

1 Introduction
The development of organic materials with optimized nonlinear optical (NLO) properties has attracted the attention of several researchers in several fields such as materials science; biophysics; energy; chemical dynamics; telecommunication sector; optics; optoelectronic devices; and atomic, molecular, and surface interface sciences. This is attributed to their specific properties, flexibility, and low cost of production compared with inorganic materials [1–3]. This kind of materials exhibits large molecular NLO responses. Metal-free organic NLO materials are considered the best NLO materials which are extensively used; this is attributed to their high electrical polarization, optical switching, and optical modulation, which make them suitable candidates to the researchers for modeling their chemical structures for large NLO properties [4,5].
Several metal-free organic dyes having a D-π-A structure have been synthesized and studied as sensitizers, especially as dye-sensitized solar cells (DSSCs) [6–9]. This structure (D-π-A) ensures the push–pull system and then promotes the intramolecular charge transfer (ICT) and charge separation upon photoexcitation [10–14]. The first hyperpolarizability (β, second-order NLO properties) of organic dyes is linked effectively to the delocalization of π electrons in the π-bond system by causing ICT from the electron-donating moiety (D) to an electron-withdrawing moiety (A) via a π-conjugated spacer [15–17]. The NLO properties of any organic dye can be controlled based on NLO properties of the basic molecular of the same dye. Following this criterion, several efforts have been made to design highly efficient organic NLO dyes [18,19]. The organic dyes containing the switching of suitable substituents (D and A groups) at the appropriate position of the π-conjugated spacer show an asymmetric electronic distribution and present a high NLO activity [20,21]. The donor moiety can enhance the spectral properties, electronic energy levels, and interfacial phenomena, while the conjugation between the π-bridge and the donor can be exploited to determine the efficiency of charge separation [22–25]. The triphenylamine moiety is widely used in organic electronics as an excellent electron source, offering extensive possibilities for controlling the energy levels, charge generation and separation, as well as light harvesting [26–28]. The π-linker that includes thiophene [28–31], ethene [32,33], or benzene moiety [34–37] is considered an excellent part of the charge transportation, which leads to a high NLO response of the studied dyes. Moreover, these dyes present smaller geometric relaxation energy upon oxidation [38–40]. Furthermore, the acceptor groups of metal-free organic dyes strongly influence the electron injection upon photoexcitation and then directly influence the photophysical properties of the NLO material. These groups are also considered responsible for anchoring the sensitizing dye on the surface of the semiconductor [41–43]. The acceptor group used for the anchoring of the sensitizing dye on the surface of the semiconductor is the carboxylic acid (−COOH); this is attributed to its relative stability, strong binding, and good electron communication between the dye and the surface of the semiconductor, forming an ester linkage [44,45].
To shift the spectral response of this chromophore toward the red region and at the same time to fine-tune the HOMO and the LUMO energy levels of the sensitizer, a series of dyes were designed theoretically (Fig. 1). In this series, the π-linker between the donor (triphenylamine) and the acceptor (cyanoacetic acid) was systematically extended to adjust the HOMO and LUMO energy levels [27], with an objective to achieve the redshift, broadening the absorption spectra. Moreover, the presence of electron-rich triphenylamine as a donor (D) and the cyanoacetic group as an acceptor moiety (A) in the D-π-A architecture allows the increase of the electronic displacement between the two poles donor and acceptor, subsequently increasing their chemical activity [46]. In this study, we present eight dyes for electronic applications by describing the possibility of improving their efficiency and stability using the D-π-A structure, and then we discuss the limitations and perspectives.
Structures of the eight dyes L1–L8.
2 Computational details
All quantum calculations were performed using Gaussian 09 package [47]. The optimization geometry of all compounds in the ground state has been carried out by the DFT approach with B3LYP exchange-correlation function combined with 6-31G(d,p) basis set [48–53]. The natural bond orbital (NBO) [54] analysis has been predicted by the calculated orbital populations using NBO 6.0 program integrated into the Gaussian 09 package program. For the absorption spectra and vertical absorption energies, the choice of a larger basis set as 6-31+G(d,p) and high-level method are essential for reliable results. Therefore, we used the TD-DFT approach with coulomb-attenuated hybrid exchange-correlation functional (CAM-B3LYP) [55]. This hybrid functional is destined to take the long-range properties into account and is considered the long-range-corrected version of B3LYP. The transition energies have been successfully estimated by using this functional as reported in the literature [14,22,56,57]. Moreover, the integral equation formalism for the polarizable continuum model (PCM) [58,59] has been used in this work with an objective to take the solvent effect on the excitation energy into account and to predict the experimental spectra with reasonable accuracy. This model (PCM) considers the solvent as a single continuous medium; in this case, two kinds of PCMs have been built: the dielectric PCM (D-PCM) in which the continuum is polarizable and the conductor-like PCM (C-PCM) in which the continuum is conductor-like, similar to conductor-like screening solvation model, in which the molecular free energy of solvation is estimated as the sum of three terms: Gsol = Ges + Gdr + Gcav, where Ges, Gdr, and Gcav are the electrostatic, dispersion-repulsion, and cavitation energy, respectively. On the other hand, Mennucci et al. [60] observed that PCM has limitations where nonelectrostatic effects dominate the solute–solvent interactions. Moreover, this team noted the following finding: “Because only electrostatic solute–solvent interactions are included in the PCM, the results lead to the conclusion that for the seven molecules studied, in cyclohexane, acetone, acetonitrile, and methanol, electrostatic effects are dominant, whereas in carbon tetrachloride, chloroform, and benzene, other nonelectrostatic effects are more important” [60]. On the other hand, the average polarizability <α> has been estimated using Eq. 1 and considering only diagonal elements [61,62].
| <α> = 1/3 (αxx + αyy + αzz) | (1) |
The output file obtained using the Gaussian software provides 10 hyperpolarizability tensors along the x, y, and z directions: βxxx, βxyy, βxzz, βyyy, βxxy, βyzz, βzzz, βxxz, βyyz, βxyz. The total first hyperpolarizability (βtot) is calculated using Eq. 2 [61,62].
| βtot = [(βxxx + βxyy + βxzz)2 + (βyyy + βxxy + βyzz)2 + (βzzz + βxxz + βyyz)2]1/2 | (2) |
3 Results and discussion
3.1 Screening of a π-conjugated bridge
The π-conjugated linkers are considered key for high-performance metal-free organic dyes with the D-π-A structure. The geometrical parameters and optimized structures of all studied dyes are illustrated in Table 1 and Fig. 2, respectively.
Brief structure of dyes L1–L8 and the calculated dihedral angles (Φ1 and Φ2 represent the dihedral angles).Image 1
| Dyes | Φ1 (°) | Φ2 (°) |
| L1 | 0.81 | 20.34 |
| L2 | 0.02 | 1.63 |
| L3 | 0.17 | 22.15 |
| L4 | 0.03 | 0.68 |
| L5 | 36.11 | 2.79 |
| L6 | 30.04 | 1.20 |
| L7 | 13.40 | 1.18 |
| L8 | 19.83 | 1.15 |

Optimized ground-state structures of the eight studied dyes Li (i = 1–8).
The dihedral angle Φ1 between π-linkers and acceptor (cyanoacetic acid) for L1–L4 dyes is close to 0°, and that for L5–L8 is 36.11°, 30.04°, 13.40°, and 19.83°, respectively, which is attributed to the electrostatic effect caused between the cyanide group and the substituted group in adjacent thiophene especially for the –CH3 group which could break up the conjugation or may be decoupled from the π-extended system of the molecule and then inhibit the ICT especially for L5 and L6. The dihedral angle Φ2 formed between the triphenylamine moiety and π-linker is 1.63°, 0.68°, 2.79°, 1.20°, 1.18°, 1.15°, and −0.02° for all dyes L1–L8 respectively. This angle is close to 0°, except for L1 (20.34°) and L3 (22.15°), which is attributed to the hydrogen-hydrogen repulsion and even for the carbon-carbon repulsion of the two adjacent rings benzene and thiophene. These repulsions can help to inhibit the close intermolecular π–π aggregation between the π-linker and triphenylamine unit and then decreases the ICT for these dyes (L1 and L3).
3.2 Electronic structure and intramolecular electron transfer
Most electronic properties of π-conjugated compounds are based on the energy level of the frontier molecular orbitals (FMOs) HOMO, LUMO, and even difference between them. Indeed, the control of these parameters of the resulting molecular materials has become progressively a major topic for the chemical engineering and of functional π-conjugated systems. The gap energy of a π-conjugated system depends on various structural factors such as bond length alternation, chain length, planarity, the presence of electron-acceptor or electron-donor moieties, and the resonance stabilization energy of the aromatic cycles.
On the other hand, the exchange-correlation functionals LDA and GGA are destined principally to describe the electronic properties and electronic transitions of π-conjugated molecules. This is attributed to the rapid decay of their long-range potential [63,64]. For these reasons, the hybrid functionals have been introduced to correct this situation by including an exact exchange portion of Hartree–Fock [65]. Indeed, it has been recently shown that the hybrid functional B3LYP correctly describes the position of the electron levels and the exciton effects in the π-conjugated molecules [66,67]. However, the accuracy of this function decreases with the increasing length of molecular chains [68,69]. Several methods have been developed to modify the asymptotic behavior of hybrid functionals and improve the description of these systems such as PBE1PBE [70], B3PW91 [71], and so on.
The calculated HOMO/LUMO energy levels of all dyes are tabulated in Table 2. We found that the energy gaps of L1, L2, L3, and L4 are 2.66, 2.47, 2.26, and 2.09 eV, respectively, which are close to those obtained experimentally, 2.64, 2.48, 2.38, and 2.35 eV, respectively. This good agreement indicates that the adopted computational methodology is appropriate to investigate the L1–L8 dyes. Through Table 2, we remark that the energy gap decreases in the following order: L1 > L2 > L3 > L4 > L5 > L6 >L7 > L8; this proves that the π-conjugated linkers used as a bridge in these dyes have a huge effect on HOMO and LUMO energy levels especially for L7 and L8. Furthermore, the dye L8 has the least value of the energy gap (1.589 eV) compared with other dyes.
Energies of the HOMO, LUMO, and gap (LUMO − HOMO) of the studied dyes obtained using DFT/B3LYP/6-31G(d,p).
| Dye | HOMO | LUMO | Gap (LUMO − HOMO) |
| L1 | −5.27 | −2.61 | 2.66 (2.64)a |
| L2 | −5.15 | −2.68 | 2.47 (2.48)a |
| L3 | −5.05 | −2.79 | 2.26 (2.38)a |
| L4 | −4.94 | −2.84 | 2.09 (2.35)a |
| L5 | −4.99 | −2.91 | 2.09 |
| L6 | −5.00 | −2.96 | 2.05 |
| L7 | −4.74 | −3.04 | 1.71 |
| L8 | −4.87 | −3.28 | 1.59 |
a Experimental values in parentheses are from the study by [52].
The weak bandgap of L7 and L8 might be due to the presence of fused benzene and pyrazine rings on the thiophene ring into the π-spacers, especially for L8 possessing two nitrogen atoms. This extended π-conjugation configuration for these latest compounds (L7 and L8) facilitates the benzene and thiophene ring to behave as an electron-withdrawing unit, which might lead to the stability of their LUMO especially for L8 (−3.28 eV) compared with others dyes (Table 2). Moreover, we remark that the LUMOs of the studied dyes L7 and L8 are close to the valence band (VB) of TiO2 (−4.0 eV), which indicates that the energy barriers of electron migration will be smaller and the injection will be efficient when we use them as DSSCs.
However, it is distinct that the energy levels of the HOMOs and the LUMOs for the dye L8 as well as its bandgap can be tuned remarkably by the introduction of nitrogen as an electron-withdrawing atom into the π-linker. The electron-withdrawing property is confirmed by electronegativity of four N atoms among others atoms in the π-linker of L8, and their charge densities are calculated in the range of −0.452 to −0.488.
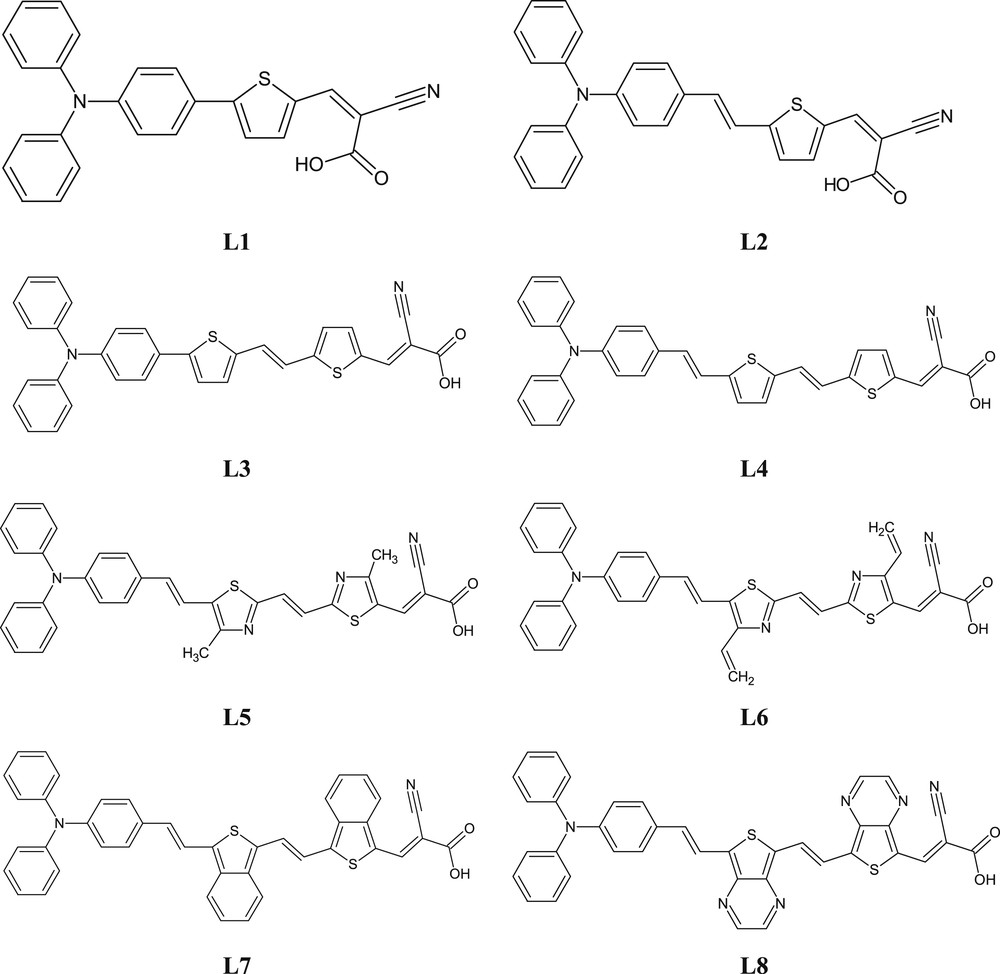
A strong intramolecular charge-transfer interaction between a donor and an acceptor via a π-linker with smaller transition energy and even the best overlap between density electronic of HOMO and LUMO orbitals play a crucial role in the smallest energy gap [72–74]. The highest value of energy gap has been observed to be 2.66 eV for L1; this energy gap was also computed higher for L2 (2.47 eV) and L3 (2.26 eV) than that of other investigated dyes. Generally, all studied dyes (L1–L8) have small energy gaps, which indicates that these studied dyes would be excellent candidates for NLO properties. The structural tailoring by modifying the π-conjugated linkers would be an excellent strategy to obtain decent NLO activity. The electron density distributions of HOMO and LUMO orbitals are represented in Fig. 3. These distributions are valuable for proficient charge transfer. From Fig. 3, it can be seen that a major portion of HOMO is localized on the donor moiety and a small portion of this density is localized on the π-conjugated linkers, whereas LUMO is mostly localized on the acceptor moiety and partially on the π-conjugated linkers. This indicates that charge transforms from the donor moiety (triphenylamine) toward the acceptor moiety (cyanoacetic acid) via the π-conjugated linker for all studied dyes. This considerable charge transfer confirms that all investigated dyes (L1–L8) would be brilliant NLO materials.

Frontier molecular orbitals of studied dyes.
In addition, through Table 3, we remark that the contribution of the acceptor group skeleton in the HOMO orbital of all dyes L1–L8 ranges from 1% to 11%, indicating that the donor moiety and π-conjugated linkers of the studied dyes disperse the electron density and make the electrons more delocalized in the acceptor system, especially for L7 and L8, and consequently decrease the energy levels of the FMOs.
Contribution of the donor and acceptor groups to the electron density of the main molecular orbitals (%) and the NBO analysis (atomic charge) of all of the compounds (in a.u.) calculated at the DFT/B3LYP/6-31G(d,p) level.
| Dye | MO | Contribution to the electron density (%) | NBO atomic charge | ||||
| Donor | π-linker | Acceptor | Donor | π-linker | Acceptor | ||
| L1 | HOMO LUMO | 81 13 | 12 37 | 7 50 | 0.08 | 0.09 | −0.17 |
| L2 | HOMO LUMO | 70 12 | 23 47 | 7 41 | 0.06 | 0.11 | −0.17 |
| L3 | HOMO LUMO | 63 5 | 33 59 | 4 36 | 0.13 | 0.05 | −0.18 |
| L4 | HOMO LUMO | 55 4 | 41 63 | 4 33 | 0.03 | 0.14 | −0.17 |
| L5 | HOMO LUMO | 63 4 | 35 67 | 2 28 | 0.04 | 0.09 | −0.13 |
| L6 | HOMO LUMO | 62 4 | 36 64 | 1 32 | 0.04 | 0.08 | −0.12 |
| L7 | HOMO LUMO | 24 7 | 65 75 | 11 16 | 0.05 | 0.18 | −0.23 |
| L8 | HOMO LUMO | 33 7 | 58 80 | 9 13 | 0.07 | 0.09 | −0.16 |
On the other hand, the contribution of the acceptor moiety skeleton in the LUMO orbital of these dyes ranges from 13% to 50% and the contribution of donor moiety in the same orbital ranges from 4% to 13%, which means that the acceptor moiety has a huge contribution to the LUMO of all studied dyes. This indicates that the electrons are pushed toward the acceptor unit from the donor unit via π-conjugated linkers, which may be attributed to the presence of nitrogen atoms as electron-withdrawing units, which enhances the charge transfer, especially from L8. Therefore, we may anticipate that the proposed π-conjugated linkers of all dyes are majorly donors of electrons to the cyanoacetic acid group which behaves as an electron-withdrawing unit; this indicates that they are more favorable to be brilliant NLO materials.
3.3 NBO analysis
NBO investigation is considered an efficient technique for studying the ICT, and it is a suitable study for examining charge transfer between the filled and vacant orbitals. It is also believed that the charge densities which transfer from a donor moiety to an acceptor moiety via a π-linker in the D-π-A structure can be elucidated by NBO analysis. However, NBO analysis has been carried out on optimized structures of L1–L8 dyes, and the calculated results are listed in Table 3. The positive value of the donor moiety represents the proficient electron-donating aptitude of the donor provided that there were effective electron-pushing (electron-donating) units. Although the negative values of NBO charges of the acceptor moiety demonstrate that the acceptor of all dyes (cyanoacetic acid) will effectively accept electrons, the positive NBO charge values of bridges (π-conjugated linkers) of all dyes exhibit that they will provide a path and facilitate the transfer of electron (without trapping them) from the donor to the acceptor unit. As a result, we remark that NBO charges of L1–L8 point out that all donors and π-conjugated linkers show positive values, whereas all acceptors show negative values. These results confirm that electrons are successfully migrated from the donor to the acceptor moiety via π-conjugated linkers, which results in the formation of charge separation state, and upon photoexcitation, the electrons are transferred from the donor to the acceptor moiety through the π-linker and then injected into the conduction band of semiconductor. As a result and based on the obtained result presented in Table 3, we remark that the architecture “push–π–pull” helps to enhance the intramolecular charge transfer from the donor to the acceptor moiety via the π-linker of studied dyes and causes more overlap between the donor and acceptor moiety, consequently enhancing their NLO responses.
On the other hand, to further confirm the ICT, the electron density variations (Δρ) between the first singlet excited state and ground state of studied dyes have been also simulated (Fig. 4), in which the electron density variation between the excited state (EX) and ground state (GS) is calculated according to the following equation:
| Δρ (r) = ρEX (r) − ρGS (r) | (3) |
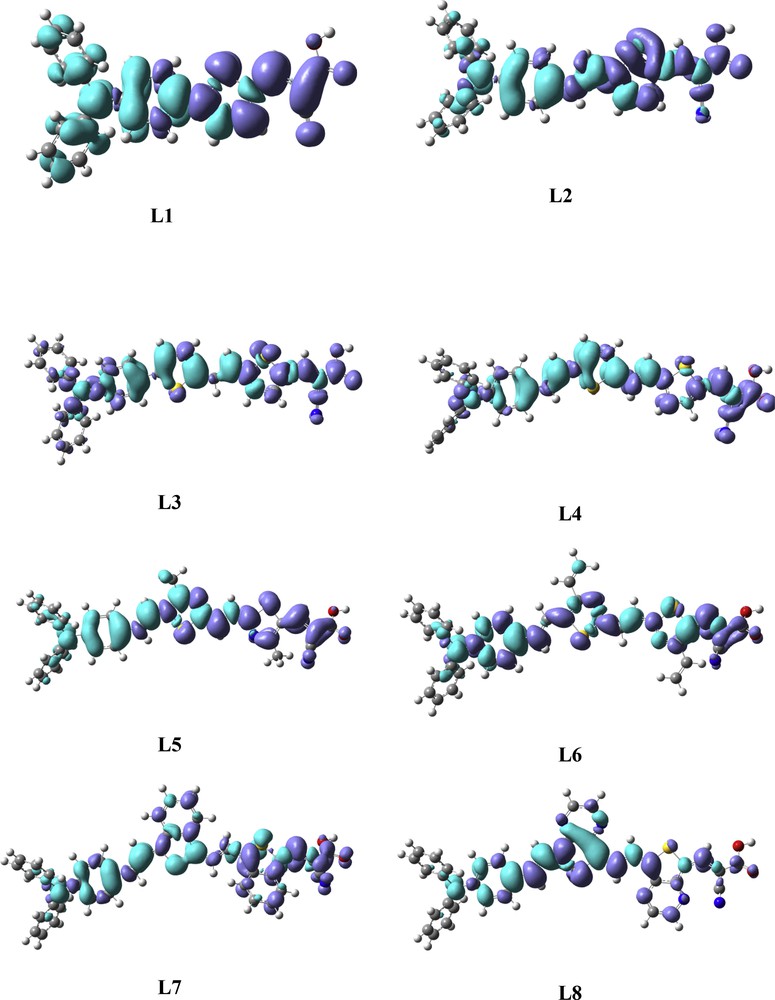
Electron density difference maps (EDDM) between the excited and ground states of the eight studied dyes. The contour thresholds for the molecular orbitals and density differences are 0.01 and 0.0004 a.u., respectively. The blue color and purple color represent the regions where the electrons are coming from and where the electrons are going to, respectively.
The decreased electron density (blue color) is mainly localized on the donor and π-linker, whereas the increased electron density (purple color) is mainly distributed on cyanoacetic acid and the π-linker. This means that the electronic transition of these studied dyes is involved in ICT, which allows rapid interfacial electron injection from the dye to CB of semiconductor. Indeed, we conclude that ICT can be effectively promoted by introducing a benzothiophene or thienopyrazine unit in the π-conjugated linker as shown in Fig. 4.
3.4 NLO properties
It is important to understand the NLO properties of any material formed by the interactions of its electromagnetic fields in various media. This study allows us to design more exceptional materials to use them in optical switches, optical memory devices, signal processing, and communication technology. The knowledge of NLO properties allows us to produce a new field which is altered in frequency, amplitude, and phase or other propagation characteristics from the incident fields [62,75]. The electronic properties responsible for the strength of the optical response of the majority of the material are found on the linear response (polarizability α) and nonlinear responses (hyperpolarizabilities, β, and γ, etc.). Hence, the NLO properties of the studied dyes L1–L8 have been evaluated. The influence of π-conjugated linkers on linear and NLO properties of the studied dyes has been examined; for this, the average polarizability values <α> were calculated for L1–L8, and the results along with the major contributing tensors are tabulated in Table 4. The linear polarizability increases with the increase of the size of the studied dyes as it is provided by the contributions of the electronic part. The calculated average polarizability of all studied dyes decreases in the following order: L8 > L7 > L4 > L6 > L5 > L3 > L2 > L1. The average polarizability value is 692.85 for L1. We remark that the modification of different π-conjugated linkers had an impact on the polarizability values for all designed dyes, especially for L7 and L8. This indicates that the dye L8 with the two rings of thienopyrazine as a linker has an average polarizability value higher than other studied dyes. The highest value of polarizability was observed in L8 for average polarizability and even for αxx tensor.
Dipole polarizabilities and major contributing tensors (a.u.) of the studied dyes (L1–L8).
| Dye | αxx | αyy | αzz | <α> |
| L1 | 756.41 | 335.59 | 161.28 | 417.76 |
| L2 | 1013.69 | 345.24 | 177.32 | 512.08 |
| L3 | 1358.51 | 412.71 | 190.35 | 653.86 |
| L4 | 1675.27 | 427.60 | 221.56 | 774.81 |
| L5 | 1598.03 | 429.27 | 256.61 | 761.30 |
| L6 | 1542.41 | 520.90 | 251.06 | 771.45 |
| L7 | 2167.90 | 562.55 | 327.35 | 1019.26 |
| L8 | 2271.03 | 590.50 | 255.99 | 1039.17 |
Dipole push–pull molecules represent the first family of molecules with strong NLO properties. They are molecules consisting of an electron-acceptor group (A) and an electron-donor group (D) connected by a polarizable π-conjugate bridge which leads to narrow band energy, and their wavelength of absorption shifts toward the red region and then makes the molecular hyperpolarizability very high. The NLO properties of this kind of molecule are characterized by a high-energy ICT. These molecules are also called rod-shaped molecules, strongly one-dimensional molecular dipole, having a large linear polarizability value which is attributed to a large hyperpolarizability value, according to experimental and theoretical NLO studies [76].
However, by using the transitions along the x- and y-directions, we can estimate the polarizability, and the dipole polarizability (along the x-direction) is expressed by Eq. 4 [62]:
| (4) |
where and are the transition moment between the ground and mth excited state and the transition energy, respectively. is a complex vector quantity. The phase factors from the ground state to the excited state have also been considered in the transition dipole moment. The polarization of the orientation of transition dipole moment has been caused by the electronic transition. Furthermore, we will also explain the interaction of the dye with an electromagnetic wave of a given polarization. As mentioned in Eq. 4, it appears that α is directly proportional to the second power of the transition moment and the transition energy has an inverse relation with it. This means that the dipole polarizability value increases with the rise of the transition moment. The second power of the transition moment explains the power of the interaction caused by the distribution of charge density contained by the system. As a result, it is considered that a system having a large value of and a small value of transition energy will possess a large hyperpolarizability value. Thus, dipole polarizabilities are estimated quantitatively with an aim to give an idea of excellent NLO activity of dyes, in which the NLO response of dyes can be calculated from their first hyperpolarizability (β). Second-order NLO properties are attributed directly to electrons which are easily polarized such as π electrons favoring the ICT from the donor–acceptor interactions via π-linkers, as observed in Fig. 3 for L1–L8 dyes. The interaction of an external electric field with electronic density alters the dipole moment and hence NLO response [77].
In this work, hyperpolarizabilities of L1–L8 have been calculated using B3LYP functional and 6-31+G(d,p) basis set, and the results of βtot values along with their major contributing tensors are provided in Table 5.
The computed first hyperpolarizabilities (βtot) and major contributing tensors (a.u) of the studied dyes (L1–L8).
| Dyes | βxxx | βxxy | βxyy | βyyy | βxxz | βxzz | βtot |
| L1 | −34503.79 | 214.28 | 779.99 | 192.34 | 111.97 | 77.27 | 33649.40 |
| L2 | −58410.29 | −193.80 | −963.74 | 199.466 | 232.20 | −79.80 | 57367.26 |
| L3 | −99745.55 | 203.67 | −1091.27 | 262.25 | 234.14 | −93.90 | 98561.73 |
| L4 | −150074.10 | 1797.93 | 966.10 | 207.91 | 1068.01 | 35.35 | 149089.52 |
| L5 | −153682.41 | 1486.11 | 899.43 | 204.01 | 224.48 | 95.60 | 152696.66 |
| L6 | −146433.37 | 4137.18 | 50.07 | −20.75 | 664.83 | 105.15 | 146336.61 |
| L7 | −149189.96 | 3260.76 | 886.78 | −340.55 | −808.86 | 315.71 | 150423.50 |
| L8 | −202248.47 | 7263.33 | 617.45 | 118.94 | −1970.32 | −239.37 | 202773.63 |
The calculated βtot decreases in the following order for all studied dyes: L8 > L5 > L7 > L4 > L6 > L3 > L2 > L1. We remark that the least value of βtot value is attributed to dye L1 [33649.40 (a.u)]. On the other hand, it was observed that the dye L8 with the two thienopyrazine rings as π-linkers provides the highest value of βtot (202773.63 (a.u)) among all compounds. This highest and lowest NLO response in case of L8 and L1, respectively, can be attributed to the effective charge transfer from the donor to the acceptor moiety via their respective π-conjugated linkers. Through Table 5, we remark that the first hyperpolarizability coefficients of all studied dyes are much high. For instance, the computed βtot value of L8 was computed 4715 times greater than the first hyperpolarizability value of urea molecule which is frequently used as a reference organic molecule [78]. Similarly, the computed βtot value of L1–L7 was found to be 782, 1334, 2292, 3467, 3551, 3403, and 3498 times of urea molecule, respectively. Furthermore, we remark that the order of βtot decreases unlike the order of the energy gap (HOMO − LUMO) and in accordance with that of average polarizability. On the other hand, we remark that the hyperpolarizability β is dominated by the longitudinal component of βxxx, which indicates that a substantial delocalization of charges has been dominated in these directions. Moreover, the overlap of interacting orbitals will be reduced for compounds having the rings connected by carbon–carbon ς bond as shown for R1, which will eventually reduce the charge transfer from donor to acceptor through the π-linker, and therefore gives a very low NLO response. The main difference between R1 and other compounds is that the rings are connected via an additional carbon atom, which will eventually stop the free rotation and then increase the ICT and subsequently increase the NLO response for other compounds compared with R1. To this end, the higher hyperpolarizability value is attributed to the movement of π electrons caused by the interaction of donor–acceptor moieties through the π-linker, which leads to the decrease in the HOMO − LUMO energy gap and stabilizes the molecules, and then shifts the absorption toward the red region of all studied dyes L1–L8.
3.5 UV analysis
To have an insight into the excited states absorption spectra, TD-DFT computations were carried out using the CPCM model in chloroform solvent at CAM-B3LYP/6-311+G(d,p) level [36,52,53,79–81]. The choice of chloroform solvent is attributed to the fact that most organic compounds are soluble in chloroform solvent. Moreover, four compounds (R1, R2, R3, and R4) are synthesized, and their solubility has been tested in this solvent. Six lowest singlet–singlet transitions were studied during TD-DFT computations. The transition energy (Eex), oscillator strength (), transition natures, maximum absorption wavelength (λmax), and the corresponding experimental values of dyes L1–L8 are summarized in Table 6, whereas absorption spectra of L1–L8 are displayed in Fig. 5. All calculations were carried out on the optimized geometries in the excited state with the chloroform solvent for all dyes. The analysis of solvent effects on the electronic structure is performed through the use of a continuum model CPCM in which the average molecule–solvent interactions are implicitly considered in the solute's Hamiltonian. All dyes show absorbance in the visible region (Fig. 5). The agreement of the calculated λmax of the four dyes L1–L4 with the experimental results has discrepancies by about 0.3 eV. This means that this good agreement confirms again the reliability of the used methods.
Absorption spectral data obtained by TD-DFT methods for studied dyes at CAM-B3LYP/6-31+G (d,p) optimized geometries.
| Compounds | λmax (nm) | (a.u) | Assignmenta | Eex (eV) | λexp (nm)b | |
| L1 | 439.82 | 1.52 | 9.59 | H→L (0.63) | 2.81 (2.64) b | 404 |
| 315.78 | 0.02 | H-1→L (0.58) | 3.92 | |||
| 283.66 | 0.04 | H→L+2 (0.61) | 4.37 | |||
| 275.82 | 0.15 | H→L+1 (0.50) | 4.49 | |||
| 272.04 | 0.17 | H −6→L (0.41) | 4.55 | |||
| 270.75 | 0.35 | H→L+3 (0.65) | 4.57 | |||
| L2 | 477.16 | 1.99 | 3.64 | H→L (0.61) | 2.59 (2.48) b | 427, 409 |
| 337.30 | 0.00 | H-1→L (0.54) | 3.67 | |||
| 296.79 | 0.20 | H→L +1 (0.54) | 4.17 | |||
| 283.26 | 0.05 | H→L+2 (0.59) | 4.37 | |||
| 274.40 | 0.11 | H-7→L (0.64) | 4.51 | |||
| 272.93 | 0.33 | H→L+3 (0.62) | 4.54 | |||
| L3 | 502.52 | 2.28 | 5.17 | H→L (0.57) | 2.46 (2.38) b | 445, 412 |
| 360.76 | 0.06 | H-1→L (0.46) | 3.43 | |||
| 319.46 | 0.19 | H→L+1 (0.46) | 3.88 | |||
| 295.70 | 0.19 | H-2→L (0.39) | 4.19 | |||
| 285.38 | 0.03 | H→L+3 (0.59) | 4.34 | |||
| 275.03 | 0.36 | H→L+4 (0.59) | 4.50 | |||
| L4 | 533.61 | 2.54 | 10.72 | H→L (0.57) | 2.32 (2.35) b | 463, 415 |
| 384.85 | 0.16 | H→L+1 (0.44) | 3.22 | |||
| 337.49 | 0.17 | H→L+1 (0.41) | 3.67 | |||
| 312.25 | 0.24 | H-2→L (0.39) | 3.97 | |||
| 285.77 | 0.05 | H→L+3 (0.47) | 4.33 | |||
| 283.64 | 0.03 | H-2→L (0.33) | 4.37 | |||
| L5 | 515.38 | 2.35 | 8.83 | H→L (0.56) | 2.40 | **** |
| 377.23 | 0.06 | H-1→L (0.42) | 3.28 | |||
| 334.54 | 0.19 | H→L+1 (0.39) | 3.70 | |||
| 316.09 | 0.34 | H-2→L (0.37) | 3.92 | |||
| 287.65 | 0.01 | H-2→L (0.37) | 4.31 | |||
| 284.70 | 0.04 | H→L+3 (0.55) | 4.35 | |||
| L6 | 506.66 | 2.18 | 9.3 | H→L (0.54) | 2.44 | **** |
| 391.39 | 0.06 | H-2→L (0.39) | 3.16 | |||
| 345.84 | 0.53 | H→L (0.37) | 3.58 | |||
| 338.26 | 0.10 | H-1→L+1 (0.37) | 3.66 | |||
| 303.46 | 0.06 | H-2→L (0.34) | 4.08 | |||
| 285.37 | 0.07 | H→L+3 (0.36) | 4.34 | |||
| L7 | 744.06 | 2.22 | 4.25 | H→L (0.67) | 1.66 | **** |
| 465.42 | 0.13 | H→L+1 (0.46) | 2.66 | |||
| 413.22 | 0.01 | H→L+1 (0.47) | 3.00 | |||
| 376.83 | 0.35 | H-2→L (0.54) | 3.29 | |||
| 327.98 | 0.26 | H→L+2 (0.47) | 3.78 | |||
| 311.73 | 0.14 | H→L+3 (0.57) | 3.97 | |||
| L8 | 779.33 | 2.08 | 10.1 | H→L (0.66) | 1.59 | **** |
| 497.06 | 0.09 | H→L+1 (0.59) | 2.49 | |||
| 439.66 | 0.06 | H-1→L (0.54) | 2.82 | |||
| 371.16 | 0.00 | H-6→L (0.53) | 3.34 | |||
| 361.27 | 0.19 | H-2→L (0.56) | 3.43 | |||
| 357.01 | 0.38 | H→L+2 (0.55) | 3.47 |
a Corresponding orbital contribution to the first excited state (H = HOMO, L = LUMO).
b Experimental absorption maximum wavelength (in nm) in chloroform and experimental values of transition energy in parentheses are from the studies by Bourass et al. [52].
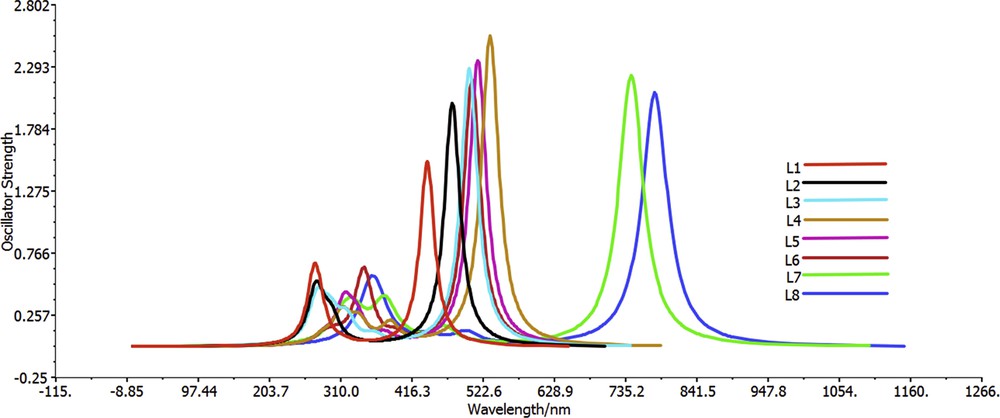
Simulated UV-visible optical absorption spectra of the title dyes using the calculated data at the TD-DFT/CAM-B3LYP/6-31G+(d,p) level in chloroform solvent.
We remark that the UV-vis absorption spectra of all dyes have two major bands. The absorption peaks in the range of 270–380 nm (Fig. 5) are attributed to the π–π* transitions in the triphenylamine group. The absorption bands at 440–780 nm were caused by the n–π* transitions in the π-conjugated linker groups and weak ICT from π-conjugated linker groups to cyanoacetic acid. Compared with L1, the dyes L7 and L8 present a redshift toward higher wavelengths, 744.06 for L7 and 779.33 for L8; this is attributed to the presence of electron donation nature of benzothiophene and thienopyrazine into π-conjugated linker groups for L7 and L8, respectively. On the other side, once the two phenylene rings fused on the two thiophene rings into π-conjugated linker of L7, a large redshift has been observed (305 nm), which may be attributed to the extended effects of π-conjugation, and stronger ICT interactions have been observed for this dye. Moreover, the fusion of the two pyridine rings on the two thiophene rings in the π-conjugated linker of L8 leads to a larger redshift (340 nm), which is attributed to the extended effects of π-conjugation and also to the presence of the four nitrogen atoms which behave as a rich source of electrons.
Generally, the interactions such as those between n and π electrons cause a redshift of the secondary and primary absorption bands of aromatic compounds (extended conjugation). Although the transition n→π* allows the formation of charges, the system then becomes deficient in electrons and the π* system acquires an extra electron, which causes a separation of charge in the molecule and stabilizes it by resonance in the π* orbital; this excited state is called charge transfer state and is easily applicable to such system. As a result, the simulation of absorption spectral data informs us that the thienopyrazine and benzothiophene are the best rings for extending the π conjugation of any system with the D-π-A structure, especially when we use them as a π-linker, and then favors the long wavelength light harvesting.
To understand the second-order NLO properties, a better clarification of the structure–property relationship is mandatory. In the context of perturbation theory sum-over-states (SOS), hyperpolarizability can be expressed as the sum of two terms (β = β2L + β3L). In this case, the term β2L is dominant, which allows us to consider that β = β2L. For this, we can apply the two-level model [82], which presents β as connected to an energy of transition E, its oscillator force f, and the variation of the dipole moment of transition according to the following Eq. 5 [62,75]:
| (5) |
The value of transition moment, oscillator strength, and excitation energy are tabulated in Table 6. From Table 6, it can be seen that for L1–L8, the factors , , and are closely related to each other and acquire the same general framework. The relationship between the first hyperpolarizability values and the corresponding two-level model values of L1–L8 are represented in Fig. 6. Fig. 6 shows that βtot and βCT values evolved in the same direction, and the appearance of the graph of the two parameters shows this finding, which indicates that they are proportional.
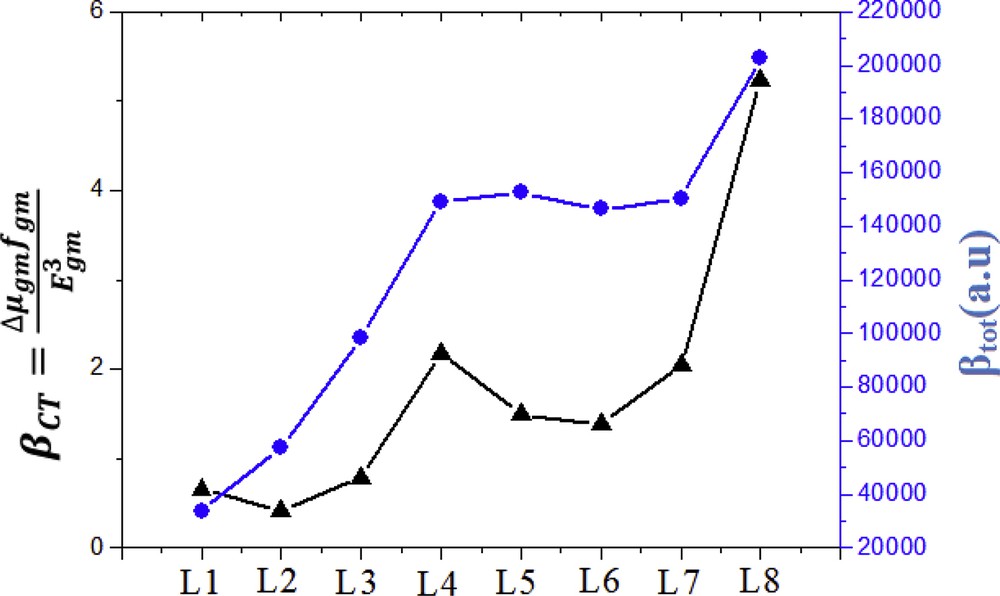
Relationship between the (blue line) values and the corresponding (black line) values for dyes (L1 to L8).
These results suggest that the controlling of π-bridges placed between donor and acceptor electron groups is a significant approach to design new molecules of the D-π-A structures having a strong dipole moment, as well as a strong ICT photo-induced by a light excitation, which corresponds to a redistribution of charge between the donor group and the acceptor group and consequently, these molecules have brilliant NLO properties. We also anticipate that the insight of the effect of π-conjugated linkers on NLO properties in D-π-A structures will be used to design new molecules, with a nonzero permanent dipole moment used as a photoelectronic material with a fine performance for photonic applications. Under excitation by electric or optical field, changes in value after a delocalization of the electron cloud; this occurs when the molecule goes into an excited state where delocalization of electrons is further amplified, and then there is a strong difference between dipole moment in the ground and in the excited state.
The general approach for designing NLO compounds possessing strong NLO response consists of pairing of a strong electron donor (D) and a strong acceptor (A) simultaneously to an organic π-linker to provide a strong electronic coupling between the donor moiety and the acceptor moiety and then to have a large amplitude of charge transfer; the linker must drive the electronic density without damping the electronic flux all along the molecule. Although a finite amount of D-A coupling is essential for a significant first hyperpolarizability, the pairing facilitated by the bridge must not be so strong to avoid the removal of the electronic asymmetry provided by the donor and acceptor moieties.
4 Conclusions
In this work, the electronic structures, the absorption spectra, and the first hyperpolarizability values have been calculated to predict the NLO response of the studied metal-free organic dyes. The influence of different π-linkers on the difference in spectral and NLO properties has been analyzed and discussed for all studied dyes L1–L8. We remark that the band π–π* of all studied compounds is shifted toward the red region with a high transition moment and low transition energy; this is due to the presence of a strong electron donor and a strong acceptor substituent and even a strong π-linker in the studied dyes, which have the effect of increasing the character of charge transfer of the transition π–π *, especially for L7 (744.06 nm) and L8 (779.33 nm). The NBO charge analysis shows that electrons are successfully migrated from the donor to acceptor through a π-conjugated linker of all dyes, which provides a strong charge separation state. The electron density distribution of HOMO is localized over the triphenylamine moiety and first π-conjugated linker, whereas LUMO is localized over the cyanoacetic acid moiety, and this is illustrated by the electron density variations (Δρ) between the excited and ground state that provide a decreased electron density on the donor and π-linker and the increased electron density on cyanoacetic acid and π-linker which promote the ICT from triphenylamine to the cyanoacetic acid segment through π-conjugated linker and thus play a crucial role in the large NLO response of L1–L8. Generally, all dyes (L1–L8) present a large NLO response ranging from 33649.40 to 202773.63 (a.u); L8 especially presents the highest <α> [1039.17 (a.u)) and βtot (202773.63 (a.u)] values. The titled compounds exhibited good NLO property because of their β values and were much greater than that of urea. We can conclude that the titled molecule is an attractive object for future studies of NLO properties and important parameters influencing β generally are donor and acceptor substituents, nature of substituents, and conjugated π system. Furthermore, βtot values of L1–L8 were found proportional to the corresponding / values in good concurrence suggested by the two-level model. This work also describes that structural modeling of π-linkers in the D-π-A architecture of the studied dyes is an important approach used for designing a new appealing NLO compound. Metal-free organic dyes are a very hot area of research, and this theoretical framework provides new ways for experimentalists to design high-performance NLO materials for optics and electronics. In the conceptual design of possible high-performance NLO materials, the proposed dyes should be targeted for further synthetic investigations.