


 CC-BY 4.0
CC-BY 4.0
1. Introduction
Current cancer treatments include surgery, radiotherapy and chemotherapy, the latter causing many side effects such as the killing of cancer cells along with healthy ones [1]. Photodynamic therapy (PDT) is an interesting alternative which allows cytotoxic species to be produced in situ after administration of a photosensitizer (PS) followed by light irradiation of the tumor area [2]. The most frequently studied PS are tetrapyrrolic compounds such as porphyrins, phthalocyanines, chlorins, and bacteriochlorins and some of them have been clinically approved [3, 4]. However, the use of these molecules suffers from two major limitations. Most PS are highly hydrophobic and thus are sparingly soluble in water; they also exhibit a poor selectivity toward tumor cells. Several approaches have been developed to bypass these drawbacks. Among the most promising approaches is the use of water soluble nanoparticles associated with PS [5]. These particles increase the solubility of hydrophobic PS, while particles smaller than 100 nm with long circulation times will preferentially accumulate in the tumor tissue thanks to the enhanced permeability and retention (EPR) effect [6, 7, 8]. Many organic and inorganic carriers of photosensitizers have already been studied, including silica nanoparticles (SiNPS) [9, 10, 11, 12]. Silica nanoparticles possess several advantages, notably biocompatibility, tunable size, large surface area and ease of functionalization [13]. In order to increase the circulation time of inorganic nanoparticles in the blood stream and to reduce their opsonization, it is possible to cover them with a hydrophilic polymer [14] such as polyethylene glycol [15] or polysaccharides such as chitosan [16] or dextran [17].
We have recently developed core–shell hybrid nanoparticles consisting of a silica core coated with xylan carrying a porphyrin shell. In vitro biological evaluations of this composite nanoparticle have shown a significant gain in efficiency against HCT-116 and HT-29 colorectal cancer cell lines as compared with the free porphyrin [18, 19]. In order to further increase the efficiency of this system, we focused our work on mitochondrial targeting. Mitochondria are well known to play crucial roles in cell life and death [20]. Mitochondrial targeting of drugs can be achieved by the covalent binding of the triphenylphosphonium (TPP+) group which is a moiety of MitoTrackers such as MitoSOXTM Red reagent [21]. We report herein the synthesis and characterization of silica/xylan/porphyrin-TPP nanoparticles, along with xylan/porphyrin-TPP. The latter was included in our study in order to address the controversy surrounding the use of silica nanoparticles. Preliminary phototoxicity studies of these platforms have been carried out against the HCT-116 and HT-29 colorectal cancer cell lines.
2. Experimental section
2.1. Materials
For a list of materials, see Supplementary Information (SI).
2.2. Analytical methods
FTIR analyses were performed on a PerkinElmer FTIR Spectrum 1000 spectrometer using KBr pellets (1–2 wt%). NMR analyses were carried out on a Bruker Avance III HD 500 MHz NMR spectrometer. UV–vis spectra were recorded on a AnalytikaJena SPECORD 210 double beam spectrophotometer, using 10 mm quartz cells. MALDI-TOF spectra were performed on a Shimadzu AXIMA confidence using dithranol as matrix. High resolution electrospray ionization mass spectrometry (HR ESI–MS) was performed at the ICOA/CBM platform (Orléans University) on a Bruker Q-TOF maXis mass spectrometer, coupled to an Ultimate 3000 RSLC chain (Dionex). Purifications were performed with Combiflash Rf 100® from Teledyne Isco. The stationary phase consisted of an 80 g silica column. The products to be purified were solubilized in a minimum amount of solvent and fixed on Florisil (60–100 mesh, VWR). For Scanning Electron Microscopy (SEM) studies, a droplet of nanoparticle suspension was lyophilized on a sample holder. The sample was metalized by sputtering of platinum using an Agar Sputter Coater. Images were taken with a LEO 1530 VP instrument. Particle size distribution was analyzed through Dynamic Light Scattering (DLS) using a Zetasizer Nano-ZS (Malvern, UK). NPS suspension was diluted in absolute ethanol and analyzed at 20 °C at a scattering angle of 173°. The mean diameter of NPS was expressed as the average value of two measurements, each one comprising 15 runs.
2.3. Syntheses
2.3.1. Synthesis of 5,15-bis(4-hydroxyphenyl)-10,20-bisphenylporphyrin (1)
4-Hydroxybenzaldehyde (7.02 g, 57.5 mmol) and benzaldehyde (6.10 g, 57.5 mmol) were dissolved in 300 mL of propionic acid and heated till reflux. Once the aldehydes were dissolved, pyrrole (7.71 g, 115 mmol, 8 mL) was added dropwise and the solution was refluxed for 90 min. Propionic acid was removed in vacuo to leave a purple oily residue. The porphyrin was purified using column chromatography (silica gel, CH2Cl2, followed by 2% MeOH in CH2Cl2 and 6% MeOH in CH2Cl2), giving 5,15-bis(4-hydroxyphenyl)-10,20-bisphenylporphyrin (1) (260 mg, 0.4 mmol, 1,4%). 1H NMR (CDCl3, 500 MHz) 𝛿H, ppm: 8.87 (d, 4H, J = 4.6 Hz, H𝛽-pyrrol), 8.83 (d, 4H, J = 4.6 Hz, H𝛽-pyrrol), 8.21 (d, 4H, J = 7.4 Hz, HAr), 8.06 (d, 4H, J = 8.4 Hz, HAr), 7.75 (m, 6H, HAr), 7.19 (d, 4H, J = 8.4 Hz, HAr), −2.78 (s, 2H, NH); MS (ESI): m∕z = C44H31N4O2, [M + H]+, 647.2435, calcd. 647.2442. UV–vis (CHCl3) 𝜆max nm (𝜀, 103 L⋅mol−1⋅cm−1): 420 (607), 519 (20), 553 (9), 591 (6), 649 (5).
2.3.2. Synthesis of tert-butyl 4-bromobutanoate (2)
Concentrated sulfuric acid (1.375 mL, 25 mmol) was added to a vigorously stirred suspension of anhydrous magnesium sulfate (12.02 g, 25 mmol) in dry CH2Cl2 (40 mL). The mixture was stirred for 15 min, after which 4-bromobutanoic acid (4.15 mL, 25 mmol) was added. Tert-butanol (11.95 mL, 100 mmol) was added last. The mixture was tightly stoppered and stirred for 48 h at room temperature. The reaction mixture was then quenched with cold saturated sodium bicarbonate solution (187 mL) and stirred until complete dissolution of magnesium sulfate. The organic phase was separated, washed with brine, dried (MgSO4), and concentrated. The crude product was purified by flash chromatography (silica gel, CHCl3) to yield tert-butyl 4-bromobutanoate as a pale yellow liquid (3.27 g, 14.65 mmol, 60%). 1H NMR (CDCl3, 500 MHz) 𝛿H, ppm: 3.45 (t, 2H, J = 6.8 Hz, Br–CH2), 2.40 (t, 2H, J = 7.2 Hz, CH2–C =O), 2.13 (qt, 2H, J = 6.8 Hz, CH2–CH2–CH2), 1.45 (s, 9H, CH3); MS (ESI): m∕z = C8H15BrO2, [M+H]+, 223.0331, calcd. 223.0328.
2.3.3. Synthesis of porphyrin (3)
5,15-Bis(4-hydroxyphenyl)-10,20- bisphenylporphyrin (1) (500 mg, 0.772 mmol) was dissolved in DMF (20 mL) and K2CO3 (2.69 g, 19.31 mmol) was added. tert-Butyl 4-bromobutanoate (2) (172 mg, 0.772 mmol) was then added to the solution and the mixture was stirred for 2 h at 110 °C. After removal of DMF with a rotary evaporator, the resulting residue was dissolved in chloroform (15 mL) and the solution was washed three times with water. After purification by preparative thin-layer chromatography (EtOH/CHCl3 = 5/95), the desired monosubstituted porphyrin 3 was obtained as a dark violet solid (279 mg, 0.347 mmol, 45% yield). 1H NMR (CDCl3, 500 MHz) 𝛿H, ppm: 8.87 (d, 4H, J = 4.7 Hz, H𝛽-pyrrol), 8.83 (d, 4H, J = 4.7 Hz, H𝛽-pyrrol), 8.21 (dd, 4H, J = 7.7 Hz,1.4 Hz, HAr), 8.11 (dd, 4H, J = 8.4 Hz, 1.8 Hz, HAr), 7.79–7.72 (m, 6H, HAr), 7.27 (d, 2H, J = 8.6 Hz, HAr), 7.26 (d, 2H, J = 8.5 Hz, HAr), 4.29 (t, 2H, J = 5.9 Hz, O–CH2), 2.60 (t, 2H, J = 7.4 Hz, CH2–CO), 2.27 (tt, 2H, J = 6.9, 6.5 Hz, CH2–CH2–CO), 1.53 (s, 9H, tBu), −2.76 (s, 2H, NH); MS (MALDI) : m∕z = C52H45N4O4, [M+H]+, 789.50, calcd. 789.34. UV–vis (CHCl3) 𝜆max nm (log𝜀 L⋅mol−1⋅cm−1): 420 (5.78), 519 (4.30), 553 (3.85), 591 (3.78), 649 (3.70).
2.3.4. Synthesis of porphyrin (4)
Porphyrin (3) (285 mg, 0.361 mmol) was dissolved in DMF (12 mL) and K2CO3 (1.24 g, 9.03 mmol) was added. 1,4-Dibromobutane (1.95 g, 9.03 mmol) was then added to the solution and the mixture was stirred for 5 h at 110 °C. After removal of DMF with a rotary evaporator, the resulting residue was dissolved in chloroform (15 mL) and the solution was washed three times with water. After purification by preparative thin-layer chromatography (CHCl3), the desired porphyrin 4 was obtained as a dark violet solid (237 mg, 0.271 mmol, 71% yield). 1H NMR (CDCl3, 500 MHz) 𝛿H, ppm: 8.88 (m, 8H, H𝛽-pyrrol), 8.21 (dd, 4H, J = 7.7, 1.4 Hz, HAr), 8.10 (d, 4H, J = 7.6 Hz, HAr), 7.79–7.70 (m, 6H, HAr), 7.27–7.23 (m, 4H, HAr), 4.37 (t, 2H, J = 5.8 Hz, O–CH2–CH2–CH2–CO), 4.27 (t, 2H, J = 6.1 Hz, O–CH2–CH2–CH2–CH2–Br), 4.26 (t, 2H, J = 4.8 Hz, CH2–Br), 2.59 (t, 2H, J = 7.3 Hz, CH2–CO), 2.41 (tt, 2H, J = 6.8 Hz, J = 6.6 Hz, CH2–CH2–CO), 2.05 (m, 4H, O–CH2–CH2–CH2–CH2–Br), 1.52 (s, 9H, tBu), −2.75 (s, 2H, NH). MS (MALDI): m∕z = C56H51BrN4O4, [M+H]+, 923.10, calcd. 923.32. UV–vis (CHCl3) 𝜆max nm (log𝜀 L⋅mol−1⋅cm−1): 420 (5.76), 517 (4.38), 551 (4.19), 591 (3.98), 649 (3.95).
2.3.5. Synthesis of porphyrin (5)
Porphyrin (4) (224 mg, 0.242 mmol) was dissolved in DMF (25 mL). Triphenylphosphine (1.03 g, 3.93 mmol) was then added to the solution and the mixture was stirred for 24 h at 110 °C. After removal of DMF with a rotary evaporator, the desired product was obtained after purification by preparative thin-layer chromatography (EtOH/CHCl3 = 5/95) as a dark violet solid (210 mg, 73% yield). 1H NMR (CDCl3, 500 MHz) 𝛿H, ppm: 8.84 (m, 8H, H𝛽-pyrrol), 8.21 (dd, 4H, J = 7.1 Hz, J = 1.3 Hz, HAr), 8.10 (dd, 4H, J = 8.5 Hz, J = 8.5 Hz, HAr), 7.95 (dd, 3H, J = 7.5 Hz, J = 1.0 Hz, HAr), 7.93 (dd, 3H, J = 7.5 Hz, J = 1.0 Hz, HAr), 7.82 (dd, 3H, J = 8.2 Hz, J = 6.6 Hz, HAr), 7.79–7.72 (m, 12H, HAr), 7.26 (d, 2H, J = 8.5 Hz, HAr), 7.20 (d, 2H, J = 8.5 Hz, HAr), 4.39 (t, 2H, J = 5.3 Hz, O–CH2–CH2–CH2–CO), 4.29 (t, 2H, J = 6.1 Hz, O–CH2–CH2–CH2–CH2–PPh3), 4.08 (m, 2H, CH2–PPh3), 2.60 (t, 2H, J = 7.3 Hz, CH2–CO), 2.42 (qt, 2H, J = 6.1 Hz, CH2–CH2–CO), 2.27 (qt, 2H, J = 6.7 Hz, O–CH2–CH2–CH2–CH2–PPh3), 2.05 (m, 2H, O–CH2–CH2–CH2–CH2–PPh3), 1.53 (s, 9H, tBu), −2.76 (s, 2H, NH); MS (ESI): m∕z = C74H66N4O4P, [M]+, 1105.4812, calcd. 1105.4816. UV–vis (CHCl3) 𝜆max nm (log𝜀103 L⋅mol−1⋅cm−1): 421 (5.77), 518 (4.35), 554 (4.09), 592 (3.82), 648 (3.73).
2.3.6. Synthesis of porphyrin (6)
Porphyrin (5) (297 mg, 0.25 mmol) was dissolved in dichloromethane (16 mL) and mixed with TFA (4 mL). After 2 h stirring at room temperature, the reaction mixture was concentrated in vacuo, and the residue was dissolved in dichloromethane (30 mL). The solution was washed with a saturated sodium bicarbonate solution (100 mL), water (3 × 50 mL), dried over magnesium sulfate, filtered, and concentrated in vacuo. The product was purified by preparative thin-layer chromatography (EtOH/CHCl3 = 15/85) to give porphyrin 6 as a dark violet solid in 94% yield (266 mg, 0.235 mmol). 1H NMR (CDCl3 + 10% TFA-D, 500 MHz) 𝛿H, ppm: 8.69 (d, 2H, J = 4.7 Hz, H𝛽-pyrrol), 8.67 (m, 4H, H𝛽-pyrrol), 8.62 (d, 2H, J = 4.7 Hz, H𝛽-pyrrol), 8.53 (dd, 4H, J = 7.5 Hz, J = 2.0 Hz, HAr), 8.48 (d, 4H, J = 7.5 Hz, HAr), 8.03 (m, 6H, HAr), 7.91 (dd, 3H, J = 8.0 Hz, J = 6.7 Hz, HAr), 7.77 (m, 6H, HAr), 7.68 (m, 6H, HAr), 7.55 (d, 2H, J = 8.5 Hz, HAr), 7.47 (d, 2H, J = 8.4 Hz, HAr), 4.44 (t, 2H, J = 5.8 Hz, O–CH2–CH2–CH2–CO), 4.39 (t, 2H, J = 5.2 Hz, O–CH2–CH2–CH2–CH2–PPh3), 3.28 (dt, 2H, J = 8.1 Hz, J = 12.9 Hz, CH2–PPh3), 2.88 (t, 2H, J = 7.1 Hz, CH2–CO), 2.41 (qt, 2H, J = 6.4 Hz, CH2–CH2–CO), 2.25 (qt, 2H, J = 6.0 Hz, O–CH2–CH2–CH2–CH2–PPh3), 2.13 (m, 2H, O–CH2–CH2–CH2–CH2–PPh3); MS (ESI): m∕z = C70H58N4O4P, [M]+, 1049.4199, calcd. 1049.4190. UV–vis (DMSO) 𝜆max nm (log𝜀 103 L⋅mol−1⋅cm−1): 422 (5.44), 518 (4.15), 554 (4.98), 593 (3.78), 649 (3.74).
2.4. Preparation of xylan/porphyrin 6 conjugate
After solubilizing porphyrin 6 (43 mg, 0.038 mmol) in 8 mL DMSO, N,N-carbonyldiimidazole (CDI) (37 mg, 0.23 mmol) was added and the mixture was stirred at 60 °C for 24 h. This solution was then added to 100 mg of xylan and allowed to react under stirring at 80 °C for 48 h. The product was precipitated with absolute ethanol and was washed three times with ethanol and three times with chloroform to remove unreacted starting material and then dried under vacuum. 106 mg of xylan/porphyrin 6 conjugate were obtained.
2.5. Preparation of the hybrid nanoparticles SiNPS@Xylan/Porphyrin-TPP
The hybrid nanoparticle SiNPS@Xylan/Porphyrin-TPP was prepared according to a previously published procedure [18]. Xylan/porphyrin 6 conjugate (10 mg) was dissolved in 20 mL of distilled water. Then, an ethanolic suspension of 100 mg of silica nanoparticles functionalized with APTES (3-aminopropyltriethoxysilane) was added dropwise and the mixture was ultrasonicated during addition and during the five following minutes. This mixture was then stirred for 15 min. The resulting SiNPS@Xylan/Porphyrin-TPP were centrifuged for 30 min at 8000 rpm and subjected to washing and centrifugation, three times in water and two times in absolute ethanol.
Mass concentrations and molar concentrations of porphyrins in the final solutions containing the self-assembled porphyrin nanoparticles of the three different samples, Ac-Xyl 4-Porphyrin, Ac-Xyl 5-Porphyrin and Ac-Xyl 4-Porphyrin-TPP
| Ac-Xyl 4-Porphyrin | Ac-Xyl 5-Porphyrin | Ac-Xyl 4-Porphyrin-TPP | |
|---|---|---|---|
| Amount of xylan used (mg) | 20 | 20 | 15 |
| Mass concentration (g/L) | 8.7 | 5.5 | 6.6 |
| Porphyrin concentration (mol/L) | 1.24 × 10−4 | 1.59 × 10−5 | 1.26 × 10−5 |
2.6. Acetylation of xylan
To a solution of xylan (200 mg, 1.51 mmol of xylose unit) in 20 mL of DMSO were added 10 mL of N-methylimidazole (125.43 mmol). The mixture was magnetically stirred at 80 °C for 1 h under nitrogen atmosphere. Then the mixture was cooled down to room temperature, and 0.25 to 4 equivalents of acetic anhydride were added. After 2 h stirring at room temperature, acetylated xylans were precipitated by addition of absolute ethanol, recovered by filtration, washed, and dried in vacuo. Seven acetylated xylans (Ac-Xyl 1 to Ac-Xyl 7) were obtained with DS values ranging from 0.04 to 1.10. IR: 897 cm−1 (𝛽-glucosidic linkages), 1045 cm−1 (C–O stretching in C–O–C linkages), 1220 cm−1 (C–O stretching), 1370 cm−1 (–C–CH3), 1735 cm−1 (C =O ester), 3400 cm−1 (O–H stretching).
2.7. 5-(4-(3-carboxypropoxy)phenyl)-10,15,20-triphenylporphyrin grafting to acetylated xylan Ac-Xyl 3, Ac-Xyl 4 and Ac-Xyl 5
5-(4-(3-Carboxypropoxy)phenyl)-10,15,20-triphenylporphyrin (108 mg, 0.15 mmol), whose synthesis has been previously described [18], was solubilized in 20 mL DMSO, then six equivalents of CDI (147 mg, 0.903 mmol) were added. After stirring at 60 °C for 24 h, 100 mg of acetylated xylan (0.757 mmol of xylose residue) were added into the mixture and allowed to react under stirring at 50 °C for 24 h. The product was precipitated out in absolute ethanol and was washed three times with ethanol and three times with chloroform to remove unreacted starting material and then dried under vacuum.
2.8. Porphyrin 6 grafting to acetylated xylan Ac-Xyl 4
Porphyrin 6 (70 mg, 0.062 mmol) was solubilized in 12 mL DMSO, then six equivalent of CDI (61 mg, 0.37 mmol) were added. After stirring at 60 °C for 24 h, 40 mg of acetylated xylan Ac-Xyl 4 (0.31 mmol xylose unit) were added to the mixture and allowed to react under stirring at 50 °C for 24 h. The product was precipitated with absolute ethanol and was washed three times with ethanol and three times with chloroform to remove unreacted starting material and then dried under vacuum.
2.9. Preparation of self-assembled nanoparticles (NPS)
The self-assembled nanoparticles were prepared by a dialysis method. Acetylated xylan/porphyrin conjugates (20 mg or 15 mg) were dissolved in 5 mL of DMSO or DMAc (depending of their solubility). The solutions were placed in dialysis membranes (regenerated cellulose, Spectra/Por 3, molecular weight cutoff of 3500 Da) of about 10 cm length and dialyzed against 500 mL of deionized water for 15 h. Water was renewed five times, every 3 h. After centrifugation at 10,000 rpm for 30 min the supernatant was removed. The obtained nanoparticles were redispersed in distilled water, then two washing/centrifugation cycles were carried out. Nanoparticles were redispersed in 5 mL of water and stored at +4 °C. 1 mL of this suspension of nanoparticles was freeze-dried to determine the mass concentration and then re-solubilized in DMSO prior to UV–vis spectrophotometry analysis. Porphyrin concentrations in the final suspensions containing the self-assembled porphyrin nanoparticles were determined at 420 or 422 nm thanks to the molar extinction of the free porphyrin (444,347 L⋅mol−1⋅cm−1) or porphyrin-TPP (274,815 L⋅mol−1⋅cm−1). The mass concentrations and the porphyrin concentrations of the three different samples, Ac-Xyl 4-Porphyrin, Ac-Xyl 5-Porphyrin and Ac-Xyl 4-Porphyrin-TPP are listed in Table 1.
2.10. Determination of degree of substitution in acetyl groups and porphyrin
NMR is a common method to determine the degree of substitution of a polymer [22, 23].
The formula to calculate the degree of substitution in acetyl groups per anhydroglucose unit is expressed as follows:
The formula to calculate the degree of substitution in porphyrins per repeat unit composed of 10 xylose units and one 4-O-methyl glucuronic acid (MeGlcA) is expressed as follows:
2.11. Determination of porphyrin-TPP concentration in SiNPS@Xylan/Porphyrin-TPP
The amount of porphyrin-TPP attached to the SiNPS was determined by UV–visible assay. SiNPS@Xylan/Porphyrin-TPP were dispersed in absolute ethanol at a concentration of 10 mg/mL and diluted with water to 0.5 mg/mL. Absorbance of this sample was measured at 420 nm, and absorbance of SiNPS was substracted. The Xylan/Porphyrin-TPP content of nanoparticles was calculated from a calibration curve constructed with different concentrations of free Xylan/Porphyrin-TPP in distilled water (Figure S9). The concentration of porphyrin-TPP in mol per gram of silica was calculated according to the following equation:
DSPorphyrin-TPP = 0.1, calculated from the 1H NMR spectrum;
C(Xylan∕Porphyrin-TPP) = concentration of Xylan/ Porphyrin-TPP attached to SiNPS, calculated from the standard calibration curve = 0.09 g/L;
MWrepeat of xylan (xyl/MeGlcA = 10:1) = 1511 g/mol; MWPorphyrin-TPP = 1163.25 g/mol.
2.12. In vitro phototoxicity of hybrid SiNPS@Xylan/Porphyrin-TPP and self- assembled nanoparticles
Human colorectal cancer cell lines, HCT-116 and HT-29, were purchased from American Culture Type Collection (LGC Standards) Middlesex, UK. HCT-116 and HT-29 cells were respectively grown in RPMI-1640 and DMEM, supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin and 100 μg∕mL streptomycin. For all experiments, cells were seeded at 4 × 103 cells∕well and 7 × 103 cells∕well for HCT-116 and HT-29 cells, respectively, and maintained in a humidified atmosphere with 5% CO2 at 37 °C. Photocytotoxicity was determined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays. Cells were seeded in 96-well culture plates and grown for 24 h in culture medium prior to exposure to free porphyrin or nanoparticles. Stock solutions of porphyrins and nanoparticles were diluted in culture medium to obtain the appropriate final concentrations. The same amount of vehicle (percentage of ethanol did not exceed 0.6%) was added to control cells. After 24 h incubation, the culture medium was replaced by phenol red-free DMEM medium, then the cells were either irradiated or not irradiated with a 630–660 nm CURElight lamp (PhotoCure ASA, Oslo, Norway) at 42 mW∕cm2 for 30 min until a fluence of 75 J∕cm2. MTT assays were performed 48 h after irradiation and cell viability was expressed as relative absorbance (570 nm) as compared with cells maintained in the dark. Results are expressed as the mean ± standard error of the mean (SEM) of two separate experiments.
2.13. Intracellular localization
Cells were seeded in 6-well culture plates and were grown for 24 h prior to exposure to SiNPS@Xylan/ Porphyrin or SiNPS@Xylan/Porphyrin-TPP at the same concentration (1 μM in porphyrin). After 24 h incubation, porphyrin fluorescence (excitation/ emission: 405/650 nm) was determined by AMNIS® imaging flow cytometry analysis and studied with IDEAS software (Merck). To determine SiNPS@Xylan/ Porphyrin and SiNPS@Xylan/Porphyrin-TPP localizations, cells were seeded and treated as described above and co-treated at 37 °C with 150 nM MitoTracker during 45 min. SiNPS@Xylan/Porphyrin and SiNPS@Xylan/Porphyrin-TPP localizations were determined by AMNIS® imaging flow cytometry and studied with IDEAS software using porphyrin fluorescence (excitation/emission: 405/650 nm) and MitoTracker fluorescence (excitation/emission: 490/516 nm). The same protocol was used for confocal microscopy analysis and photos were taken with a laser Zeiss LSM 510 Meta—×1000 confocal microscope.
3. Results and discussion
3.1. Synthesis of the triphenylphosphonium-monosubstituted porphyrin (6)
The different steps of triphenylphosphonium-monosubstituted porphyrin (6) synthesis are summarized in Scheme 1. First, 5,15-bis(4-hydroxyphenyl)-10,20-bisphenylporphyrin (1) was synthesized according to the Little’s method [24]. After purification, the expected compound was obtained in 1.4% yield [25]. Tert-butyl 4-bromobutanoate (2) was synthesized according to the method described by Wright et al. [26]. This method, which involves the reaction between 4-bromobutanoic acid, in presence of magnesium sulfate and a catalytic amount of sulfuric acid, with isobutylene formed in situ by dehydration of tert-butanol, led to ester 2 in 60% yield. The subsequent Williamson ether reaction between porphyrin 1 and one equivalent of ester 2 in presence of potassium carbonate led to the formation of monosubstituted porphyrin 3 (45% yield). The triphenylphosphonium cation was then covalently bound to the second phenolic function in a very simple way, as described by Lei et al. [27]. Porphyrin 3 was reacted with 1,4-dibromobutane via a second Williamson reaction, affording 4 in 71% yield. This compound was then reacted with excess triphenylphosphine to generate porphyrin derivative 5 in 73% yield. The tert-butyl ester function was hydrolyzed by action of trifluoroacetic acid to produce porphyrin 6 with free carboxylic acid function in 94% yield. The chemical structures of each one of these compounds were confirmed by mass spectrometry and 1H NMR spectroscopy.
Synthesis of triphenylphosphonium-monosubstituted porphyrin 6.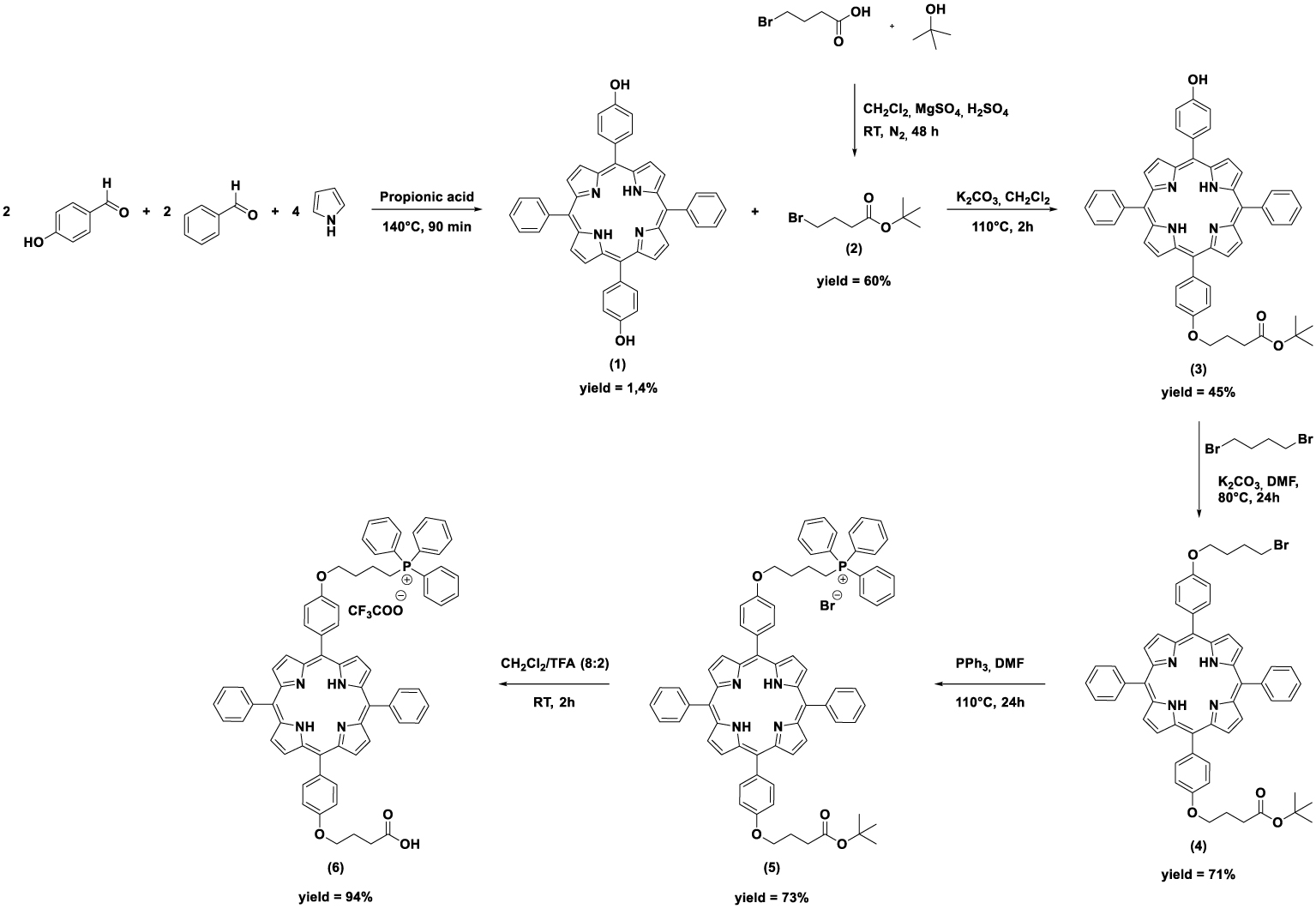
3.2. Synthesis of the hybrid nanoparticles SiNPS@Xylan/Porphyrin-TPP
The hybrid nanoparticles were prepared according to our previous work [18] (Scheme 2). Porphyrin 6 was then covalently bound to xylan by esterification. The synthesis of porphyrin-TPP-xylan was carried out by reaction of porphyrin 6 with CDI for 24 h at 60 °C to give the corresponding acyl-imidazole which was then coupled with xylan. 1H NMR was used to calculate the degree of substitution (DS) of xylan, by measuring and comparing the integral area of the aromatic protons of porphyrin with those corresponding to the anomeric protons of xylan. On the average, one of every ten xylose residues was found to be substituted by porphyrin-TPP (DS = 0.1). A previously described method [18] was used to prepare core–shell hybrid nanoparticles. An ethanolic suspension of APTES-functionalized SiNPS (size = 80 nm) was added dropwise to a solution of Xylan/Porphyrin-TPP in water. The resulting nanoparticles were then recovered by centrifugation and washed several times with water to remove unbound polymers. Functionalization of SiNPS with Xylan/Porphyrin-TPP was confirmed by UV–visible analysis (Figure S10, SI). A standard calibration curve obtained from different concentrations of Xylan/Porphyrin-TPP diluted in water was used to measure the concentration of Xylan/Porphyrin-TPP bound to SiNPS. A concentration of 1.1 × 10−5 mol Porphyrin-TPP per gram silica was determined.
Preparation of the SiNPS@Xylan/Porphyrin-TPP hybrid nanoparticles.
3.3. Preparation and characterization of Xylan/Porphyrin and Xylan/Porphyrin-TPP nanoparticles (NPS)
SiNPS are subject to controversy due to their potential negative impact on human health [28]; this is why we decided to create nanoparticles without the silica core. It has recently been reported that conjugates consisting of hydrophilic xylan backbone and hydrophobic segments can form self-assembled nanoparticles in aqueous solution [29, 30, 31, 32, 33]. As a first step, we evaluated the self-assembly of different conjugates of xylan and 5-(4-hydroxyphenyl)-10,15,20-triphenylporphyrin with DS varying from 0.3 to 1.1. The preparation of these conjugates has been previously reported [18]. DLS and Scanning Electron Microscopy (SEM) showed that particles in the 1–2 μm size range were obtained. These particles were too large to penetrate tumor cells. The hydrophobic–hydrophilic balance backbone governs the formation of nanoparticles [34]. The DS in grafted porphyrin is probably too low to induce sufficient hydrophobicity to obtain nanoparticles; it is therefore necessary to introduce more hydrophobic groups. Xylan acetylation, practiced for many years to modify these properties, especially hydrophobicity, seemed appropriate [35]. Acetylation is most often carried out using acetic anhydride in presence of pyridine as a catalyst [36, 37]. More recently, Zhang et al. published a milder method of xylan acetylation using DMSO/N-methylimidazole (NMI) mixture where NMI replaces pyridine as a catalyst, but also acts as a solvent [38]. Hence, following these conditions, seven acetylated xylans with different degrees of substitution ranging from 0.04 to 1.10 have been obtained (Scheme 3).
Acetylation of xylan with different molar ratios of acetic anhydride in DMSO/NMI at room temperature.
The ability of these seven acetylated xylans to form nanoparticles was then evaluated by the dialysis method, that is replacing the organic solvent, in which the acetylated xylans have been dissolved, by water, by dialysis. The DLS method was used to characterize nanoparticle size; sizes were comprised between 80 to 120 nm for Ac-Xyl 5, 60 to 105 nm for Ac-Xyl 6 and 50 to 90 nm for Ac-Xyl 7 (Figure S12). As expected, increasing the amount of hydrophobic groups allowed to obtain smaller nanoparticles. However, SEM images showed that the nanoparticles with the highest DS (Ac-Xyl 7, DS = 1.10) no longer possess a spherical shape (Figure S13).
To obtain photosensitive nanoparticles of spherical shape and of nanometric size, acetylated xylans, Ac-Xyl 3, Ac-Xyl 4 and Ac-Xyl 5 with DS value of 0.21, 0.34, and 0.68, respectively, were chosen to be functionalized with porphyrins, which will consequently increase hydrophobicity of xylan. The two-step grafting procedure is illustrated in Scheme 4. 5-(4-(3-Carboxypropoxy)phenyl)-10,15,20-triphenylporphyrin [18] (TPPOH) was activated by reaction with CDI to furnish the corresponding acyl-imidazoles, which were then coupled with Ac-Xyl-3, Ac-Xyl 4 and Ac-Xyl 5 to give Ac-Xyl 3-Porphyrin, Ac-Xyl 4-Porphyrin, Ac-Xyl 5-Porphyrin, respectively. DS in porphyrin, calculated from 1H NMR spectra, were 0.71, 0.52 and 0.13 for Ac-Xyl 3-Porphyrin, Ac-Xyl 4-Porphyrin, Ac-Xyl 5-Porphyrin, respectively. Logically, the higher the acetyl DS of xylan, the harder it is to further graft porphyrin moieties.
Grafting of TPPOH or porphyrin 6 onto acetylated xylans.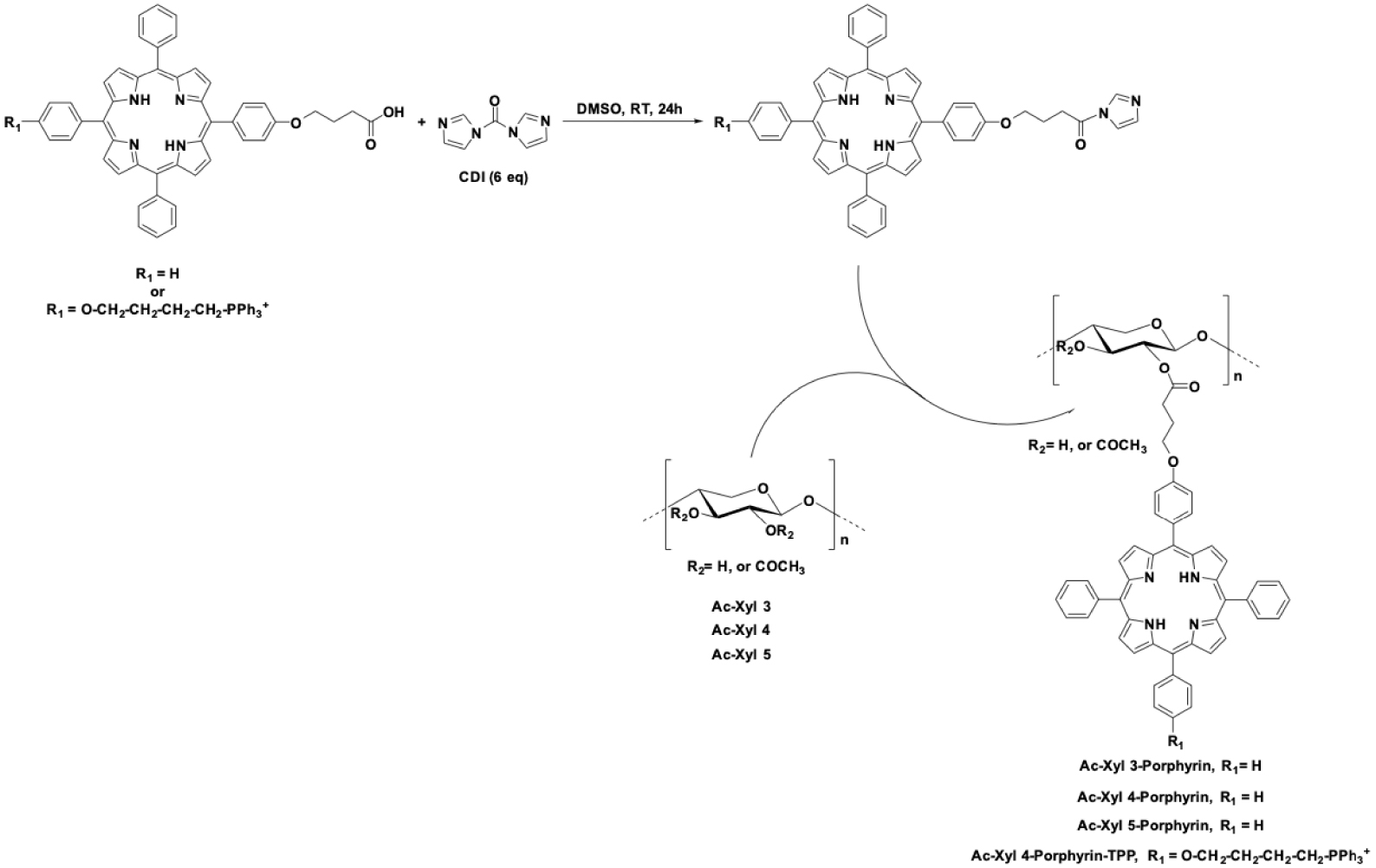
The formation of nanoparticles from Ac-Xyl 3-Porphyrin, Ac-Xyl 4-Porphyrin and Ac-Xyl 5-Porphyrin was then investigated by the dialysis method. SEM images providing information on the morphology and size of the nanoparticles are shown in Figures 1B, D and F. Dialysis of Ac-Xyl 3-Porphyrin led to the formation of polymer aggregates instead of nanoparticles. On the other hand, Ac-Xyl 4-Porphyrin and Ac-Xyl 5-Porphyrin actually formed spherical nanoparticles. The mean hydrodynamic volumes of nanoparticles in water as measured by DLS (Figures 1A, C and E) were 128 ± 48 nm for Ac-Xyl 4-Porphyrin and 90 nm ± 35 nm for Ac-Xyl 5-Porphyrin. Ac-Xyl 4-Porphyrin and Ac-Xyl 5-Porphyrin nanoparticles display polydispersity index (PDI) values of 0.221 and 0.235, which indicated a moderately polydisperse distribution [39]. It therefore appears that an acetylation ratio above 0.21 is essential for the formation of nanoparticles and that the higher the ratio, the smaller the nanoparticles, provided that the ratio does not exceed 0.8.
Particle size and morphology of nanoparticles: (A) DLS size measurement of Ac-Xyl 3-Porphyrin particles; (B) SEM image of Ac-Xyl 3-Porphyrin particles; (C) DLS size measurement of Ac-Xyl 4-Porphyrin particles; (D) SEM image of Ac-Xyl 4-Porphyrin particles; (E) DLS size measurement of Ac-Xyl 5-Porphyrin particles; (F) SEM image of Ac-Xyl 5-Porphyrin particles.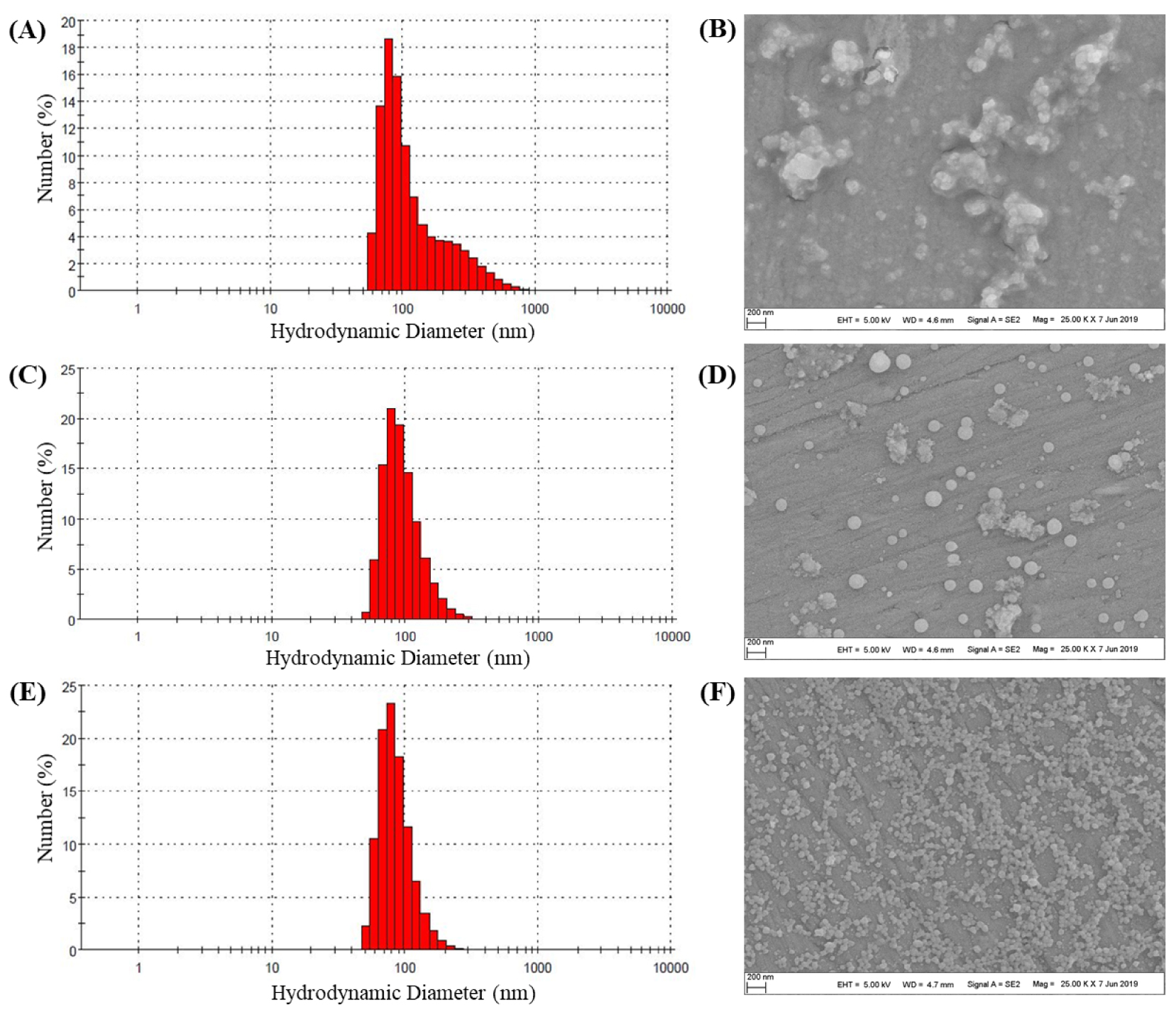
In order to evaluate the effects of mitochondrial targeting, self-assembled xylan nanoparticles bearing porphyrin-TPP were prepared. Xylan polymer bearing acetyl groups was functionalized with porphyrin (6) bearing the triphenylphosphonium cation. Acetylated xylan Ac-Xyl 4 was chosen because it has made it possible to reach a porphyrin DS of 0.52, compared with the corresponding 0.13 value of Ac-Xyl 5. Triphenylphosphonium-monosubstituted porphyrin 6 was activated by reaction with CDI to furnish the corresponding acyl-imidazole, which was then coupled with Ac-Xyl 4 (Scheme 4). The degree of substitution of the product (Xyl-Ac 4-Porphyrin-TPP) was calculated from 1H NMR data. Unexpectedly, a DS of 0.1 in porphyrin (6) was obtained. This degree of substitution is five times lower than that of xylan Ac-Xyl 4-Porphyrin (DS = 0.52), obtained in the same experimental conditions. The presence of the TPP positive charge on porphyrin 6 could limit its reactivity, by allowing ionic interaction with glucuronic acid substitutions or the hydroxyl groups of xylan.
Ac-Xyl 4-Porphyrin-TPP was then dissolved in DMSO and dialyzed against water for 15 h. After washing and centrifugation, nanoparticles were characterized using SEM and DLS (Figure 2). Spherical nanoparticles were observed in SEM image and the mean hydrodynamic size of particles in water as measured by DLS was 151 ± 25 nm with a PDI value of 0.332, which indicated a moderately polydisperse distribution type of these nanoparticles.
In vitro photocytotoxicity of hybrid SiNPS@Xylan/Porphyrin-TPP and organic Xylan/Porphyrin and Xylan/Porphyrin-TPP nanoparticles in human colorectal cancer cells
| HCT-116 | HT-29 | |||
|---|---|---|---|---|
| Dark | Illumination | Dark | Illumination | |
| TPPOH [18] | Not determined | 2943 ± 102 | Not determined | 5959 ± 430 |
| Porphyrin-TPP (6) | >5000 | 960 ± 430 | >5000 | 1660 ± 40 |
| SiNPS@Xylan/Porphyrin [18 ], a | >1000 | 72.6 ± 2.8 | >1000 | 550.2 ± 7.5 |
| SiNPS@Xylan/Porphyrin-TPPa | >1000 | 25 ± 15 | > 1000 | 157.5 ± 17.5 |
| Xylan/Porphyrin NPSa | > 5000 | 1045 ± 136 | >5000 | 2280 ± 190 |
| Xylan/Porphyrin-TPP NPS | >5000 | <2000 | >5000 | 1240 ± 180 |
a IC50 nM concentrations are directly corresponding to the amount of porphyrins.
Particle size and morphology of nanoparticles: (A) DLS size measurement of Ac-Xyl 4-Porphyrin-TPP particles; (B) SEM image of Ac-Xyl 4-Porphyrin-TPP particles.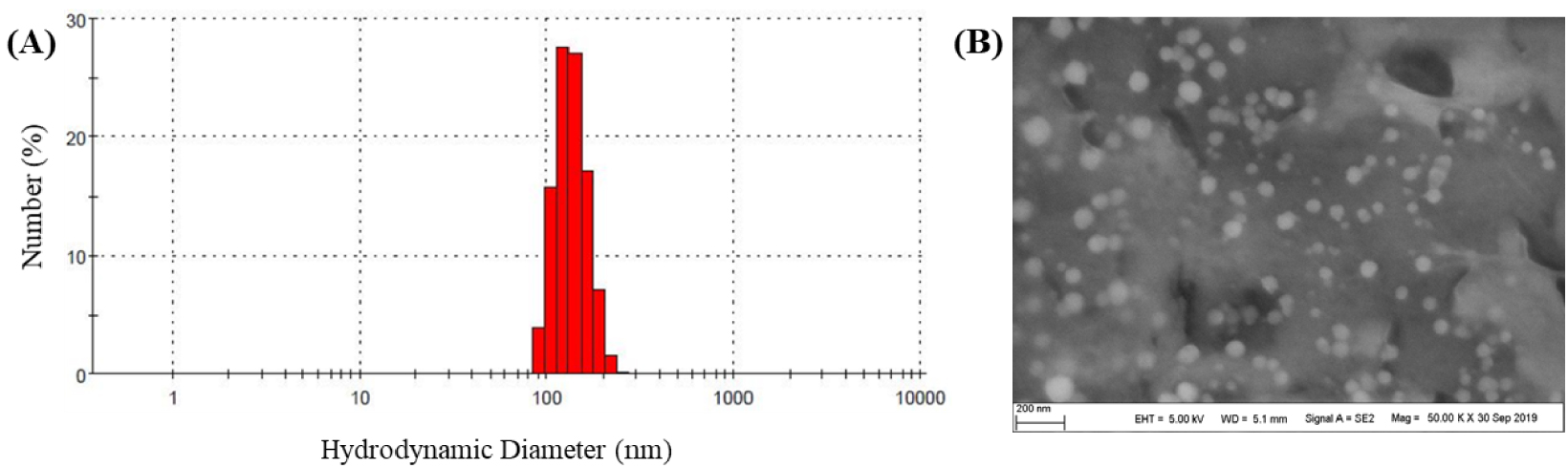
The intracellular localization of SiNPS@Xylan/Porphyrin and SiNPS@Xylan/Porphyrin-TPP in HT-29 cells with MitoTracker Green. Images were merged to indicate the overlap in fluorescence.
3.4. In vitro evaluation of nanoparticles against human colorectal cancer cells
The effect of hybrid SiNPS@Xylan/Porphyrin-TPP and Xylan/Porphyrin and Xylan/Porphyrin-TPP nanoparticles on cell viability was evaluated against HCT-116 and HT-29 colorectal cancer cells, via MTT assays (Table 2). This evaluation was carried out in the presence of photosensitizers free or with nanoparticles ranging from 0.01 to 5 μM during 48 h in the dark and under illumination. Within the concentration range tested, nanoparticles showed no or low cytotoxicity in the dark. Under illumination, all nanoparticles showed obvious cytotoxicity on HCT-116 and HT-29 at concentrations lower than 2 μg∕mL. By comparison, organic nanoparticles showed significantly less photocytotoxicity than hybrid nanoparticles. One possible explanation is that the organic nanoparticles self-organize in such a way that the hydrophobic porphyrins tend to gather inside the nanoparticle while the hydrophilic parts of xylan lie on the solvent-exposed periphery of the shell. Accordingly, the spatial promiscuity of porphyrins leads to self-quenching of the stacked PSs inside the nanoparticles. The lower fluorescence quantum yield of Xylan/porphyrin nanoparticles (𝛷F < 0.01 in D2O) compared to hybrid SiNPS@Xylan/Porphyrin nanoparticles (𝛷F = 0.04 in EtOH) tends to confirm 𝜋-stacking interaction. Such an arrangement would justify the reduction in phototoxicity of the organic nanoparticles. However, their photocytotoxicities were still much stronger than those of free porphyrins. Interestingly, addition of the triphenylphosphonium cation resulted in reduction by factors of 2.8–3.5 the IC50 values of the hybrid nanoparticles.
Confocal microscopy was used to examine the subcellular localization of SiNPS@Xylan/Porphyrin and SiNPS@Xylan/Porphyrin-TPP in HT-29 cancer cells (Figure 3). Our previous investigations revealed that SiNPS@Xylan/Porphyrin localized predominantly in lysosomes [19]. Based on the yellowish color arising from the overlay of red fluorescence of SiNPS@Xylan/Porphyrin-TPP and the green fluorescence of the organelle marker, it can be concluded that SiNPS@Xylan/Porphyrin-TPP was localized in mitochondria. These first in vitro results are very encouraging and suggest that mitochondrial targeting of nanoparticles would increase their photocytotoxicity.
4. Conclusion
Hybrid nanoparticles with a silica core coated with xylan substituted with porphyrins bearing a triphenylphosphonium cation have been constructed for the first time. Addition of the triphenylphosphonium cation allowed to enhance threefold the photocytotoxicity of the nanoparticles against colorectal cancer cell lines HCT-116 and HT-29. The controversy surrounding the use of silica nanoparticles has led us to build xylan nanoparticles devoid of the silica core. The self-organization of xylan in water required an increase in its hydrophobicity which has been achieved by means of acetylation. Post-functionalization of acetylated xylans with porphyrins, whether bearing the triphenylphosphonium cation or not, followed by nanoparticle shaping, has made it possible to obtain photosensitive organic nanoparticles. These organic nano-objects showed lower toxicity than hybrid nanoparticles, probably due to the self-quenching effect of the porphyrins inside the nanoparticles. However, organic nanoparticles retained a strong photocytotoxicity, much higher than that of the corresponding free porphyrin. This strategy therefore appears promising for the design and development of photosensitizer nanocarriers for oncological photodynamic therapy.
Acknowledgments
We thank Dr. Cyril Colas for ESI-HRMS analyses. We acknowledge financial support from GDR MAPYRO and the regional council of Nouvelle-Aquitaine, and Dr. Michel Guilloton for his help in manuscript editing.
Supplementary data
Supporting information for this article is available on the journal’s website under https://doi.org/10.5802/crchim.108 or from the author.