


 CC-BY 4.0
CC-BY 4.0
1. Introduction
Polyoxometalates (POMs), a large family of transition metal oxygen anion clusters with d0 electronic configurations, exhibit interesting photocatalytic properties. Upon light irradiation, electrons are promoted from an oxygen-centered 2p orbital to an empty metallic d-orbital, generating a highly reactive charge-separated state [1]. The impregnation of polyvinylidene fluoride-based solid polymer electrolyte by POM was also shown to improve the performance of dye sensitized solar cell. The fabricated cell generated high open circuit voltage of 426 mV and short circuit current of 3.90 mA upon illumination with visible light [2]. The above-mentioned excellent properties and pioneering studies indicate that POMs are promising candidates to optimize the photocurrent generation efficiency.
Furthermore, POMs are also of interest for oxidative photodegradation of organic compounds in water purification technology [3].
Metal recovery is also a topic of great concern from economic and environmental aspect. Since many metals are either valuable or toxic, the development of methods that aim to recover them could contribute to save the limited resources and resolve the environmental problems.
Metallic nanoparticles can be produced using various methods, such as thermal decomposition [4], electrochemical techniques [5, 6], sonochemical synthesis [7, 8] radiolysis [9], microwave irradiation [10] and photocatalysis [11].
In particular, polyoxometalates (POMs) can be used in photocatalytic processes for the recovery of metals or the synthesis of nanoparticles. Indeed, POMs are also excellent electron reservoirs as they exhibit variable oxidation states and the possibility of multiple reductions making the POMs an excellent candidate as photocatalysts for the reduction of metal ions. In the process, illumination at the O→M charge-transfer band (UV region), renders POM strong oxidants able to extract electrons from organic electron donors. POMs display remarkable activity and selectivity which can be adjusted by the choice of POM with suitable redox potential, while operational parameters such as the concentration of POM, organic substrate (sacrificial electron donor such as propan-2-ol), and metal ions should affect the photocatalytic efficiency.
Top: representation of (A) zinc-𝛽-octaethylporphyrin ZnOEP, (B) free base 5,15-ditolylporphyrin H2T2P and (C) Lindqvist-type polyoxometalate [V6O13{(OCH2)3CNHCO(4 − C5H4N)}2]2− (Py-V6O13-Py). Bottom: electropolymerization scheme of H2T2P in the presence of Py-V6O13-Py giving the copolymer poly-V6O13-H2T2P.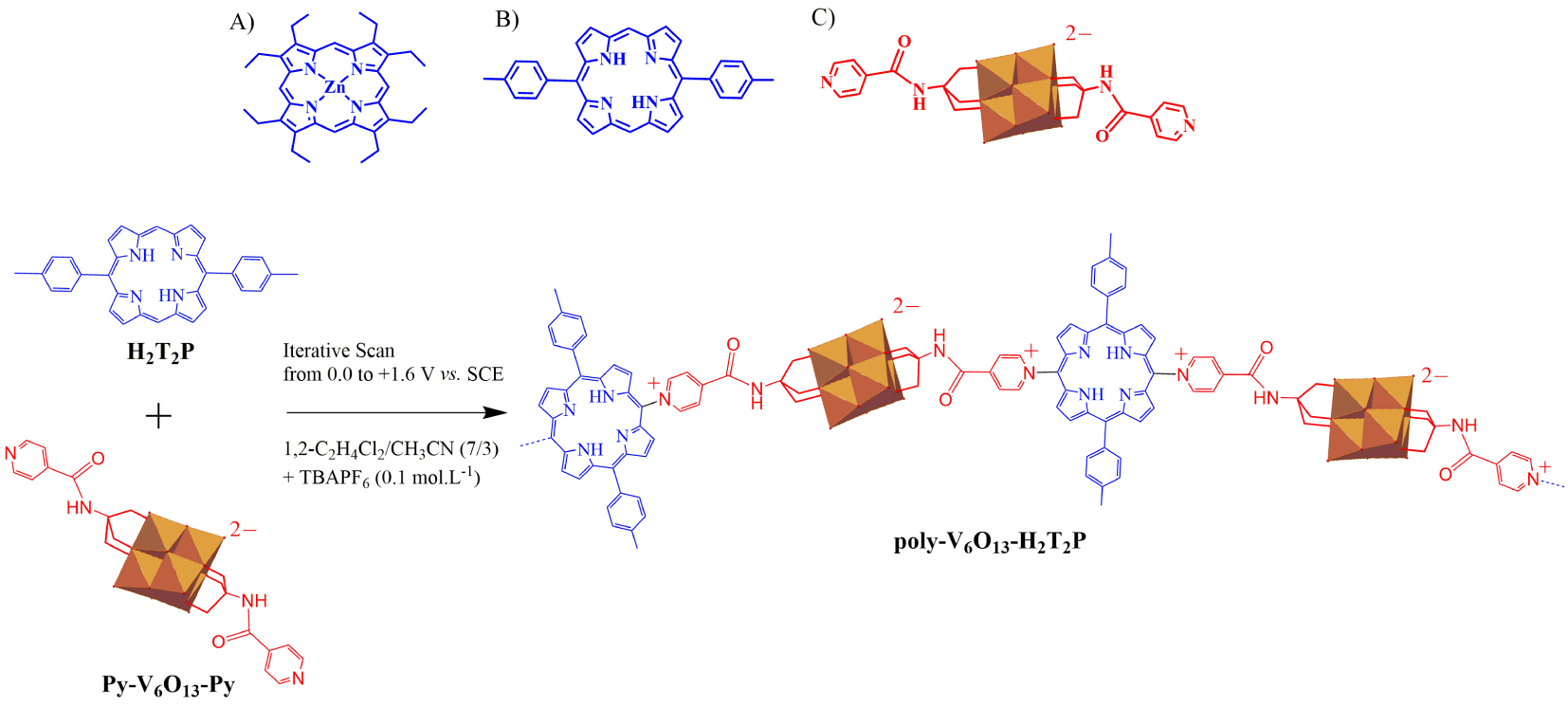
Recovery of metallic ions from aqueous solutions can be obtained through a homogeneous photocatalytic process in the presence of a sacrificial electron donor such as propan-2-ol that undergoes photolysis under UV illumination. In these conditions, POMs are quantitatively reduced and this leads to the reduction of metallic ions. This procedure is an useful alternative for synthesis and recovery of metallic nanoparticles [12, 13].
To expand the practical application range, their association to a light-harvesting antenna is so far necessary, since POMs themselves are mainly photoactive only in the UV part of the solar spectrum. Among photosensitizers, porphyrins offer the advantages of strong absorption coefficients in the visible domain. Synthesis of covalently bonded POM–porphyrin hybrid system [14, 15] can be proposed in order to obtain efficient photoinduced intramolecular electron transfer from the porphyrin ring to the POM cluster. Formation of covalently bonded POM–porphyrin copolymers can be similarly proposed. Then the reduced POMs can be used for the reduction of metallic ions. The electrochemical synthesis of the POM–porphyrin copolymers uses the previously published nucleophilic substitution on porphyrins via an E(ECNECB)nE process [16, 17, 18, 19, 20, 21]. A polarization of a working electrode at the first ring-oxidation potential of porphyrins in the presence of pyridine induces a nucleophilic attack and leads to the attachment of the pyridyl nitrogen to the meso positions of the porphyrin. If the applied potential corresponds to the second ring-oxidation potential of porphyrins in the presence of bipyridine, copolymer with viologen spacers can be obtained. In the present work, the nucleophile pyridine groups came from the functionalized POM (Py-POM-Py) [22, 23, 24], resulting in the formation of {POM–porphyrin}n copolymers.
Preliminary study concerning the use of photosensitized systems coupled with POMs for the elaboration of metallic silver nanosheets and nanowires (heterogeneous photocatalysis) has been reported. In this case, the copolymers were composed of porphyrin and Anderson-type POM subunits [22]. Precisely, the hybrid copolymer was obtained by electropolymerization of zinc 𝛽-octaethylporphyrin (ZnOEP) or zinc 5,15-dipyridinium-octaethylporphyrin in the presence of a functionalized Anderson-type polyoxometalate bearing two pyridyl groups [MnMo6O18{(OCH2)3CNHCO (4 − C5H4N)}2]3−(Py − MnMo6O18 − Py) [1, 24]. The photocatalytic reduction of using this copolymer was conducted under visible light illumination and aerobic conditions in the presence of propan-2-ol. Quantitative formation of metallic Ag0 nanowires as well as triangular nanosheets was observed.
The main goal of this work is to demonstrate that other hybrid porphyrin–POM copolymers are still working as photocatalysts for the reduction of metallic ions. By changing the nature of the porphyrin and of the polyoxometalate, a change in the photoreduction kinetics as well as in the shape of the nanoparticles is expected. In the present paper, we use copolymers obtained by electropolymerization of metalloporphyrin-type zinc-𝛽-octaethylporphyrin (ZnOEP) or free-base-type 5,15-ditolyl porphyrin (H2T2P) with the functionalized Lindqvist polyoxovanadate bearing two pyridyl groups [V6O13{(OCH2)3CNHCO(4 − C5H4N)}2]2− (Py-V6O13-Py) (Scheme 1). The photocatalytic reduction of as well as the H2PtIVCl6 is studied under visible irradiation in the presence of propan-2-ol, acting as sacrificial donor, at the 2D interface between water and the copolymeric films deposited on quartz substrate.
2. Results and discussion
2.1. Electrochemical synthesis of the copolymers
The syntheses of the copolymers were achieved using our electropolymerization method, as reported earlier [25, 26]. It corresponds to the addition of the dipyridyl-substituted Lindqvist-type polyoxovanadate [V6O13{(OCH2)3CNHCO(4 − C5H4N)}2]2− (Py-V6O13-Py) to an electrogenerated dicationic 5,15-ditolylporphyrin (H2T2P) or zinc-𝛽-octaethylporphyrin (ZnOEP) obtained by iterative scans between 0 V and +1.60 V versus SCE (Scheme 1) [27].
Using this method, the poly-Py-V6O13-Py-H2T2P and the poly-Py-V6O13-Py-ZnOEP hybrid copolymers were prepared as described previously [27].
(A) UV–Vis absorption spectra of poly-V6O13-ZnOEP and poly-V6O13-H2T2P obtained after 20 iterative scans between 0.00 and +1.60 V versus SCE on ITO. (B) Thickness of poly-V6O13-ZnOEP and poly-V6O13-H2T2P measured by AFM versus different numbers of iterative scans. Tapping mode AFM topography and section analysis of the aggregate marked by a blue line of (C) and (C′) poly-V6O13-H2T2P, and of (D) and (D′) poly-V6O13-ZnOEP (film obtained after 20 scans, between 0.00 V and +1.60 V).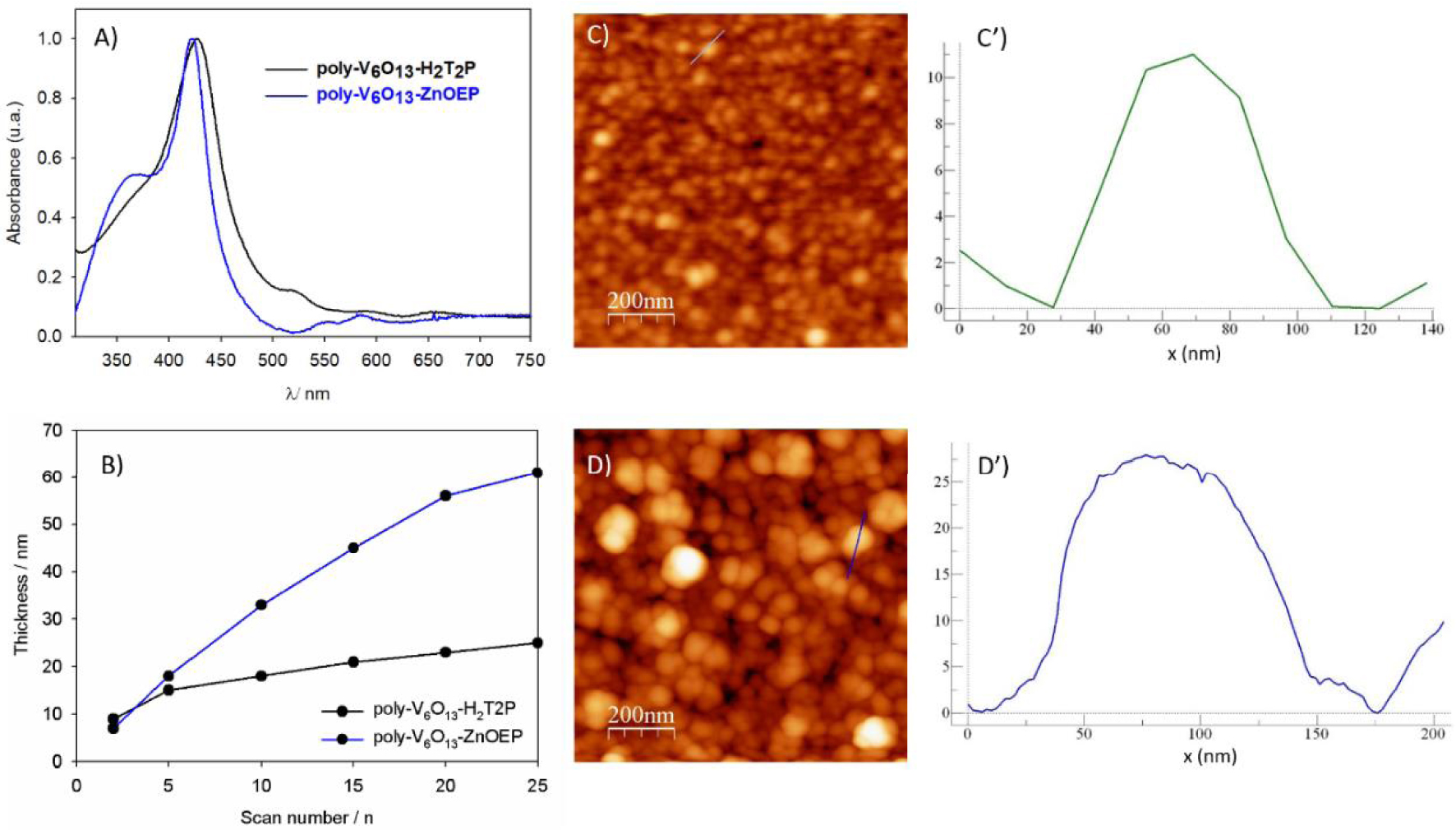
Note that the free base meso-5,15-ditolylporphyrin (H2T2P) presents only two meso positions occupied by one substitutable proton at positions C10 and C20 while the zinc-𝛽-octaethylporphyrin (ZnOEP) presents four substitutable meso positions at C5, C10, C15 and C20 (top of Scheme 1). The described electrochemical synthesis of the copolymers uses the previously reported E(ECNECB)nE process of nucleophilic substitution on porphyrins [17, 18, 19, 26, 27, 28, 29]. As soon as the iterative scans were performed at an anodic potential sufficiently high to allow the formation of the porphyrin dication, the formation of a copolymer coating the working electrode was observed.
The mechanism of the formation of the copolymer can be described such as: first, the porphyrin (abbreviated Porph) radical cation (Porph∙+, electrochemical step E) and dication (Porph2+, electrochemical step E) are electrogenerated. Then, the dication porphyrin Porph2+ can be attacked by a two pendant isonicotinate groups of the pyridyl-substituted Lindqvist-type polyoxovanadate [V6O13{(OCH2)3CNHCO(4 − C5H4N)}2]2− (abbreviated Py-V6O13-Py) at meso-carbon position to yield an isoporphyrin (chemical step CNmeso). This later intermediate can be oxidized (electrochemical step E) and the hydrogen atom originally situated on the meso-carbon is released inducing the rearomatization of the porphyrin (chemical step CB) which leads to the monosubstituted porphyrin Porph-meso-Py+ − V6O13 − Py. At this stage, monosubstituted porphyrin is obtained with one pyridinium covalently connected to the porphyrin and one pendant pyridyl group which is still active for nucleophilic attack. This pendant pyridyl group can further attack oxidized porphyrin continuing the growth of the copolymer. Thus, the polarization of a working electrode at the porphyrin’s second ring-oxidation potential in the presence of Py-V6O13-Py, leads to the formation of the two hybrid copolymer films with general formula (bottom of Scheme 1), namely poly-V6O13-H2T2P and poly-V6O13-ZnOEP.
UV–visible spectra of both copolymers on ITO electrodes presented similar characteristics [27]. They exhibit a large Soret absorption band (Figure 1A), which was red shifted by 15 and 20 nm compared to the ZnOEP and H2T2P monomer respectively [27]. It can be explained by intra- or intermolecular excitonic interactions between the porphyrin subunits [17, 30]. The red shifts observed are in agreement with the presence of the disubstituted porphyrin ring by two pyridinium groups [31] in the copolymer.
The films were also examined by scanning atomic force microscopy (AFM) showing tightly packed coils with average diameters of 40–60 nm and a height of 5 nm in the case of poly-V6O13-H2T2P (Figures 1C and C′). The rms surface roughness of the film was 3.5 nm for 1 mm2 area. For poly-V6O13-ZnOEP the diameter and the height of the coils were almost two times larger (Figures 1D and D′) [27].
Change in the UV–Vis absorption spectra of a deaerated aqueous solution of 8.0 × 10−5 mol⋅L−1 Ag2SO4 and 0.13 mol⋅L−1 propan-2-ol containing a slide of quartz modified with (A) poly-V6O13-ZnOEP, (B) poly-V6O13-H2T2P film under illumination. Inset: plot of the absorbance at (A) 𝜆 = 421 and (B) 𝜆 = 429 nm versus the time of irradiation.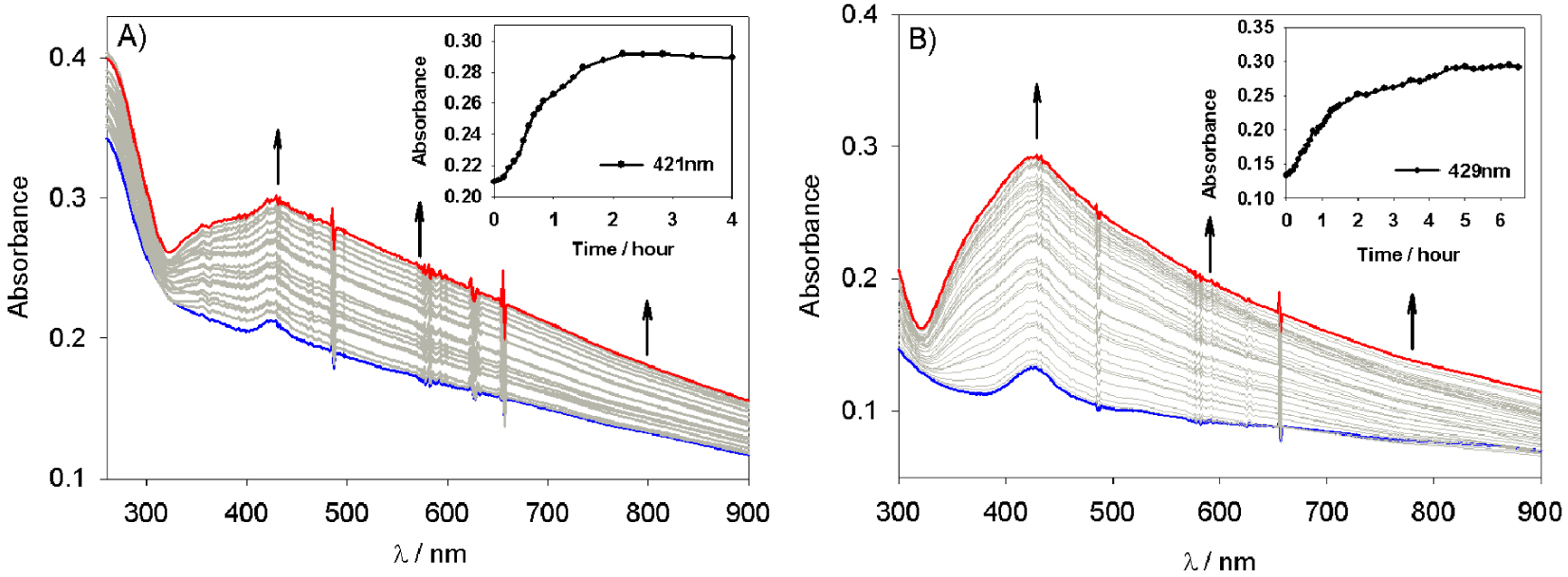
Thickness of the deposited copolymeric film was measured using AFM by scratching the film with a metallic tip. The thickness increased upon the number of iterative scans as shown in Figure 1B. The values obtained after 20 scans between 0.00 V and +1.60 V were about 23 nm for poly-V6O13-ZnOEP and 56 nm for poly-V6O13-H2T2P.
2.2. Photocatalytic recovery of silver
The electrochemically deposited poly-V6O13-ZnOEP or poly-V6O13-H2T2P copolymers were dissolved and removed from ITO with dimethyl formamide (DMF). Subsequently, the copolymer in DMF solution was deposited on a quartz slide, and the DMF solvent was evaporated in air. Then, the quartz slide was plunged in an optical cell containing a deaerated aqueous solution with 8 × 10−5 mol⋅L−1 Ag2SO4 and 0.13 mol⋅L−1 propan-2-ol. The whole sample was illuminated under visible light with a 385 nm cutoff filter to prevent POM photoexcitation allowing only the excitation of the porphyrin. Figure 2 presents the absorption spectra recorded during the experiment performed with the poly-V6O13-ZnOEP (Figure 2A) or poly-V6O13-H2T2P (Figure 2B) films. The absorbance increases in the whole visible domain during illumination and the solution becomes slightly yellow which reveals the formation of silver nanoparticles [22, 32]. The initial rate of Ag+ reduction is 3.4 × 10−5 mol⋅L−1⋅min−1 for poly-V6O13-ZnOEP and 3.1 × 10−5 mol⋅L−1⋅min−1 for poly-V6O13-H2T2P. After almost 95 min (for poly-V6O13-ZnOEP) and 105 min (for poly-V6O13-H2T2P) of visible light irradiation, the absorption spectrum does not evolve anymore which indicates the end of the reaction.
After removal of the quartz slide covered with the film, the UV–visible spectrum of the solution exhibits a large plasmon band in the whole visible domain with a maximum around 410 nm (Figure 3). This can be attributed to the presence of silver nanoparticles in the solution. No change in the spectrum is observed for one week indicating the good stability of the silver nanoparticles. Moreover, it has been found that the slides of quartz can be reused at least five times with almost no change in the efficiency of the photocatalysis. Silver ions Ag(I) are reduced quantitatively at the interface between water and the copolymeric films without poisoning the surface (as checked by AFM analysis, data not shown).
UV–Vis absorption spectra of the silver nanoparticles solution before and after the photocatalysis using the poly-V6O13-ZnOEP (red curve) or the poly-V6O13-H2T2P (blue curve) films.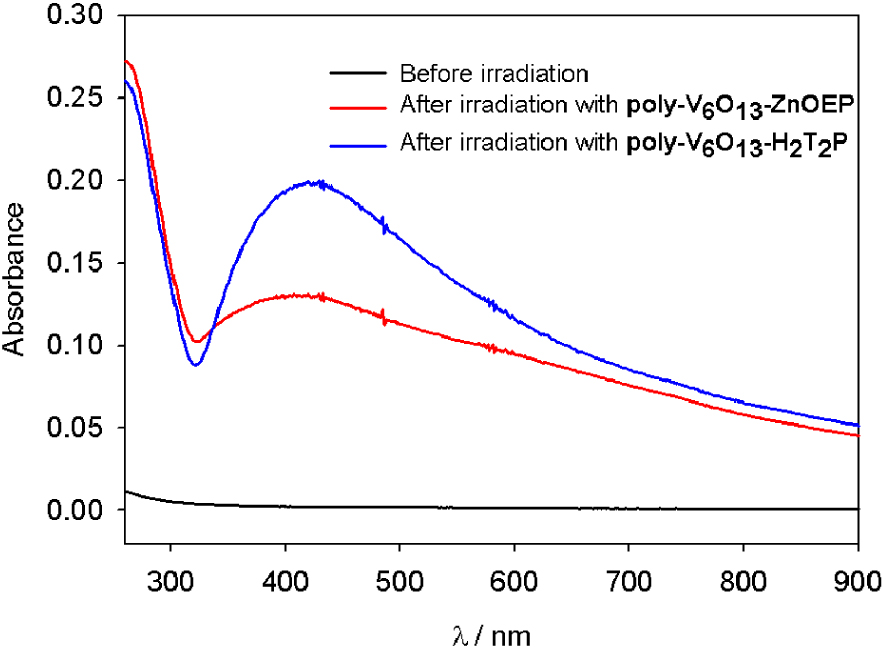
The TEM micrographs confirmed the formation of silver particles (Figures 4 and 5). The nanoparticles obtained with poly-V6O13-ZnOEP have an average diameter of 20 nm but are agglomerated (Figure 4).
(A–D) TEM images of the silver nanoparticles obtained with the poly-V6O13-ZnOEP film in deaerated solution of 8.0 × 10−5 mol⋅L−1 Ag2SO4. (E) Selected-area electron diffraction pattern of the silver nanoparticles. The inner spots (circled) corresponded to the formally forbidden 1/3 {422} reflections. The second spots (squared) could be indexed to the {220} reflections. (F) EDS spectrum showing the presence of Ag in the sample.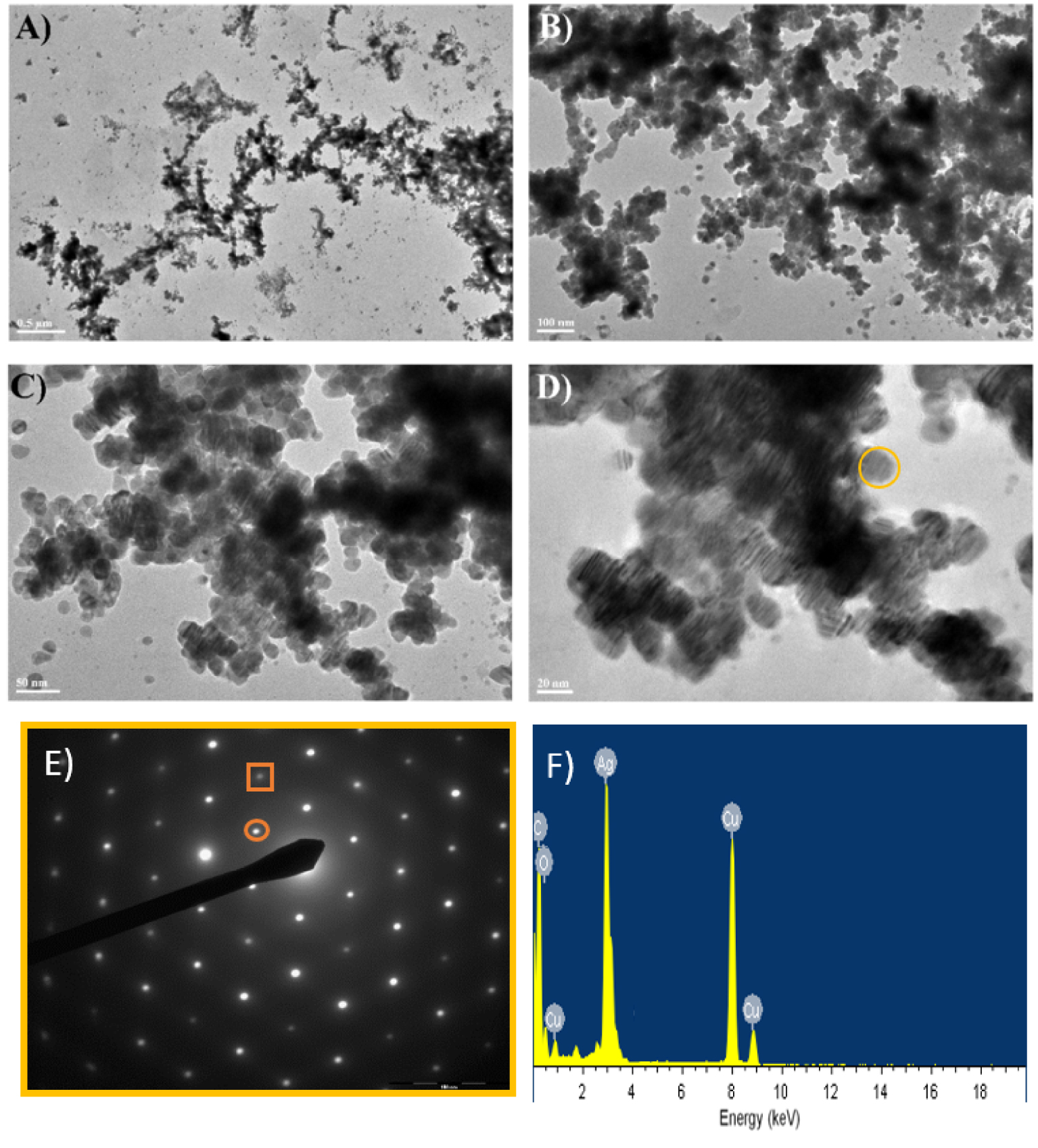
The presence of Moiré pattern in TEM images of silver particles is due to the presence of metallic silver crystal planes with different spacing and/or orientations. Each silver nanoparticles seems to be crystalline as shown by electron diffraction analysis (Figure 4E).
The 6-fold rotational symmetry displayed by the diffraction spots implied that the faces represented the {111} planes. The first set of spots could be indexed to the formally forbidden 1/3 {422} reflections of face-centered cubic (fcc) silver with a corresponding lattice spacing of 2.48 Å. The second set corresponded to Bragg diffraction from the {220} planes of fcc silver with a lattice spacing of 1.44 Å (1.445 Å in JCPDS file 04-0783). These observations were in agreement with the usual published indexes for silver nanosheets [33, 34]. We explain the observation of 1/3 {422} reflections that are formally forbidden for a perfect fcc structure by the presence of stacking faults [35]. These stacking faults could be caused by bending, which explains the presence of Moiré pattern in TEM images [36]. The EDS spectrum in Figure 4F reveals the presence of metallic Ag in the sample, the C and Cu detected belonging to the TEM grid.
Conversely, the nanostructures obtained with poly-V6O13-H2T2P exhibited larger dispersion in size and shape, because the samples presented not only spherical, elongated particles but also long linear silver nanowires (thickness ca. 20–40 nm and length ca. 200–500 nm) and large silver nanosheets (Figure 5). The EDS spectrum in Figure 5C reveals the presence of metallic Ag in the sample.
(A) TEM images of the silver nanomaterials obtained with the poly-V6O13-H2T2P film in deaerated solution of 8.0 × 10−5 mol⋅L−1 Ag2SO4. (B) Selected-area electron diffraction pattern of the silver nanoparticles. The inner spots (circled) corresponded to the formally forbidden 1/3 {422} reflections. The second spots (squared) could be indexed to the {220} reflections. (C) EDS spectrum showing the presence of Ag in the sample.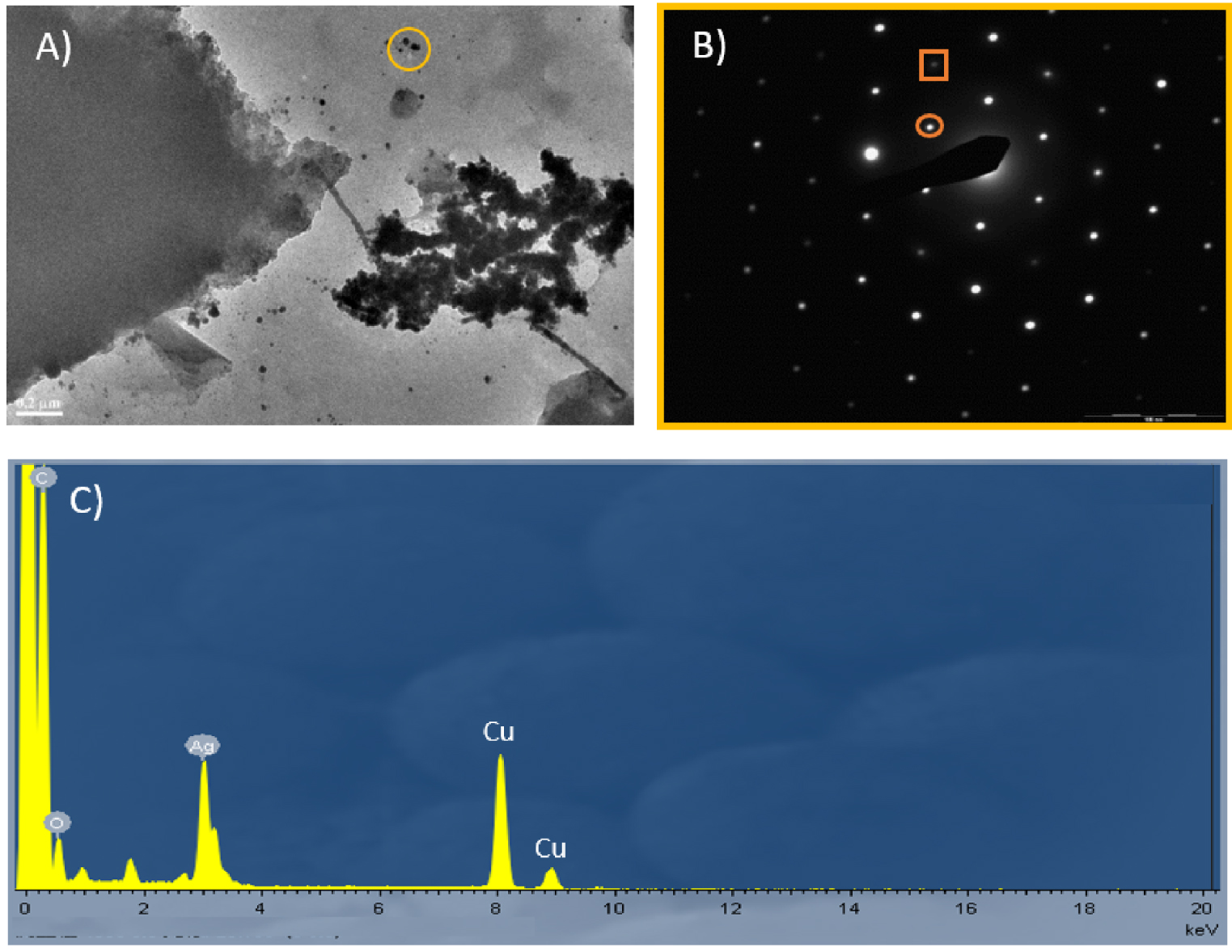
Two possible mechanisms can be involved to describe the formation of AgNPs. As shown in Figure 6A, the first mechanism consists in the reduction of the excited porphyrins by propan-2-ol, followed by the cascade electron transfer via the pyridinium to the POM subunit {V6O13}2− giving the reduced {V6O13}3−. Then, {V6O13}3− can in turn reduce silver ions. A complexation step between alcohol radical and silver ions can initiate the formation of silver clusters (reactions (4)–(7)) before a thermodynamically possible direct reduction with the reduced POM ({V6O13}3−) or with the alcohol radical (CH3)2C∙OH (when the nuclearity of initial silver aggregates is sufficient).
(A) and (B) Two possible mechanisms for the photoreduction of silver ions by the use of the poly-V6O13-H2T2P copolymer. Similar mechanism can be proposed for poly-V6O13-ZnOEP. Bottom: scale of apparent standard redox potentials of couples involved in this mechanism in the case of H2T2P. ZnOEP gives similar tendencies. Note that the reduction of the pyridinium group is achieved at a redox potential very close to that of the reduction of {V6O13}.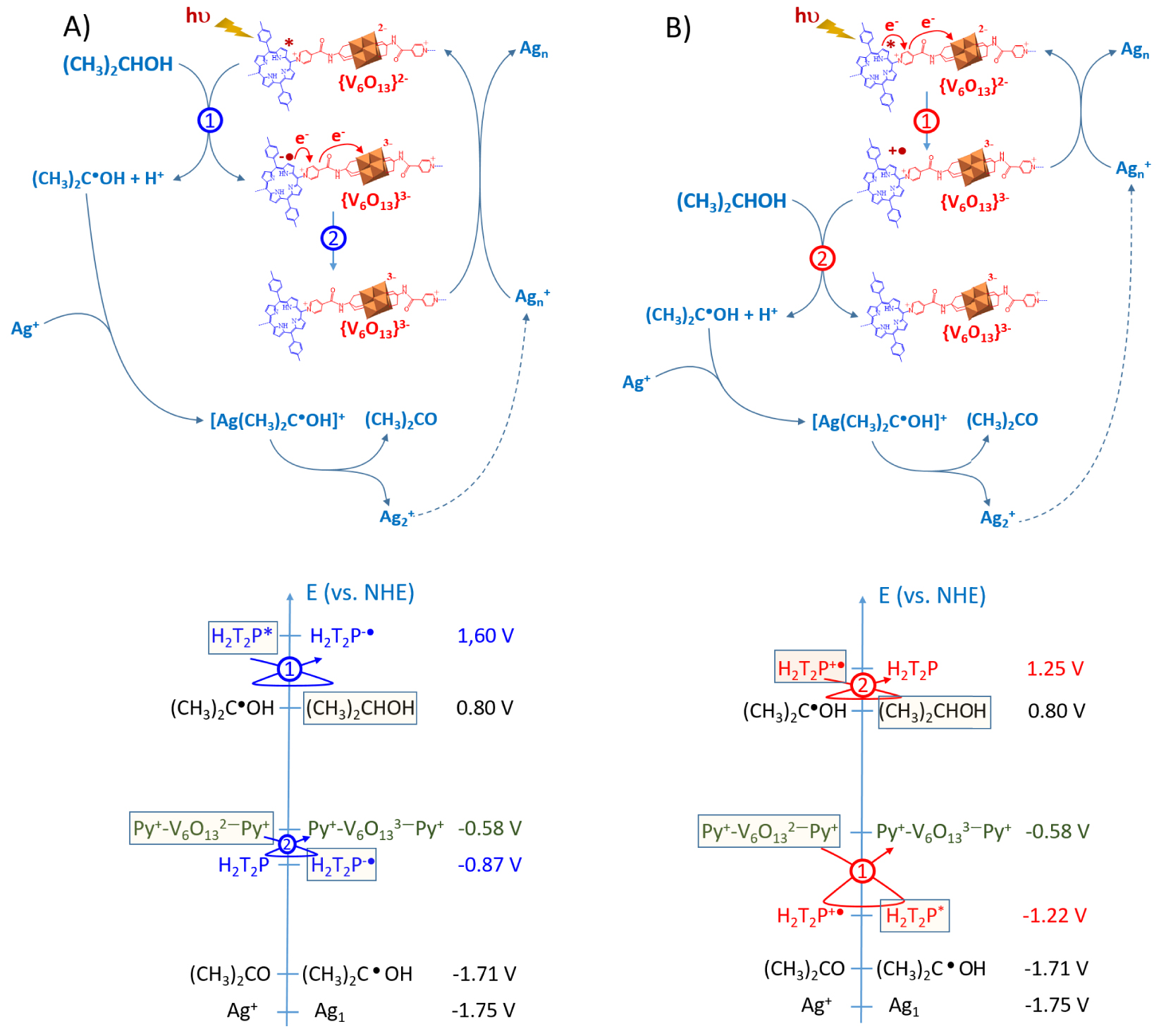
The photoreduction processes should involve the reduction of the excited porphyrin (Porph∗) by propan-2-ol leading to the formation of the reduced porphyrin (Porph∙−) and the alcohol radical according to the global reaction (1):
| (1) |
This reaction is thermodynamically favorable. Indeed, the reduction potentials of the excited porphyrins are evaluated according to the relation:
| (2) |
The calculated E(Porph∗∕Porph∙−) values are higher than the reported values for (CH3)2C∙OH∕ (CH3)2CHOH (E = 0.80 V versus NHE [37]).
The second mechanism (Figure 6B) corresponds to a direct intramolecular electron transfer from the excited porphyrins to polyoxovanadate via the reduction of the pyridinium subunit which acts as relay of electron followed by the reduction of silver ions by {V6O13}3−. A fast intramolecular electron transfer agrees with fluorescence quenching of the films observed and is also thermodynamically favorable.
Indeed, the reduction potentials of the excited porphyrins are evaluated according to the relation:
| (3) |
It should be mentioned that in the two proposed mechanisms, the simple consideration of the redox potential of the radical alcohol (CH3)2C∙OH), the reduced porphyrin or the reduced {V6O13}3− does not allow to explain the subsequent photoreduction of Ag+ ions. Indeed, the redox potential of the (Ag+∕Ag1) couple is too low (E° = −1.75 V versus NHE [38]) to allow the direct reduction of Ag+ to a single Ag1 atom either by the reduced porphyrin (E(H2T2P∗∕H2T2P−∙) = 1.60 V versus NHE) or the reduced {V6O13}3− (E({V6O13}2−∕{V 6O13}3−) = −0.58 V versus NHE).
Referring to radiolytic studies, the reduction of the Ag+ might be enabled by a complexation step between alcohol radical and silver ions that initiates the formation of silver clusters (4)–(7) [13, 38, 39]:
| (4) |
| (5) |
| (6) |
| (7) |
Another reasonable explanation is the formation of by reaction between Ag+ and the alcohol radical (CH3)2C∙OH even if the potential (E((CH3)2CO∕(CH3)2C∙OH) = −1.71 V versus NHE [40]) is slightly higher than the one the couple Ag+∕Ag1. This process cannot be excluded.
Finally, as the redox potential of the silver cluster, E(Agn+∕Agn), increases with the nuclearity n, the direct reduction of large clusters by the reduced porphyrin or the alcohol radical becomes thermodynamically feasible.
(A) Change in the UV–Vis absorption spectra of a deaerated aqueous solution of 1.6 × 10−4 mol⋅L−1 H2PtCl6 and 0.13 mol⋅L−1 propan-2-ol containing a slide of quartz modified with poly-V6O13-ZnOEP film under illumination. Inset: plot of the intensity of the absorbance at 𝜆 = 201 and 262 nm versus the time of irradiation. (B) Spectrum of the platinum solution before and after photocatalysis. Inset: photo of the obtained solution in the cuvette after the irradiation.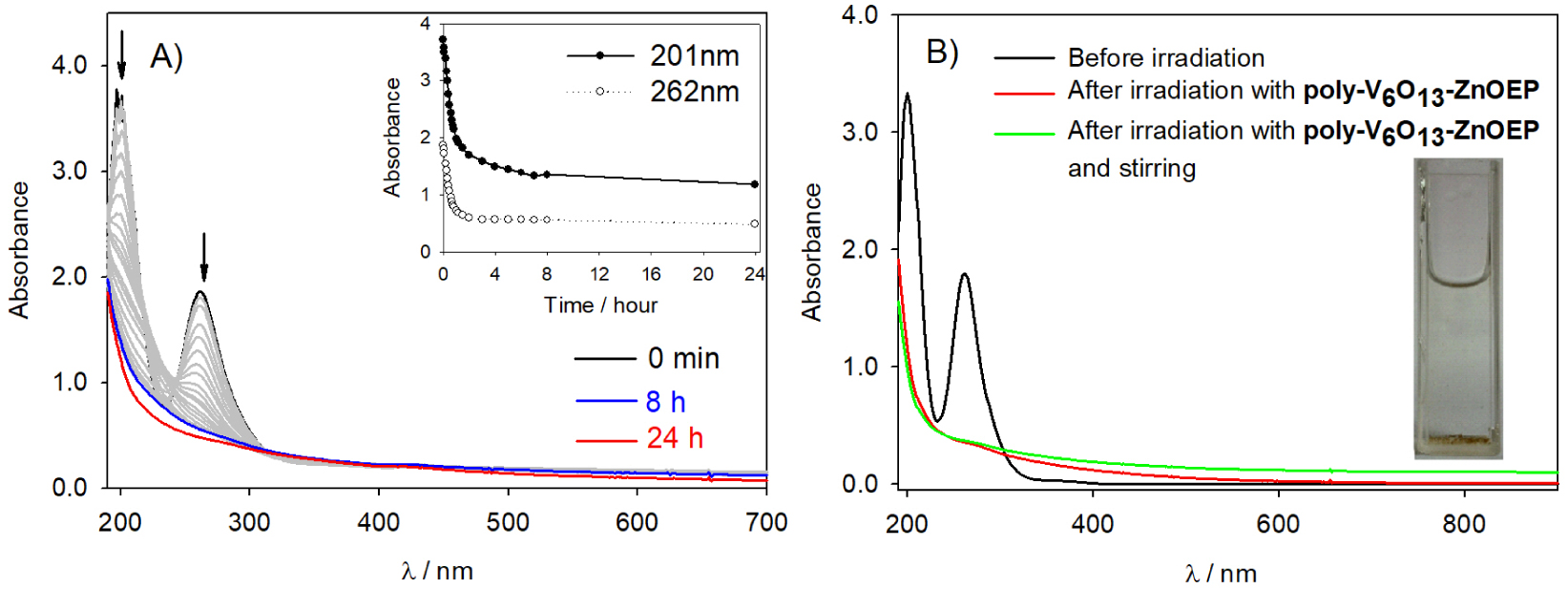
Note that the absence of reduced polyoxovanadate at the end of the reaction is in favor of the mechanism (B). It can be explained by an intramolecular back electron transfer between the reduced {Py+-V6O -Py} and the porphyrins oxidized Porph+∙ which is thermodynamically favorable.
To justify this electron transfer in the case of the mechanism B (Figure 6B), we can calculate the 𝛥r G of this process using the Rehm–Weller equation: 𝛥G = E(Porph∙+∕Porph) − E(POM∕POMred) − ES0–S1 where E(Porph+∙∕Porph) corresponds to the first oxidation potential of the porphyrin (E(Porph+∙∕Porph) = +1.25 V versus NHE for H2T2P) and E(POM∕POMred) corresponds to the first reduction potential of the POM (E({V6O13}2−∕{V6O13}3−) = −0.58 V versus NHE) [27]. ES0–S1 corresponds to the energy of the lowest electronic transition of the porphyrin estimated from its absorbance spectrum in solution. This value leads to negative 𝛥G (− 61.5 kJ⋅mol−1), indicating that the electron transfers between the excited porphyrins and the POMs subunit {V6O13}2− are thermodynamically possible. These electronic transfers lead to oxidized porphyrins (porph+∙) and reduced POMs (POMred, namely {V6O13}3−) via probably a relay including the reduction of the pyridium. Then, oxidized porphyrins porph+∙ are regenerated by reaction with propan-2-ol which leads to the formation of alcohol radicals (CH3)2C∙OH. This reaction is also really plausible, since the redox potential of the couple (CH3)2C∙OH∕(CH3)2CHOH is estimated to 0.80 V versus NHE [37], that is lower than the oxidation potential of the porphyrin.
However, the potential of the couple {V6O13}2−∕ {V 6O13}3−) is too high to permit the reduction of silver ions (E(Ag+∕Ag1) = −1.75 V versus NHE), corresponding to the redox potential of a single Ag(0) atom, different from the one corresponding to metallic silver which is 0.799 V versus NHE. We can also exclude the possibility of a photoinduced electron transfer from the excited porphyrins to the silver ions, because the 𝛥G value of such transfer is positive and is estimated to + 77.2 kJ⋅mol−1.
2.3. Photocatalytic recovery of platinum
To extend the photocatalytic application, we have explored the photocatalysis of the reduction of anions using the two covalent porphyrin–Lindqvist-type POM copolymers. The study is conducted under deaerated medium in the presence of 1.6 × 10−4 mol⋅L−1 H2PtIVCl6 (to keep the same concentration of metal ions) and 0.13 mol⋅L−1 propan-2-ol. Figure 7 illustrates the change in the UV–Vis absorption spectrum recorded during the visible light irradiation of a quartz slide covered with poly-V6O13-ZnOEP. The initial absorbance of anions decreased during illumination and disappeared completely after 100 min, however, because the Pt nanoparticles do not possess plasmon band or other type of UV–visible absorbance, it is impossible to monitor the creation of the Pt nanoparticles from the UV–visible spectra.
After 24 h of illumination, a yellow sediment on the bottom of the cuvette is obtained, and after stirring, the absorbance in the whole domain increases.
The TEM micrographs confirm the formation of the Pt nanoparticles. The diameter of the basic nanoparticle subunit is 1.0–1.5 nm which forms the closed-packed nanoparticles with a diameter of 10–20 nm (Figure 8). The EDS spectrum in Figure 8E indicates the presence of merely pure Pt in the sample.
(A–D) TEM images of the platinum nanomaterial obtained with the poly-V6O13-ZnOEP in a deaerated solution of 1.6 × 10−4 mol⋅L−1 H2PtCl6. (E) EDS spectrum showing the presence of Pt in the sample.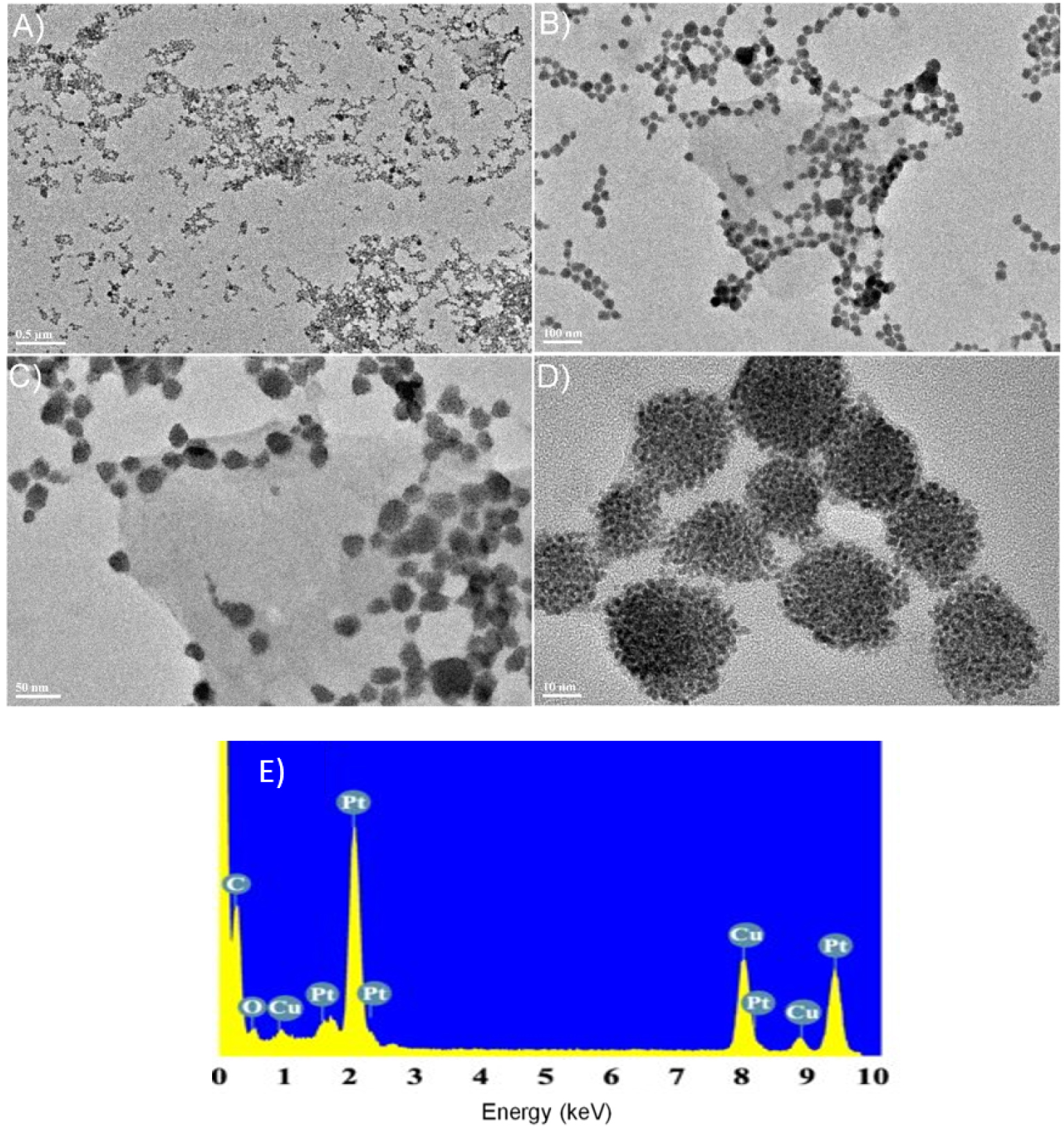
Figure 9 presents the absorption spectra of a deaerated aqueous solution containing 1.6 × 10−4 mol⋅L−1 H2PtCl6 and 0.13 mol⋅L−1 propan-2-ol with poly-V6O13-H2T2P covered quartz slide during visible light irradiation. The initial absorbance of anions disappeared completely after 110 min which is a little slower than for poly-V6O13-ZnOEP. After 24 h of illumination, we obtained a uniform and faint yellow solution which is different than the sediment obtained when using poly-V6O13-H2T2P. The TEM micrographs of the obtained Pt nanostructures are presented in Figure 10. The Pt nanoparticles showed again closed-packed spherical structure similar to the ones obtained using poly-V6O13-ZnOEP film. Nevertheless, such structures are also mixed with large nanosheets. The obtained Pt nanomaterial is in this case inhomogeneous in size and shape. The EDS spectrum in Figure 10E shows the presence of Pt nanoparticles in the sample.
(A) Change in the UV–Vis absorption spectra of a deaerated aqueous solution of 1.6 × 10−4 mol⋅L−1 H2PtCl6 and 0.13 mol⋅L−1 propan-2-ol containing a slide of quartz modified with poly-V6O13-H2T2P film under illumination. Inset: plot of the intensity of the absorbance at 𝜆 = 201 and 262 nm versus the time of irradiation. (B) Spectrum of the platinum solution before and after photocatalysis. Inset: photo of the obtained solution in the cuvette after the irradiation.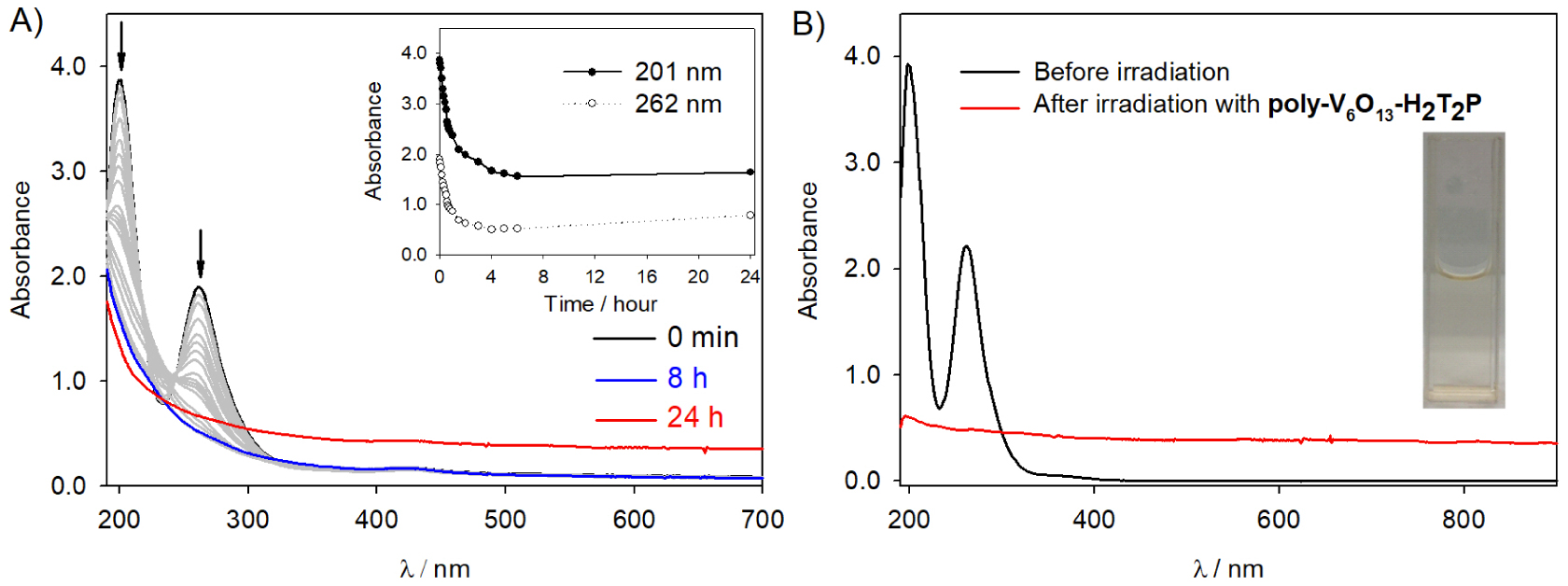
After almost 3 h for poly-V6O13-ZnOEP and 4 h for poly-V6O13-H2T2P of visible light irradiation, the absorption spectrum does not evolve anymore which indicates the end of the reaction. The duration of the photocatalytic reduction is longer in comparison to the silver recovery which can be explained by the number of electron implied: 4 for the Pt(IV) versus 1 in the case of the Ag(I).
We assume that the mechanism for the photocatalysis Pt(IV) is much more complicated than the one proposed for the reduction of Ag(I) ions probably due to the fact that 4 electrons must be transferred between the catalysts and the metallic ion. We will therefore not provide a mechanism in this present case although there is a good chance that the first step is similar to that of the reduction of Ag(I) ions, that is the electron transfer from the excited porphyrin via the pyridinium to the polyoxovanadates (mechanism B).
Last but not least, the heterogeneous photocatalysis can also be repeated at least five times while keeping the same efficiency. Platinum ions are reduced quantitatively and no deposition at the surface of the slide was observed (as checked by AFM analysis, data not shown).
(A–D) TEM images of the platinum nanostructures obtained with the poly-V6O13-H2T2P film in a deaerated solution of 1.6 × 10−4 mol⋅L−1 H2PtCl6. (E) EDS spectrum showing the presence of Pt in the sample.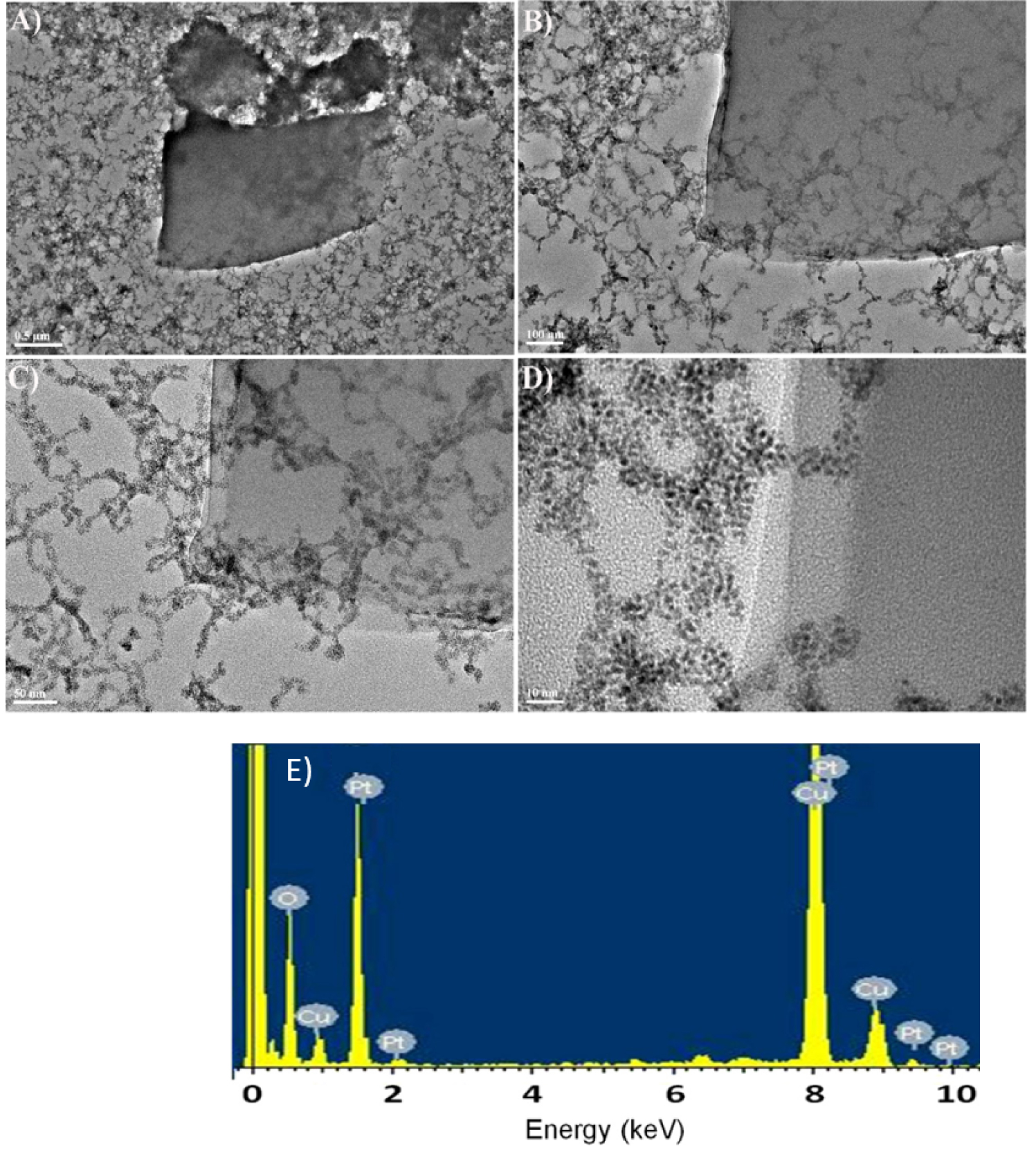
3. Conclusion
We demonstrated the efficiency of hybrid polyoxovanadate–porphyrin copolymers (poly-V6O13-H2T2P and poly-V6O13-ZnOEP) in the photocatalytic reduction of Ag(I) and Pt(IV) using visible light. Under ambient conditions, or H2PtIVCl6 were reduced quantitatively at the interface between water and the copolymeric films without poisoning the surface. The heterogeneous catalysis can be repeated at least five times while keeping the same efficiency at room temperature and under mild reaction conditions.
This process verifies our starting hypothesis that the copolymerization of photosensitizers such as porphyrin and POMs yields photoactive materials that are useful for photocatalytic reduction in general. The obtained silver nanoparticles form aggregates while the Pt nanoparticles showed closed-packed spherical structures and some large sheets.
4. Experimental section
Most common laboratory chemicals were reagent grade, purchased from commercial sources and used without further purification.
The 5,15-ditolylporphyrin (H2T2P) was purchased from SAS PorphyChem® and zinc-𝛽-octaethylporphyrin (ZnOEP) was purchased from Sigma-Aldrich. Lindqvist-type polyoxovanadate TBA2[V6O13{(OCH2)3CNHCO(4 − C5H4N)}2] (abbreviated Py-V6O13-Py) was synthesized according to the previous publications [24, 41].
The corresponding copolymers poly-Py-V6O13-Py-H2T2P and poly-Py-V6O13-Py-ZnOEP were prepared as described previously [27].
Electropolymerization have been carried out under an argon atmosphere using a 0.1 mol⋅L−1 solution of 0.1 mol⋅L−1 TBAPF6 in 1,2-C2H4Cl2∕CH3CN (7/3) containing 0.25 mmol⋅L−1 of ZnOEP or H2T2P and 0.25 mmol⋅L−1⋅mmol⋅L−1 of Py-V6O13-Py (Scheme 1) between 0 and 1.6 V versus SCE. ITO electrodes, with a surface of 1 cm2, were used as working electrode. For each copolymer, the number of iterative scans (n) was 20. After electropolymerization, the modified working electrodes were washed with CH3CN and then with CH2Cl2 in order to remove the monomers and the conducting salt present on the deposited films.
The electrochemically deposited poly-V6O13-ZnOEP or poly-V6O13-H2T2P films were dissolved and removed from ITO electrode with DMF. The operation is repeated six times. Subsequently, the DMF solution containing the copolymer was deposited on a quartz slide, and the DMF solvent was evaporated in air.
Water was obtained by passing through a Milli-RO4 unit and subsequently through a Millipore Q water purification set.
Irradiation was performed using a 300 W Xe arc lamp (Lot, Quantum design) with intense focused output beams (50 mm beam diameter) equipped with a water cell filter to absorb the IR radiation. A spherical reflector collects the output from the rear of the lamp and focuses it on or near the arc for collection by the condenser. One condenser is positioned for compensating focal length change due to dispersion and to produce a beam converging to the photochemical cell. According to the supplier, the irradiance of the lamp from 320 to 790 nm was around 50 mW⋅m−2⋅nm−1. The samples consisted of 4 mL of aqueous solutions with propan-2-ol, the quartz slide covered by the copolymer Py-V6O13-Py-H2T2P and poly-Py-V6O13-Py-ZnOEP and, Ag2SO4 or H2PtCl6 contained in a spectrophotometer quartz cell of 1 cm path length.
Deaerated solutions were obtained by bubbling with argon (Ar–U, from Air Liquide) before illumination. All experiments were carried out at room temperature.
We checked that the temperature of the solution did not increase by more than 1 degree during light illumination.
UV–Vis absorption spectra were recorded with a single beam Hewlett-Packard HP 8453 diode array spectrophotometer operated at a resolution of 2 nm.
Atomic force micrographs (AFM) measurements have been conducted directly on the ITO surfaces using a Veeco Dimension 3100 apparatus in the tapping mode under ambient conditions. Silicon cantilevers (Veeco probes) with a spring constant of 300 N/m and a resonance frequency in the range of 120–139 kHz have been used. The scanning rate was 1.0 Hz.
Transmission electronic microscopy (TEM) observations were performed with a JEOL 100 CXII TEM instrument operated at an accelerating voltage of 100 kV. Samples for TEM analysis were prepared by dropping the solution on carbon-coated copper TEM grids.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgments
The authors are grateful to the University of Strasbourg and the CNRS for constant financial support. The authors also thank the Overseas Study Program of Guangzhou Elite Project (GEP) for the Ph.D. grant of Zhaohui Huo.