the peaks of curves a and b approve successful linkage of organic pyridine ligands and coordination of Cu2+ ions to the hybrid silica support.
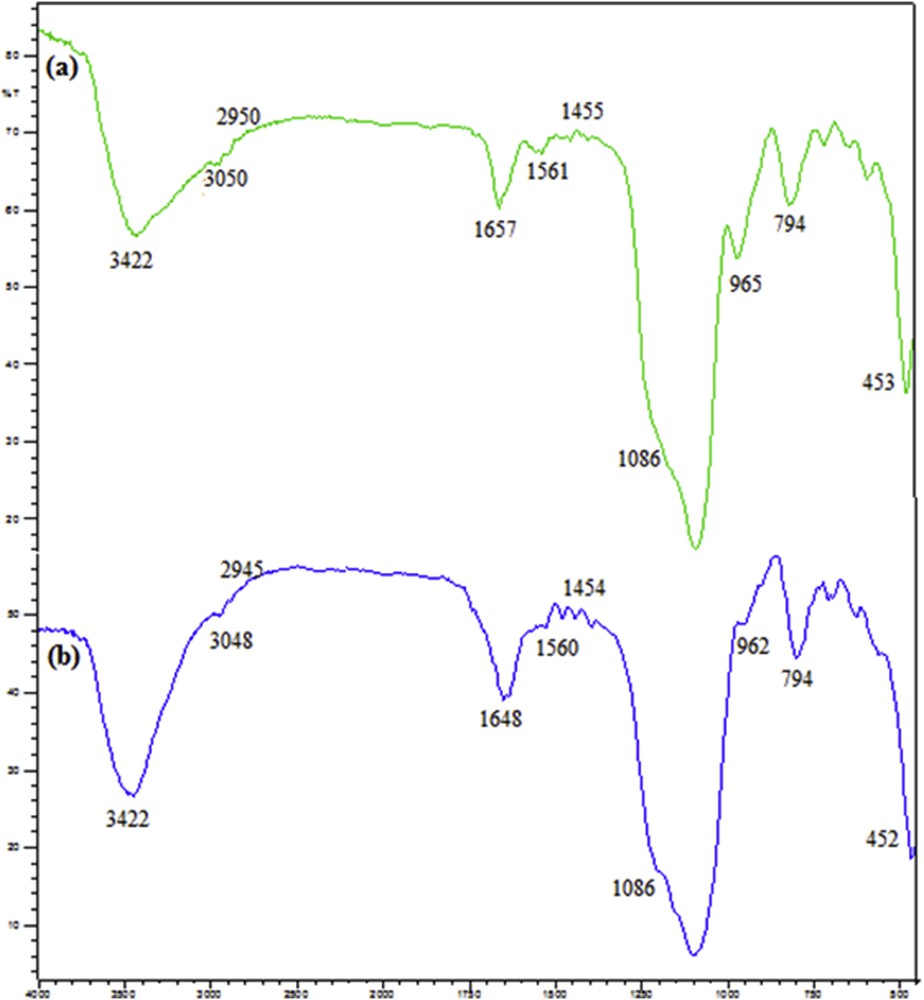
FTIR spectra of (a) SiO2/CCPy, and (b) SiO2/CCPy/Cu(OAc)2.
Particle size and the spherical structure of the silica NPs were revealed by transmission electron microscopy (TEM) (Fig. 2). The TEM image of SiO2/CCPy/Cu(OAc)2 shows that particle size of the synthesized catalyst is in the nanometer range and the catalyst retains the morphology of the silica support after functionalization. Furthermore, the TEM results demonstrate a nearly uniform size distribution for the SiO2/CCPy/Cu(OAc)2 particles and clarify that the silica NPs with 10–15 nm dimension are self-aggregated throughout the sample. Also, some randomly distributed micropores, that is, the white spots, are clearly visible in each porous silica NP.

TEM image of SiO2/CCPy/Cu(OAc)2.
The EDX spectrum of the catalyst approved successful functionalization of the silica particles and outlined the existence of C, N, and Cu elements in addition to Si and O (Fig. 3). Moreover, scanning electron microscopy (SEM) of the silica NPs was performed. In the obtained microgram (Fig. 4), the observed particles are mostly spherical. Also, self-aggregation of the particles is evident throughout the sample. Therefore, it can be stated that the self-aggregation nature of the silica NPs is conserved even after modification of their surfaces with organic functional groups.
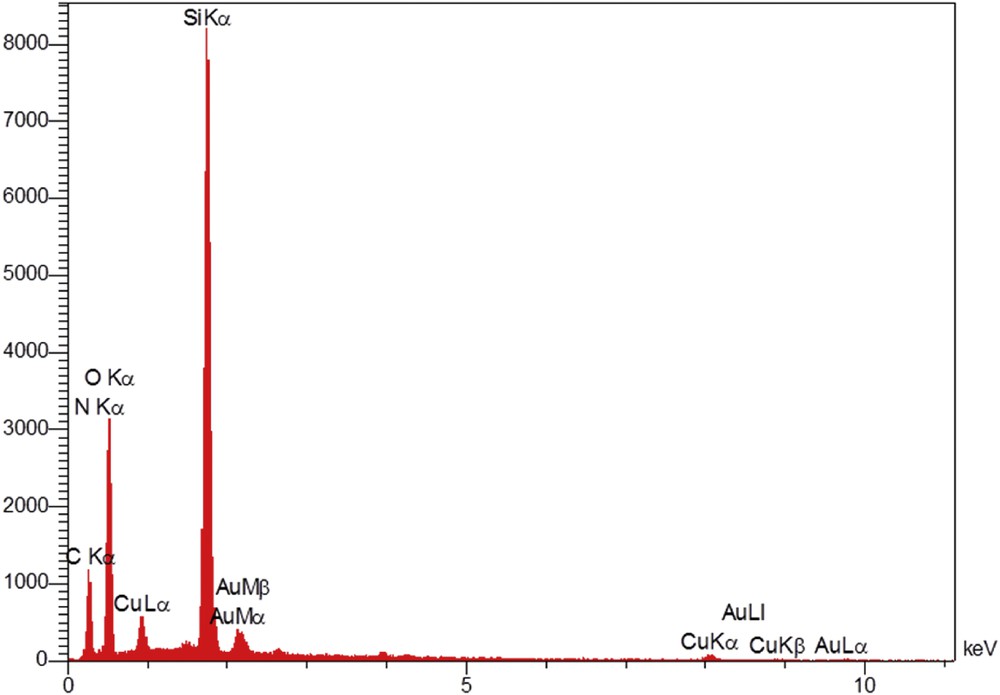
EDX spectrum of SiO2/CCPy/Cu(OAc)2.

FESEM image of SiO2/CCPy/Cu(OAc)2.
Formation of a bond between the silica NPs and the Cu catalyst can be inferred from thermogravimetric analysis (TGA) (Fig. 5). TGA curve of the SiO2/CCPy/Cu(OAc)2 catalyst displays two steps of weight loss above 220 °C. The weight loss observed at temperatures less than 200 °C is attributed to desorption of physisorbed solvent molecules and surface hydroxyl groups. One the other hand, the organic groups desorb at temperatures greater than 230 °C. Therefore, the observed weight loss, that is, about 11.3% from 230 to 500 °C, has resulted from decomposition of the organic spacer grafted on the silica surface.
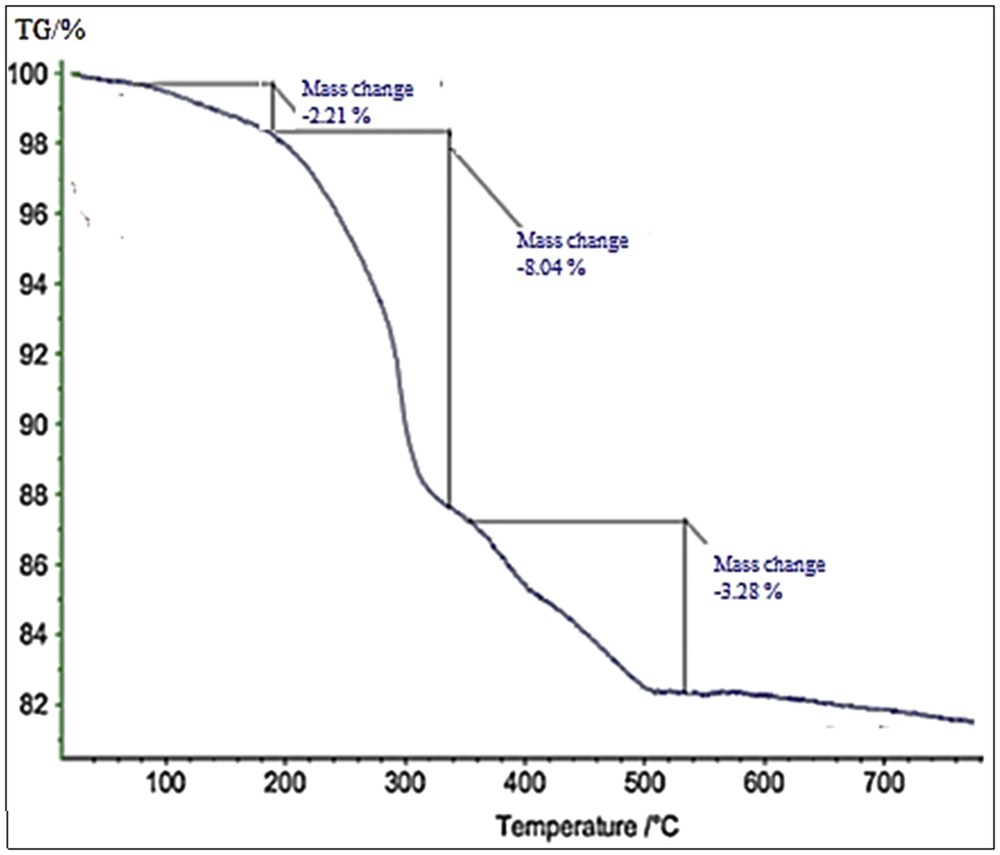
TGA diagram of the catalyst.
Nitrogen adsorption isotherms of the as-synthesized SiO2/CCPy/Cu(OAc)2 catalyst were evaluated to calculate its porosity and specific surface area, which suggested that the sample is mostly microporous (Fig. 6a). The obtained adsorption/desorption isotherms can be related to type I microporous materials [36]. Moreover, at a relatively low pressure of nitrogen gas, the slopes of the curves are very small, which indicate existence of micropores. However, upon increase in relative pressure, the slopes of the curves increase dramatically. Also, some loops can be seen in the curves, which indicate the presence of interparticle mesopores [37]. On the basis of the results, surface area and pore volume of SiO2/CCPy/Cu(OAc)2 are 302.31 m2 g−1 and 0.7 cm3 g−1, respectively, and the catalyst presents a wide distribution of pore size peaking around ca. 100 Å (Fig. 6b).
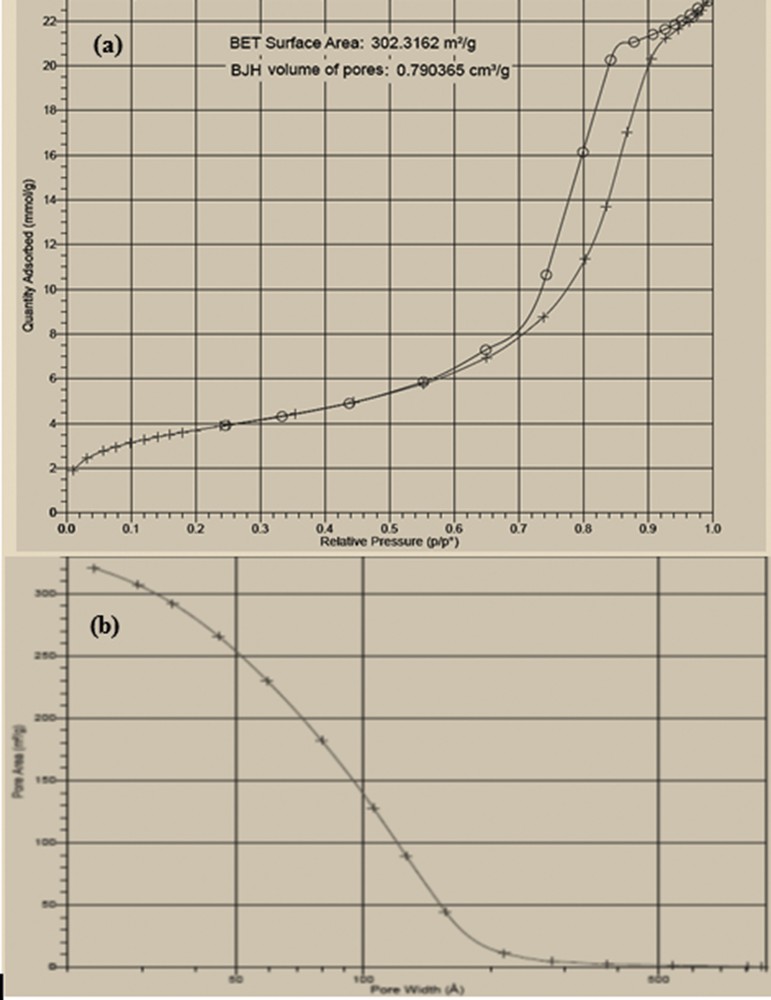
(a) N2 adsorption isotherm, (b) pore size distribution of SiO2/CCPy/Cu(OAc)2.
The crystallite structure of the catalyst was investigated by X-ray diffraction (XRD) spectroscopy. No crystalline impurity was detected in the wide-angle XRD spectra (Fig. 7). The broad XRD peaks belong to the silica matrix. In addition, electron paramagnetic resonance (EPR) spectroscopy was used to probe the heterogeneous copper catalyst. When conducting EPR evaluation on SiO2/CCPy/Cu(OAc)2, a strong EPR signal was observed (Fig. 8), which is consistent with the literature [38]. This result implies that single electrons exists in the catalyst, which means that the Cu sites are in their Cu(II) status.
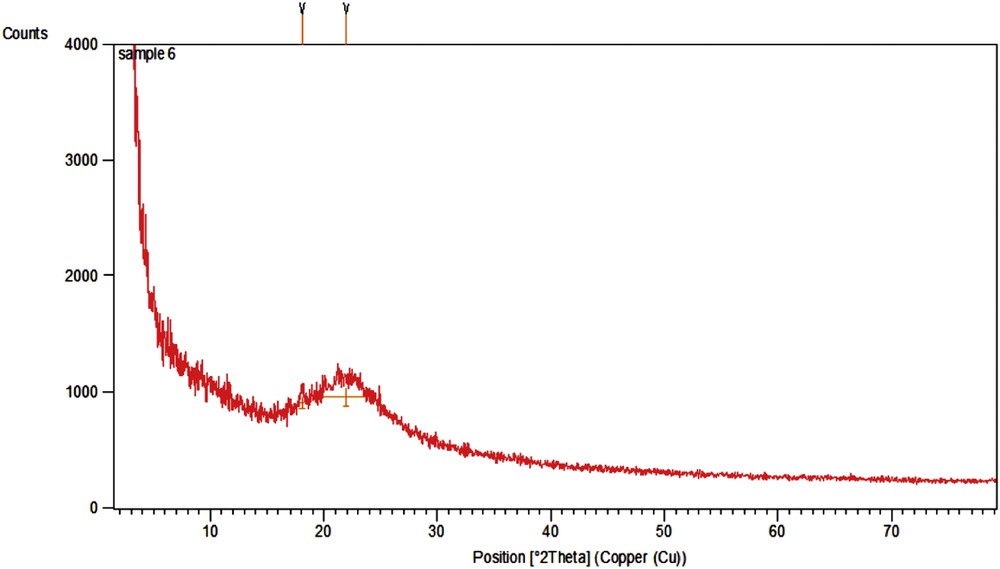
Wide-angle XRD pattern of SiO2/CCPy/Cu(OAc)2.
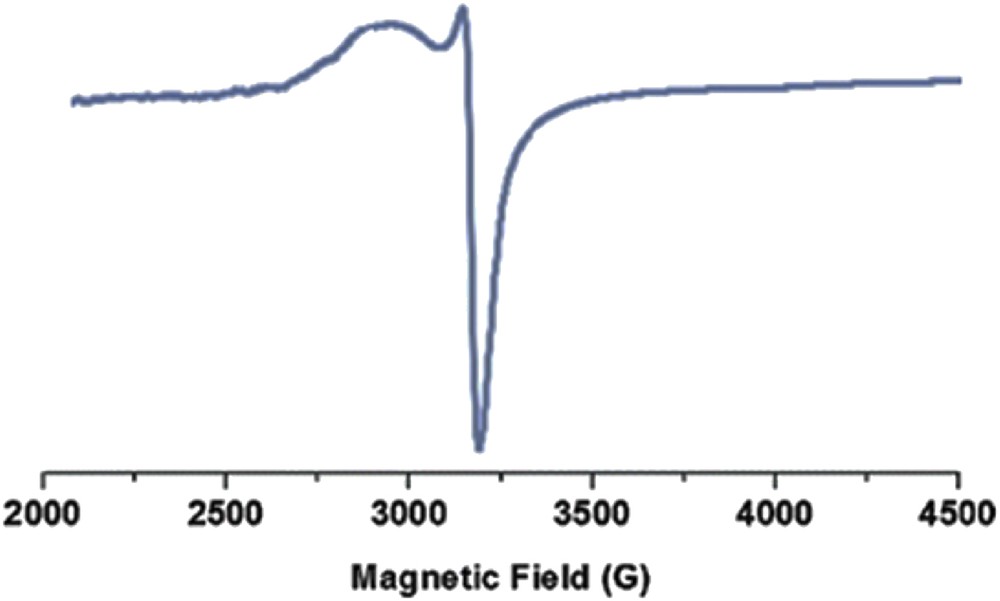
EPR spectrum of SiO2/CCPy/Cu(OAc)2.
After structural characterization of SiO2/CCPy/Cu(OAc)2, as a novel nanocatalyst, its catalytic activity was evaluated in the Ullmann coupling reaction of amines with aryl halides under aerobic conditions (Scheme 2). To optimize the reaction conditions, the reaction between bromobenzene and indole was chosen as a model reaction (Table 1). Then, the effects of different parameters on this model reaction were considered to get the best possible combination of various effects imposed by the solvent, reaction temperature, base, catalyst loading, and reaction period.
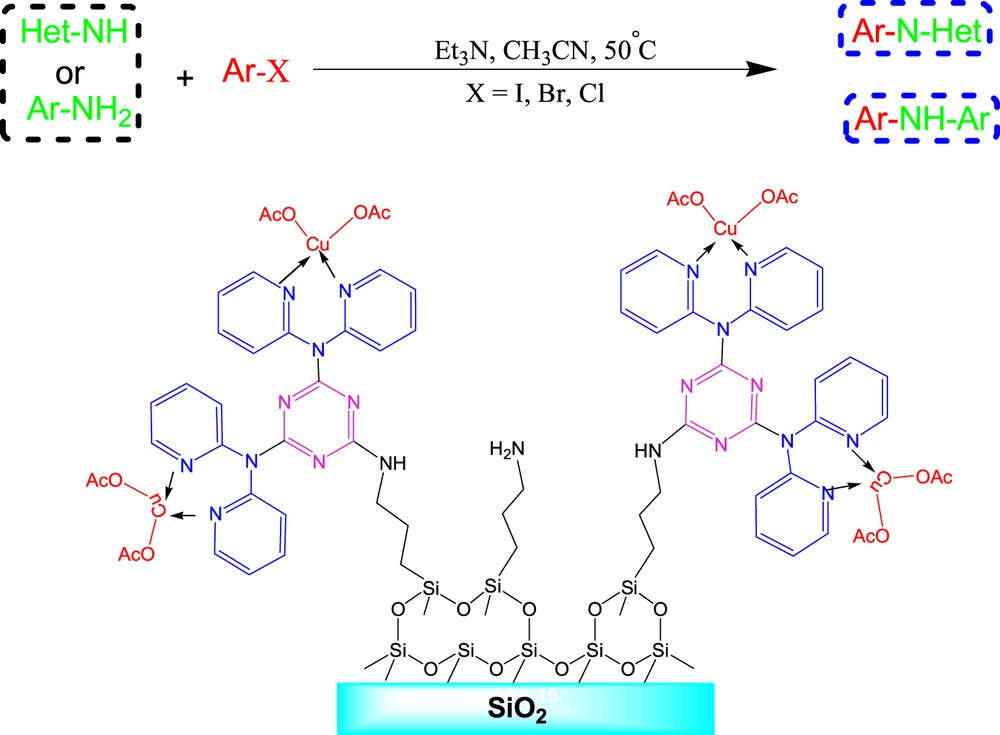
SiO2/CCPy/Cu(OAc)2 catalyzed N-arylation of amines.
Optimization of reaction condition for the N-arylation of indole with bromobenzene.a
| Entry | Catalyst (mol %) | Base | Solvent | T (°C) | t (h) | Yield (%)b |
| 1 | 0.5 | Et3N | CH3CN | 50 | 4 | 90 |
| 2 | 0.5 | Et3N | EtOH | 50 | 6 | 55 |
| 3 | 0.5 | Et3N | Toluene | 50 | 6 | 75 |
| 4 | 0.5 | Et3N | H2O | 50 | 6 | 20 |
| 5 | 0.5 | Et3N | CH2Cl2 | 50 | 6 | 35 |
| 6 | 0.5 | Et3N | DMF | 50 | 5 | 88 |
| 7 | 0.3 | Et3N | CH3CN | 50 | 5 | 60 |
| 8 | 0.7 | Et3N | CH3CN | 50 | 1 | 96 |
| 9 | 0.7 | – | CH3CN | 50 | 24 | Trace |
| 10 | – | Et3N | CH3CN | 50 | 24 | 0 |
| 11 | 0.7 | K2CO3 | CH3CN | 50 | 2 | 60 |
| 12 | 0.7 | Na2CO3 | CH3CN | 50 | 2 | 55 |
| 13 | 0.7 | KOH | CH3CN | 50 | 2 | 75 |
| 14 | 0.7 | NaHCO3 | CH3CN | 50 | 2 | 60 |
| 15 | 0.7 | Et3N | CH3CN | 25 | 2 | 35 |
| 16 | 0.7 | Et3N | CH3CN | 80 | 1 | 96 |
| 17 | 0.8 | Et3N | CH3CN | 60 | 1 | 96 |
a Reaction conditions: indole (1.0 mmol), bromobenzene (1.1 mmol), catalyst, base (2.0 mmol), solvent (3.0 mL).
b Isolated yield.
As the first factor, solvent effect was examined by using toluene, CH3CN, H2O, DMF, CH2Cl2, and ethanol in the presence of 0.7 mol % Cu(II) and 2 equiv Et3N, at 50 °C (Table 1). The reaction was considerably influenced by the nature of the solvent (entries 1–6, Table 1) and the highest yield was acquired in CH3CN (entry 1; 90%). Then, the impacts of Et3N, KOH, Na2CO3, NaHCO3, and K2CO3 bases were explored. Et3N resulted in the highest product yield (entries 1 and 11–14, Table 1). Furthermore, it was found that the presence of a base is necessary because a low yield can be obtained without applying any base (entry 9, Table 1). Also, variation in the reaction temperature posed a major influence on the model reaction (entries 8, 15, and 16, Table 1). Accordingly, reducing temperature from 50 to 25 °C impacts product yield negatively (entry 15, Table 1), meantime temperature increases to 80 °C make no difference (entry 16, Table 1). In addition, the effect of a catalyst amount was investigated and increasing its amount from 0.7 to 0.8 mol % afforded an excellent yield (entry 17; 96%) whereas using 0.3 mol % of the catalyst decreased reaction yield to 60% (entries 6–8, Table 1). Moreover, it should be emphasized that N-arylation of indole did not occur in the absence of the Cu(II) catalyst (entry 10, Table 1). Consequently, it was decided to continue the catalytic studies using CH3CN solvent, Et3N base, and 0.7 mol % SiO2/CCPy/Cu(OAc)2 at 50 °C under aerobic conditions, as the optimal conditions (entry 8, Table 1).
The optimum conditions were set and the catalyst was applied to a broader range of aryl halides and indole. In general, the conducted Ullmann coupling reactions between aryl iodides/bromides and indole were clean and gave high yields (entries 1–5, Table 2). However, lower yields were gained when chloride was used as the leaving group (entries 3 and 6, Table 2). On the basis of the results, the following order of reactivity rules this catalytic reaction: RI > RBr > RCl.
SiO2/CCPy/Cu(OAc)2 catalyzed coupling of aryl halides and amines.a
| Entry | Aryl halides | Amines | Time (h) | Yield (%)b | Reference |
| 1 | Ph-I | Indole | 0.5 | 96 | [39] |
| 2 | Ph-Br | Indole | 1 | 96 | [39] |
| 3 | Ph-Cl | Indole | 12 | 75 | [39] |
| 4 | 4-Me-Ph-I | Indole | 0.5 | 94 | [39] |
| 5 | 4-Me-Ph-Br | Indole | 1.5 | 92 | [39] |
| 6 | 4-Me-Ph-Cl | Indole | 12 | 70 | [39] |
| 7 | Ph-I | 1H-Imidazole | 1 | 96 | [40] |
| 8 | Ph-Br | 1H-Imidazole | 4 | 80 | [40] |
| 9 | Ph-Cl | 1H-Imidazole | 12 | 65 | [40] |
| 10 | Ph-I | 1H-Pyrazole | 2 | 92 | [41] |
| 11 | Ph-Br | 1H-Pyrazole | 4 | 85 | [41] |
| 12 | Ph-Cl | 1H-Pyrazole | 24 | 60 | [41] |
| 13 | Ph-I | Benzimidazole | 3 | 90 | [4d] |
| 14 | Ph-Br | Benzimidazole | 5 | 80 | [4d] |
| 15 | Ph-I | Benzyl amine | 0.5 | 98 | [41] |
| 16 | Ph-Br | Benzyl amine | 0.5 | 96 | [41] |
| 17 | Ph-Cl | Benzyl amine | 4 | 90 | [41] |
| 18 | Ph-I | Aniline | 1 | 96 | [40] |
| 19 | Ph-Br | Aniline | 2 | 90 | [40] |
| 20 | Ph-Cl | Aniline | 12 | 75 | [40] |
| 21 | Ph-I | Morpholine | 2 | 90 | [42] |
| 22 | Ph-Br | Morpholine | 3 | 80 | [42] |
a Reactions were carried out under aerobic conditions in 3 mL of CH3CN, 1.1 mmol aryl halides, 1.0 mmol amines, and 2 mmol Et3N in the presence of the Cu(II) nanocatalyst (20 mg, 0.7 mol %) at 50 °C.
b Isolated yield.
To explore activity of the proposed catalyst in a broader scope, Ullmann-type CN coupling reaction of other amines, including imidazole, pyrazole, benzimidazole, benzyl amine, aniline, and morpholine, with aryl halides was investigated (entries 7–22, Table 2). The target products were achieved in moderate to high yields. The most important observation was that the reaction is very selective and gives N-arylated products, only. However, C-arylation of the amines was never observed.
To determine if the entire SiO2/CCPy/Cu(OAc)2 complex acts as the catalyst or the Cu(II) complex separates from the support during the reaction, acts as a homogeneous catalyst, and returns to the support in the end, the reaction between bromobenzene and indole was terminated after 0.5 h at 60% conversion. Then, the catalyst was filtered and the reaction was continued with the filtrate for an additional 0.5 h. After this hot filtration process, the reaction yield remained constant. Therefore, it can be inferred that the Cu(II) catalyst has not detached from the SiO2/CCPy support, at 50 °C, and the reaction process is of heterogeneous nature.
Reusability of heterogeneous catalysts is an important issue, specifically in commercial applications. As a consequence, recyclability of SiO2/CCPy/Cu(OAc)2 was investigated by considering the reaction of bromobenzene with indole under the optimum reaction conditions. After completion of the reaction, the reaction mixture was cooled down to room temperature. Then, EtOAc was added to the solution and the catalyst mixture was centrifuged, washed with ethanol and warm water, and reused several times. The obtained data are displayed in Fig. 9 and imply that the catalyst can be reused up to five times with negligible loss of its catalytic activity. Reusability and high stability of SiO2/CCPy/Cu(OAc)2 should be due to the chelated pyridine groups on Cu(II) and porosity of the SiO2support.
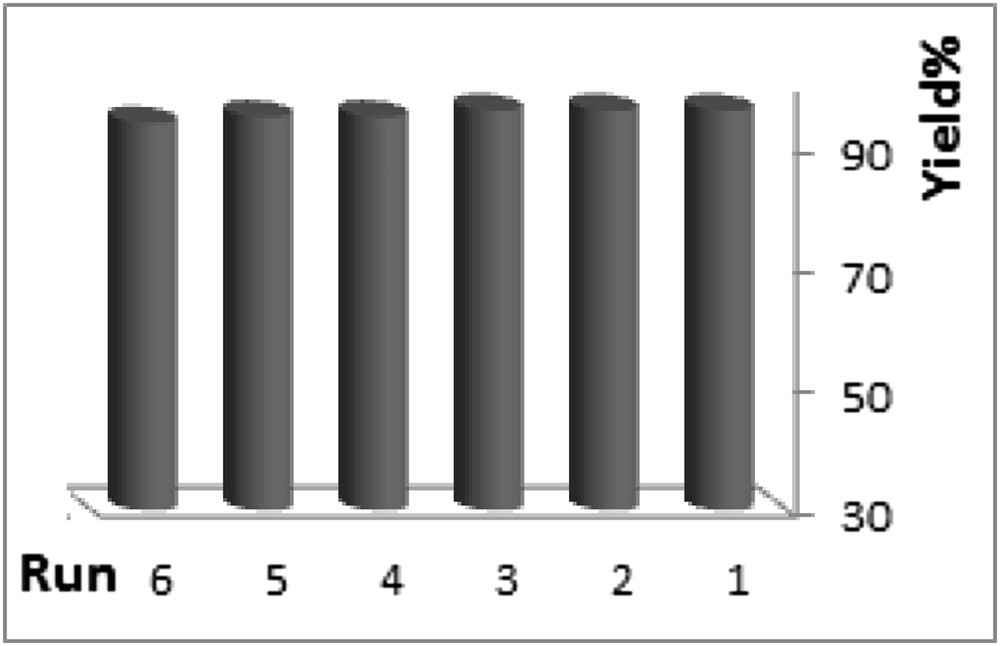
The recycling of the SiO2/CCPy/Cu(OAc)2.
Again, TEM, SEM, and EDX measurements were performed on the SiO2/CCPy/Cu(OAc)2 nanocatalyst, which was reused five times, to verify that its nanostructure has not altered. The results demonstrated that the size of the catalyst is the same as the fresh catalyst and its morphology and composition have not changed (Fig. 10).
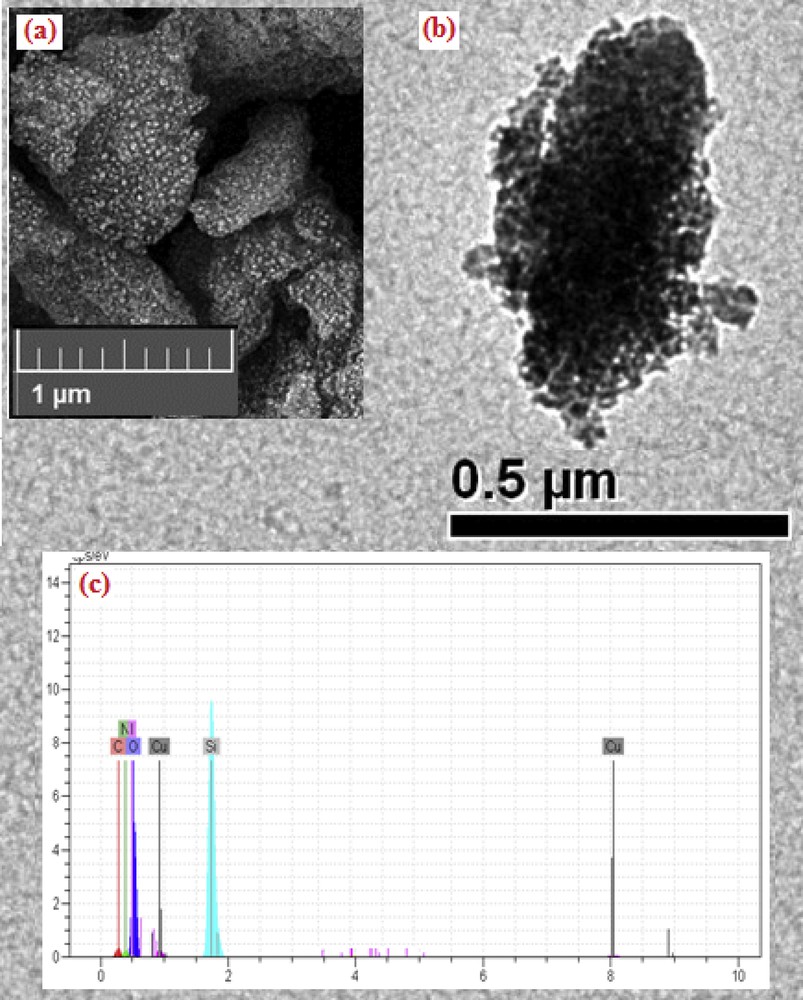
SEM (a), TEM (b), and EDX (b) images of recovered SiO2/CCPy/Cu(OAc)2.
4 Conclusions
This study introduces and prepares a modified silica supported copper catalyst for efficient N-arylation of N(H)-heterocycles, aniline, and benzyl amine by aryl halides, under mild conditions. TEM, FESEM, EDX, TGA, XRD, BET, and ICP characterization techniques confirmed the structure of the SiO2/CCPy/Cu(OAc)2 nanocatalyst. Also, the results revealed that the catalyst can be conveniently separated by filtration and reused for several more reaction cycles without any significant changes in its activity. The results demonstrated the advantages of the synthesized catalyst, that is, low catalyst loading, high product yield, experimental simplicity, broad substrate scope, and short reaction time.
Acknowledgments
We are thankful to Payame Noor University (PNU) and Kurdistan University of Iran for financial supports.