

 CC-BY 4.0
CC-BY 4.0
1. Introduction
One of the major challenges facing mankind nowadays is to produce green fuels that overcome the huge greenhouse gas emissions due to the valorization of fossil fuels. Hence, hydrogen is considered as an interesting alternative decarbonized energy vector as it can be produced from various energy sources [1, 2]. Despite being considered as a potential future fuel [3], hydrogen has also some challenges to tackle along the three main steps of the process chain: production, storage and use [4]. Different processes have been developed to replace the conventional hydrogen production technology such as Steam Reforming of Methane (SRM), Dry Reforming of methane (DRM) [5] or combined processes such as tri-reforming with a mixture of CO2, H2O and CH4 [6, 7]. One of the alternatives under study is the thermocatalytic decomposition of methane which seems to have several advantages over SRM [8, 9, 10]. Catalytic Decomposition of Methane (CDM) produces no CO∕CO2 and therefore eliminates the need for water gas shift and CO2 removal. This process may present a lower energy and carbon footprint than SRM, leads to the formation of carbon oxide free hydrogen, and could become economically competitive [11, 12]. This reaction is also an important step in SRM and DRM processes [13, 6]. The temperature required for non-catalytic methane decomposition is close to 1200 °C. Then, one of the biggest issues for the industrial implementation of CDM is the development of a catalyst that can operate without being rapidly deactivated due to the deposition of the carbon produced. It must be noticed that the regeneration of catalysts to remove the carbon deposit can affect the CO2 footprint of the process [14].
The major types of catalysts tested for this heterogeneous reaction are the metallic and carbonaceous ones [15, 16, 17, 18, 9]. Various transition metals, specifically those of group VII, were tested for methane decomposition [19]. Their order of increasing activity is as follows: Co, Ru, Ni, Rh > Pt, Re, Ir > Pd, Cu, W, Fe, Mo [20]. Despite the formation of a considerable amount of coke compared to other noble metal catalysts, nickel is considered as the most efficient catalyst for methane cracking on industrial scale [21] due to its high catalytic activity, its relatively low cost and the larger available resources compared to precious metals or to cobalt. Therefore, numerous studies have focussed on nickel-based catalysts which have then been studied for not only CDM but also for DRM processes [10]. Several oxide supports have been used including TiO2, SiO2, MgO, ZnO2, Al2O3 [22, 23] but also hydroxyapatite [24]. SiO2 has been widely studied and often leads to good performance [25]. Several studies have focussed on the doping of catalysts and supports by nickel in order to improve their activity and stability during the reaction [26, 27]. Carbon-based materials have advantages over metallic catalysts deposited on mineral supports for methane decomposition, namely low cost, high temperature resistance, good tolerance to potentially harmful impurities and the absence of formation of CO2 and metal carbides [28, 9]. Moreover, the carbon produced could be used as electrode material, sorbent or in soils. In the literature, most of the articles dealt with activated carbon (AC) and carbon black for their good activities and stabilities [29]. These studies highlighted the influence of the operating parameters (temperature, velocity, catalyst charge etc.) and the contribution of the physicochemical properties of catalysts (texture, structure, surface chemistry), that allowed obtaining kinetic expressions that describe the mechanism of this process [30, 31, 32, 33, 34, 28, 35]. AC exhibits a higher efficiency compared to carbon black because of their high surface area and surface reactivity, but they undergo a more rapid deactivation [30, 36].
For further improvement of the activity of catalysts during methane decomposition, metals supported over carbon materials have been used to benefit from the advantages of both materials [37, 21, 38, 39]. As Ni is an efficient metal and AC is the best carbon support, several studies have focussed on this kind of catalysts [40, 41, 42]. Prasad et al. [40] tested the performance of nickel supported on AC based on coconut prepared by the wet impregnation method with different Ni loads (8.6 to 30.0 wt%). The supported catalysts exhibit different performances as a function of reaction time depending on the Ni load. The total amount of methane converted for a reaction time of 4 h increased with Ni content until 23% of nickel, while the amount of carbon formed reached a plateau above this value. This could be explained with the evolution of the textural properties of the AC after the wet impregnation and then during the reaction. X-ray diffractograms showed the increase of nickel particle size, leading to an increase of crystallinity and a decrease of dispersion. Bai et al. [41] used a commercial AC as support for nickel. They observed that the Ni/AC had a higher catalytic activity than the original AC. They also attributed the deactivation of the catalyst to the increase of the crystal size but also to the formation of nickel carbides Ni3C. In addition, the authors reported that a high methane conversion rate and an acceptable stability of the catalyst may occur at a temperature of 750 °C. Scanning Electron Microscopy (SEM) analysis confirmed the presence of carbon filaments following the decomposition of methane. Zhang et al. [42] compared the activities of Ni supported over oxides and coals in methane decomposition and found that the latter had higher activity and stability at 850 °C. They confirmed also that the Ni content and the particle size may have a significant effect on methane decomposition over coal char-supported catalysts.
Despite all these extensive studies on CDM over Ni/carbon catalysts, the effect of AC preparation from the same carbon precursor has not yet been studied systematically. For this reason, in the present work we have produced two ACs (physically and chemically activated) from the same biomass (olive stones) and we have tested their properties for CDM with different Ni loads and methane partial pressures. Kinetics parameters of methane catalytic conversion have been determined. The textural and structural characterizations of both fresh and deactivated catalysts have been also done. To the best of our knowledge, we present here the first work comparing physically and chemically ACs as supports for CDM over Ni/carbon catalysts.
2. Materials and methods
2.1. Catalyst preparation
2.1.1. Carbonaceous catalysts
Catalysts used in this work were prepared via physical and chemical processes [43]. The first process involves the carbonisation of olive stones under nitrogen atmosphere at 600 °C for 2 h and the activation of the char at 850 °C using steam for 8 h [44]. The final product was labeled CAGOP. The second AC was prepared by chemical activation which consists of the impregnation of the olive stones with a solution of phosphoric acid (50%) for 9 h at 110 °C, then the carbonization of the filtered olive stones was done under nitrogen atmosphere at 410 °C for 2 h 30 min. This sample was labeled CAGOC [45].
2.1.2. Supported Ni catalysts
CAGOP and CAGOC previously prepared were used as supports for the Ni catalysts. These samples were ground and sieved to particle size of 100 to 250 μm. The supported Ni catalysts were prepared by wet impregnation: the appropriate amount of the precursor Ni(NO3)2.6H2O for the targeted nickel content on ACs (namely 2, 4 and 6 wt%) was dissolved in distilled water and mixed with the support particles. These nickel contents were lower than the ones tested in some previous studies [40], it was expected to obtain a good dispersion and a lower ripening of the metal at high temperature with less than 10% of nickel. The contact between the metal precursor and the porous supports was maintained through magnetic stirring for 24 h at ambient temperature. The impregnated samples were then filtered and dried overnight in an oven at 110 °C. Finally, the supported catalysts were calcined in a vertical stainless steel reactor at 550 °C under an inert atmosphere (N2) for 2 h. The produced samples were labelled: CAGOP–Nix and CAGOC–Nix, where x is related to nickel mass percentage. The bulk contents of Ni in these catalysts were determined using Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES—Thermo Scientific iCAP 6000 Series) revealing actual nickel amounts close to the targeted values, that is 2.5, 4.6 and 5.5 wt% for CAGOP–Ni, and 1.6, 4.7 and 6.5 wt% for CAGOC–Ni, respectively.
2.2. Methane decomposition experiments
The individual gases, methane and argon (99.999% purity), were used without further purification and supplied by Air Liquide (France). The composition of the gas mixture fed into the system was controlled by mass flow rate regulators (Brooks Instrument). The bed of catalyst under study for methane decomposition was placed inside a quartz tubular reactor heated by a vertical cylindrical furnace. The temperature was controlled by a thermocouple positioned inside the catalyst bed. All the experiments were conducted at a total pressure P = 1.013 × 105 Pa for 1 h. The internal diameter of the reactor was 1 cm. Before every run the catalyst bed was heated to the operating temperature under argon flow (100 cm3∕min at 20 °C). The studied reaction temperatures ranged from 750 to 850 °C and the total gas flow rate was 100 cm3∕min at 20 °C with different molar percentage of methane, namely 5, 10 and 20 mol%. Catalyst mass was kept at 0.1 g, the bed height was 6 mm, leading to a contact time ranging between 75 and 85 ms for the temperatures studied. The outflow gas was analysed with a micro gas chromatograph (μGC) equipped with two columns connected in series (Plot U and 5A molecular sieve, for back flush) and a thermal conductivity detector, hydrogen and methane were quantified. The methane molar fractional conversion, the methane decomposition rate and the stability factor were calculated as follows:
| (1) |
| (2) |
| (3) |
2.3. Catalyst characterization
Textural and structural characterizations were required for raw and used catalysts to better understand their contribution in methane decomposition. For this purpose, a Micromeritics ASAP 2020 physical adsorption apparatus was used to obtain the sorption isotherms of nitrogen at 77 K. The surface area of catalysts SBET was measured by applying the BET equation in the relative pressure range 0.01 to 0.05 as usual for microporous materials with type I isotherms. The total pore volume Vp,tot was obtained from the amount adsorbed at P∕P0 = 0.99. The micropore volume Vp,𝜇 was determined according to the Dubinin–Radushkevich method.
For structural characterization, powder XRD analyses were carried out using an XPert Pro MPD diffractometer (PANalytical) equipped with a Cu radiation tube (𝜆 = 1.5406 Å) and an X Celerator detector. The average size of nickel particles was determined by using the Scherrer equation. TEM was performed using a Jeol ARM 200 apparatus.
3. Results and discussion
3.1. Kinetics of methane decomposition
3.1.1. Initial reaction order
The equation rate of methane decomposition was expressed as a function of the methane molar concentration:
| (4) |
To obtain information regarding the reaction order n, the rates were measured with varying methane mole fractions using argon as the diluent. As the catalyst deactivated rapidly, the initial rates rCH 4,0 were estimated by extrapolating the curves obtained in Figure 1 to zero time.
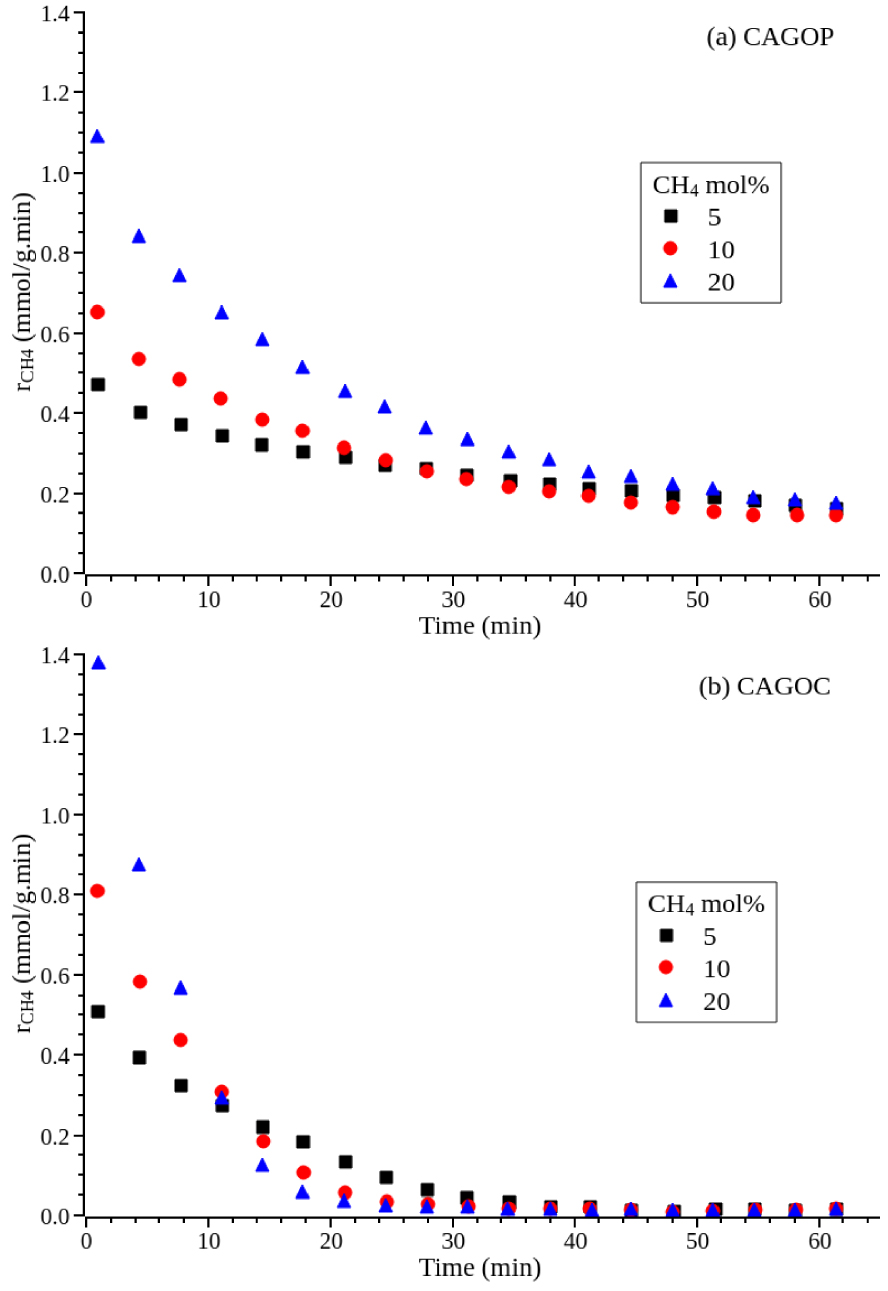
Methane decomposition rate as a function of methane partial pressure. (a) CAGOP–Ni2, (b) CAGOC–Ni2, mcat = 0.1 g, T = 850 °C, P = 1.013 × 105 Pa.
Experimental data plotted as ln(rCH 4,0) versus ln([CH4]) gave almost straight lines as shown in Figure 2 for both CAGOP–Ni2 and CAGOC–Ni2. From the slopes, the reaction order was estimated to be 0.63 and 0.74 for CAGOP–Ni2 and CAGOC–Ni2 respectively with a correlation coefficient of 0.99 for both catalysts. The reaction kinetics of methane decomposition have been extensively studied for non-doped carbon materials revealing a reaction order ranging from 0.5 to 1.0 [8, 32, 46]. Coming to metal-supported catalysts, specifically nickel metal, literature dealing with kinetics are limited. Wang et al. observed an average reaction order of 0.63 on unsupported nickel [47], however first and second orders were reported for nickel supported over oxide [48] and zeolite [49], respectively. To the best of our knowledge, this work is the first to report the order reaction for CDM over Ni/carbon.
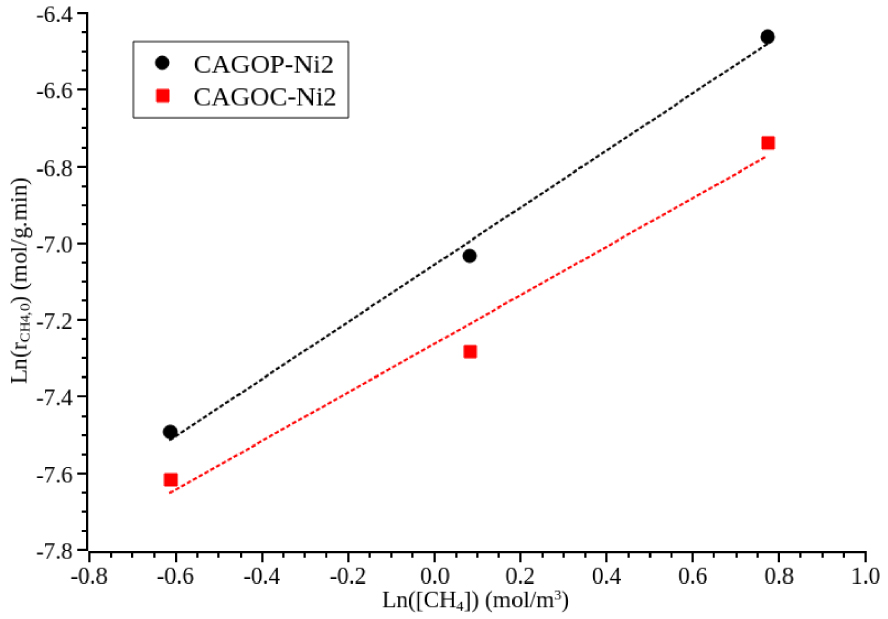
Initial methane conversion rate as a function of methane partial pressure, mcat = 0.1 g, T = 850 °C, P = 1.013 × 105 Pa.
3.1.2. Influence of temperature
The effect of temperature on the rate of carbon formation is expressed in Figure 3. Due to the endothermicity of the reaction, the methane conversion ratio increases with temperature for both catalysts. As the temperature decreases, the reaction rate decreases but the deactivation of the catalyst is significantly lowered. CAGOP–Ni2 exhibits a typical trend for the different temperatures: the thermally induced deactivation leads to a continuous decrease of the reaction rate until it reaches a low but relatively stable value. In the case of CAGOC–Ni2, the influence of temperature is far more dramatic since methane cracking rate decreases quickly down to a very low value when the temperature is higher than 800 °C. CAGOP–Ni2 then exhibits a higher stability factor than CAGOC–Ni2 as seen in Table 1. This difference between physically and chemically ACs was also observed for the corresponding carbon materials without the presence of nickel [36]. This shows the major contribution of the carbon support to the global reaction rate in this temperature range. At lower temperature, that is 750 °C, the two catalysts exhibit very similar behaviour with a continuous and slow decrease of reaction rate, the values of the reaction rates are also the same. This shows that at this temperature, the contribution of the AC to the global rate of methane decomposition is very low and the trends observed are related to the activity and deactivation of nickel.
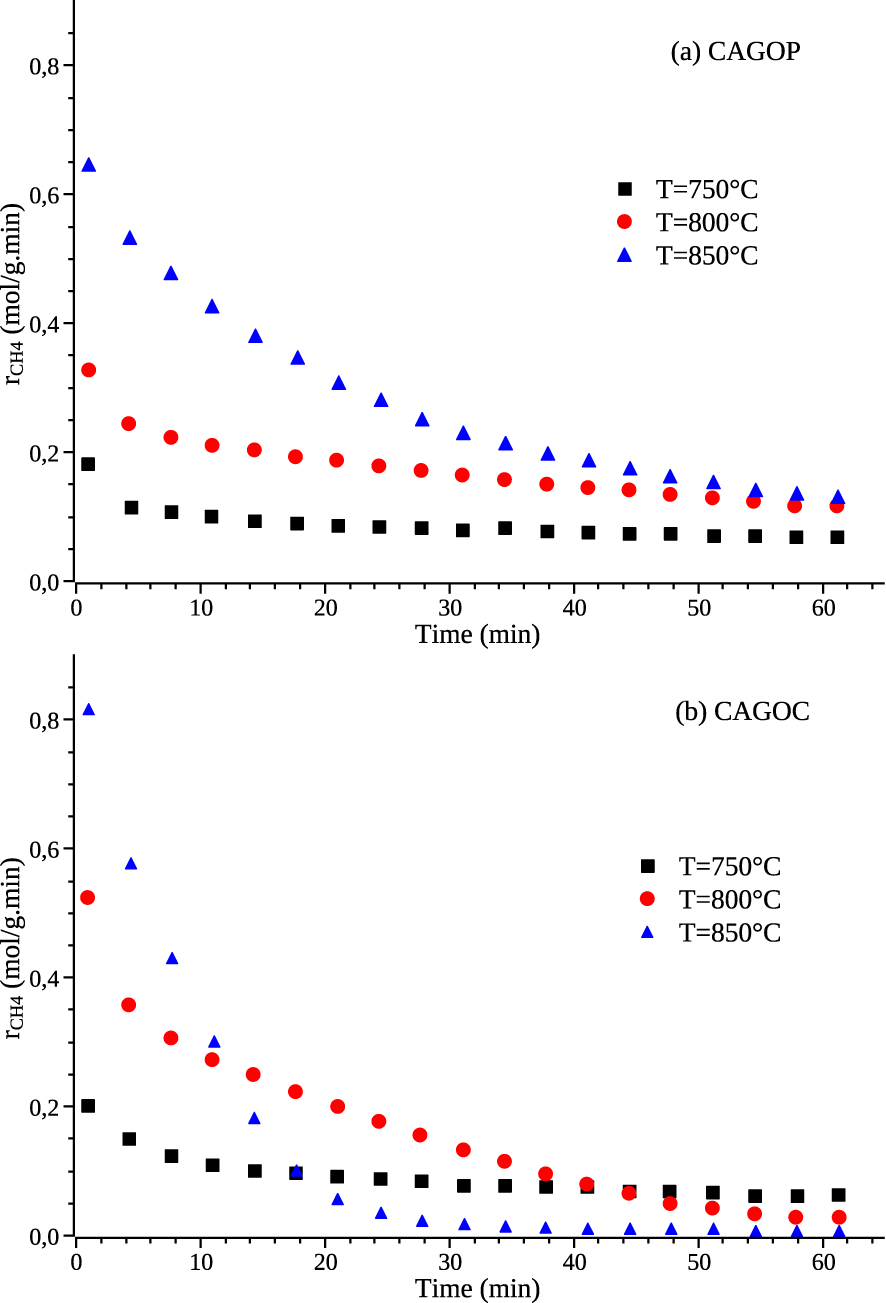
Effect of temperature on methane decomposition rate (a) CAGOP–Ni2, (b) CAGOC–Ni2, FCH 4 = 10 cm3∕min, mcat = 0.1 g, PCH 4 = 1.013 × 104 Pa. Masquer
Effect of temperature on methane decomposition rate (a) CAGOP–Ni2, (b) CAGOC–Ni2, FCH 4 = 10 cm3∕min, mcat ... Lire la suite
Stability factor of catalysts as function of temperature
| T (°C) | CAGOP–Ni2 | CAGOC–Ni2 |
|---|---|---|
| 750 | 0.38 | 0.31 |
| 800 | 0.36 | 0.05 |
| 850 | 0.21 | 0.01 |
3.1.3. Activation energy
The activation energy can be obtained from Arrhenius equation:
| (5) |
Rates were measured over the supported catalysts at different temperatures and rCH 4,0 was again extrapolated to zero time and used to calculate the rate constant k.
Activation energies obtained from the slopes of the Arrhenius plot presented in Figure 4 were 122 kJ/mol and 139 kJ/mol for CAGOP–Ni2 and CAGOC–Ni2, respectively. These values are much lower than the methane C–H bond energy 440 kJ/mol [18]. These findings are significantly different from those found in literature as the activation energy of Ni supported on metal oxide catalysts for methane decomposition ranges from 29.5 to 97 kJ/mol [18, 50]. The activation energies for bare ACs were varying over a large range depending on the raw material of the catalyst and the operating conditions of its preparation. These activation energies range from 140 to 201 kJ/mol [20]. Due to the wide range of activation energies reported, the major controlling step in methane decomposition mechanism cannot be easily determined. Then, from the comparison with published data, the apparent activation energy measured in this study are significantly higher than the one generally observed for nickel on oxide catalysts. They are more close to the values obtained for non-doped carbon materials [36]. This shows that, for short reaction times, the direct methane decomposition on carbon accounts for a significant part of the methane decomposition reaction.
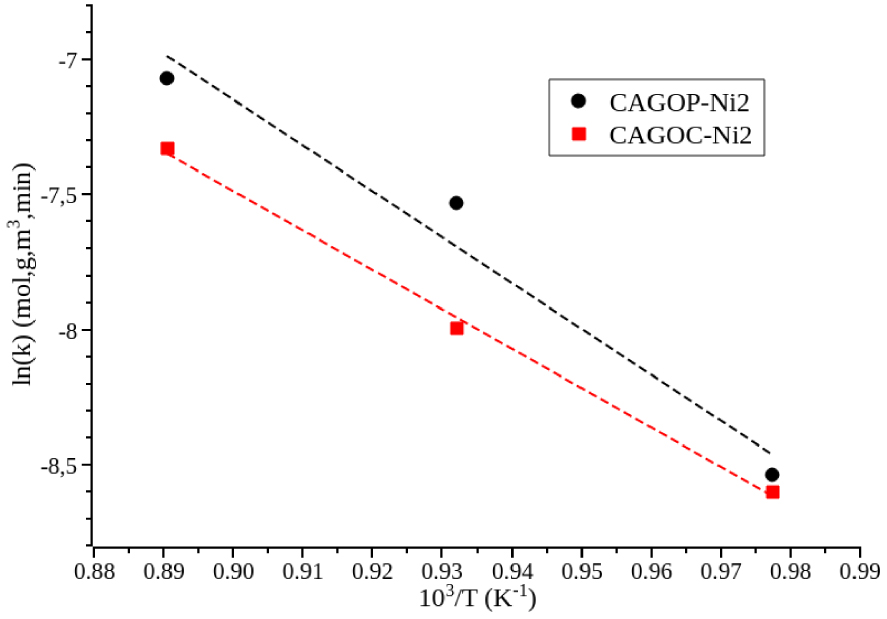
Arrhenius plot for methane decomposition over supported catalysts. FCH 4 = 10 cm3∕min, mcat = 0.1 g, PCH 4 = 1.013 × 104 Pa.
3.2. Effect of Ni load on methane decomposition
To evaluate the contribution of Ni particles on methane decomposition, a series of experiments were carried out at the same conditions with different concentrations of Ni as seen in Figure 5. Table 2 presents the initial rate and stability factor. All the catalysts revealed the same trend: a high initial rate followed by a rapid drop. It is known that ACs themselves have a good catalytic effect on methane decomposition that is enhanced with the introduction of Ni. The nickel particles contribute to the activation of the methane molecules during the induction step by generating radicals on the Ni surfaces that can migrate to the support surface where they decompose to carbon and hydrogen [51]. For CAGOP, rCH 4,0 increases from 0.6 mmol∕g⋅min for the bare carbon support to 1.0 mmol∕g⋅min for the one with 4.6 wt% Ni, corresponding to a 65% increase of reaction rate. The effect of Ni load is less significant for CAGOC as the increase resulting from Ni particles is limited to 34% for the same Ni amount.
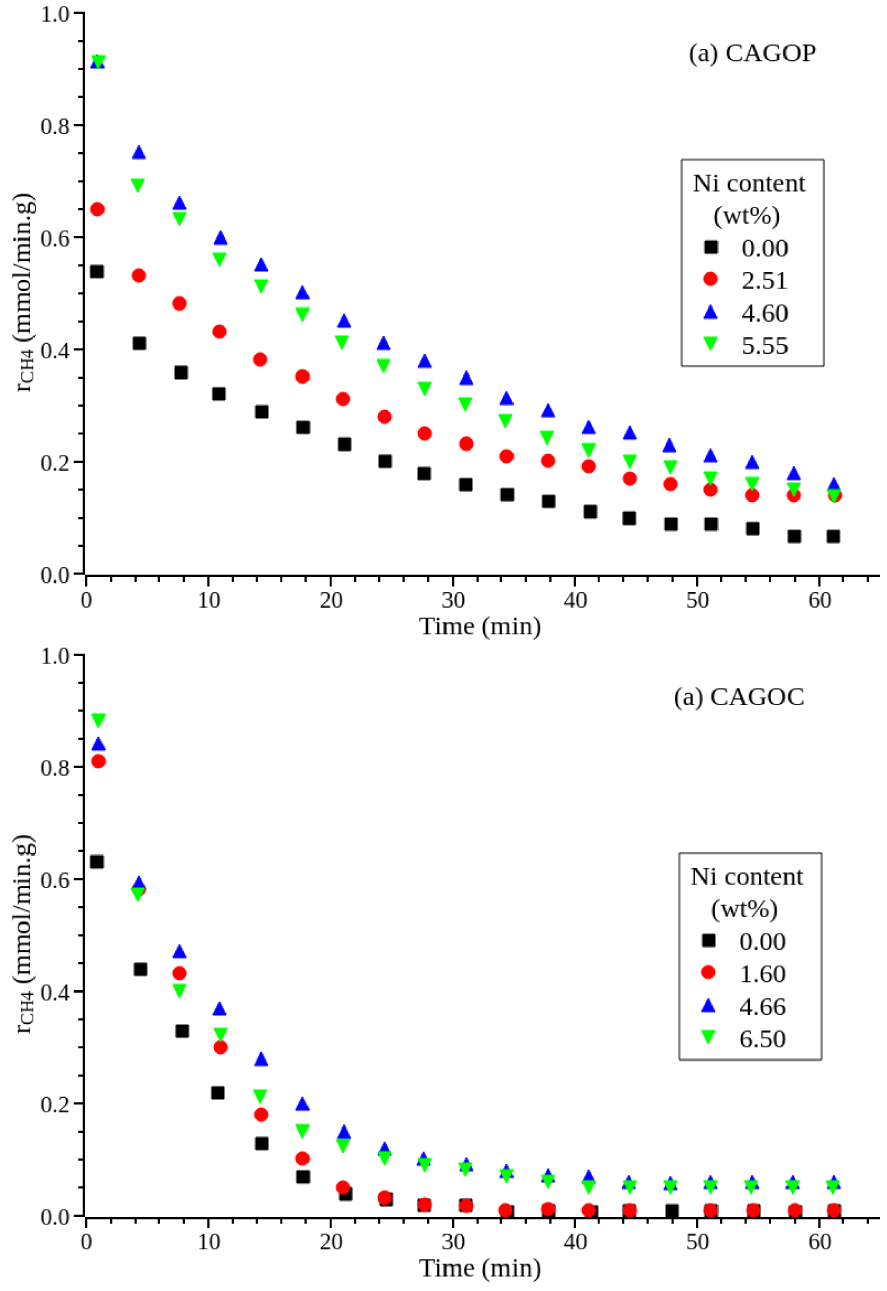
Effect of Ni loading on (a) CAGOP and (b) CAGOC during methane decomposition, FCH 4 = 10 cm3∕min, mcat = 0.1 g, PCH 4 = 1.013 × 104 Pa.
Initial rate and stability factor of catalysts
| Catalysts | Ni amount (wt%) | rCH 4,0 (mmol∕g⋅min) | K |
|---|---|---|---|
| CAGOP | 0.00 | 0.588 | 0.120 |
| CAGOP–Ni2 | 2.51 | 0.684 | 0.200 |
| CAGOP–Ni4 | 4.60 | 0.980 | 0.210 |
| CAGOP–Ni6 | 5.55 | 0.969 | 0.186 |
| CAGOC | 0.00 | 0.681 | 0.014 |
| CAGOC–Ni2 | 1.60 | 0.880 | 0.011 |
| CAGOC–Ni4 | 4.66 | 0.912 | 0.065 |
| CAGOC–Ni6 | 6.50 | 0.913 | 0.066 |
It can also be observed that whatever the carbon support, a nickel loading higher than 4.5 wt% does not lead to a further increase in methane decompostion rate. The results also show that Ni addition on the two types of ACs allows achievement of better stability compared to the the pristine carbon. These conclusions are in good agreement with those reported in literature [38, 42]. As for initial reaction rates, all CAGOP-based catalysts exhibit higher stability factors compared to catalysts based on CAGOC (Table 2). This had already been observed for methane decomposition on non-doped carbons [36].
This clearly shows the major influence of the direct reaction on the carbon support in metal/AC catalysts. These features will be further discussed in the next section based on the characterization of raw and spent catalysts.
3.3. Characterization of the pristine and spent catalysts
The results of textural characterization: BET surface area, total pore volume and micropore volumes are presented in Table 3 for the two catalysts. The initial ACs have a relatively large surface area (717 and 817 m2∕g for CAGOP and CAGOC, respectively). For the two carbons, the micropore volume is similar to the total pore volume, showing that these materials exhibit type I isotherms. NLDFT analysis showed that the pore size corresponds to that of ultramicropores (that is pores with a size below 0.7 nm, from IUPAC definition [52]). Indeed, for the two carbons, the mean micropore size was close to 0.5 nm. Such textural properties are often observed for biomass-derived carbon materials [53]. SBET decreased slightly after Ni doping and calcination, VP,tot and VP,𝜇 remained practically the same indicating that the metallic phase has no significant effect on the texture. This is explained by the small amount of nickel inserted on the surface (2 wt%), corresponding to a very low volume fraction. It should also be related to the fact that nickel particle size is expected to be larger than 10 nm after a calcination at 550 °C, it is then not expected that nickel could plug microporosity.
Textural changes of raw ACs, pristine and spent catalysts after 1 h of test at 850 °C
| Catalysts | SBET (m2∕g) | VP,tot (cm3∕g) | VP,𝜇 (cm3∕g) |
|---|---|---|---|
| CAGOP | 717 | 0.33 | 0.29 |
| CAGOP–Ni2 calcined | 708 | 0.33 | 0.27 |
| CAGOP–Ni2 deactivated | 65 | 0.06 | 0.03 |
| CAGOC | 817 | 0.34 | 0.34 |
| CAGOC–Ni2 calcined | 766 | 0.35 | 0.29 |
| CAGOC–Ni2 deactivated | 15 | 0.05 | 0.00 |
After the methane decomposition test at 850 °C, the surface area and the porosity of supported catalysts decreased dramatically (Table 3). CAGOC–Ni2 exhibits a very low pore volume and surface area, the remaining pores were in the mesoporous domain. This corresponds to an increase in average pore diameter showing that methane decomposition occurs mainly in the entries of pores in agreement with previous work [34, 36]. NLDFT analysis showed that only pores above 3 nm are detected on deactivated catalyst. Carbon deposit plugs more rapidly and more completely the entrance of micropores. In the case of CAGOP–Ni2 sample, only 10% of the pristine microporous volume was not plugged by carbon deposit. A small mesopores’ volume was also observed. This explains the higher surface area of this catalyst compared with CAGOC–Ni2 and partly its better stability since we have shown above that the direct reaction on the AC support plays an important part in the global methane decomposition reaction rate.
Therefore, in agreement with literature [20, 31, 54], we confirm here that for carbonaceous materials, the deactivation of the carbon support is strongly related to the blocking of micropores with the deposited carbon. We also observed that the decrease of textural properties is somewhat limited for physically ACs compared to the chemically activated ones. The plugging of pores exhibits the important effect of carbon nanostructures on their stability for methane conversion. For longer reaction durations, the direct reaction on AC support is minimized by the deactivation of its surface and the global reaction rate is more related to the nickel particles. Furthermore, for such supported catalysts, other parameters can interfere such as the modification of Ni particle sizes and the speciation of nickel. This was studied by XRD and TEM analysis.
From the XRD patterns of pristine and deactivated catalysts, we can notice the presence of two broad reflections near 2𝜃 = 24° and 2𝜃 = 44° corresponding to the (002) and (110) Miller indices of stacked polyaromatic carbon layers (Figure 6). The shape and position of these large reflections show that the corresponding ordered structures have a low size (a few layers, about 1 nm) and a significant density of defects leading to an interplane distance of about 0.4 nm. This was expected for such AC materials [55, 54, 36].
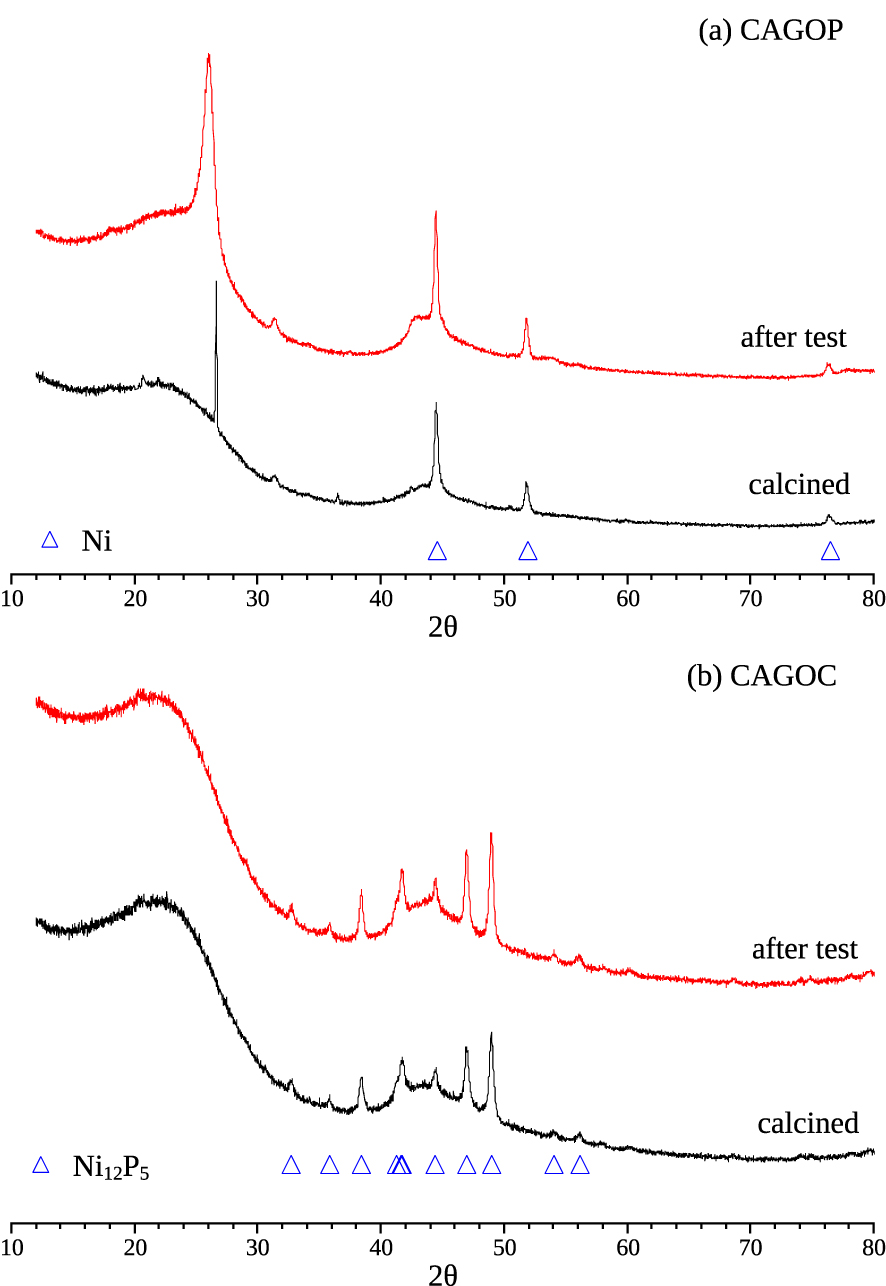
X-ray diffractograms of pristine and spent catalysts (a) CAGOP–Ni2 and (b) CAGOC– Ni2.
Further investigation of X-ray diffractograms reveals the presence of other reflections that could be identified. For catalysts obtained from CAGOP AC (Figure 6), the presence of metallic nickel particles is obvious with reflections corresponding to the Miller indices (111), (200) and (220) at 2𝜃 values of 44.55°, 51.91° and 76.48°, respectively (PDF#04-0850) [56, 57]. In the case of pristine calcined CAGOP–Ni2, a sharp reflection at 2𝜃 = 26.33° corresponds to a silica impurity coming from the fused silica reactor used for calcination. After methane decomposition, the same nickel particles are detected showing that there was no change in the metal speciation during the reaction. Besides the broad reflections of ACs which are still observed, the presence of significantly more organized carbon is attested by more intense and sharp reflections at 2𝜃 = 26.06° and 2𝜃 close to 43.5°. These values are quite close to the corresponding reflections of (002) and (110) of graphite (26.60° and 44.67°, respectively). The position of (002) reflection corresponds to a mean interlayer distance of 3.42 Å, it shows the good level of organisation of these domains. The catalytic activity of nickel for carbon graphitization has already been underlined [58].
For the pristine calcined CAGOC–Ni2, we notice the formation of Ni12P5 with the presence of all corresponding reflections for 2𝜃 values between 33° and 57° (PDF#22-1190) [59]. This corresponds to the result of a reaction of nickel with the remaining phosphorus during calcination. Although CAGOC carbon was extensively washed after its synthesis, a small amount of chemically bound phophorus remains in the final material used for impregnation and calcination. 0.4 wt% of phosphorus is enough to react with 2.0 wt% of nickel. Nickel phosphides supported on ACs have been tested as potential catalysts for hydrodeoxygenation processes [60]. It was observed that Ni12P5 often coexists with Ni2P. In the case of CAGOC–Ni2, the formation of Ni12P5 is favoured since the Ni/P ratio is high [61]. Ni12P5 is also considered as an efficient catalyst for hydrogen evolution reaction [59], for hydrogenation [62] or deoxygenation reactions [63]. Its activity towards CDM is not well known, Chen et al. observed that nickel phosphides showed significantly less cracking activity compared to nickel catalyst [64]. Further investigations are necessary for a better understanding of the effect of nickel phosphide on methane decomposition. The results presented above show that its presence leads to an improved efficiency toward methane decomposition compared to the non-doped carbon (Figure 5b). Another significant result is that no formation of graphitic carbon is observed with nickel phosphide, it then seems that it does not enhance carbon graphitisation compared to metallic nickel.
In order to study the evolution of Ni and Ni12P5 particle sizes by XRD, samples with a higher amount of nickel were synthesized (i.e. 5 wt%). The crystallite sizes of Ni for fresh and deactivated CAGOP-based materials were calculated using the Scherrer equation applied to the half-band width of the reflection corresponding to the Ni(200) planes [41]. The same analysis was done on CAGOC-based catalysts with the (022) reflection of Ni12P5. For physically AC, we observed that the nickel particle’s size increases from 17.7 to 20.6 nm after methane decomposition. In the case of chemically AC, the Ni12P5 particle size increased from 17.4 to 18.3 nm. The particle size of nickel phosphide in the pristine catalyst is then similar to the one of nickel particles in the corresponding CAGOP-based material. The variation of particle size during the reaction is limited, this could be related to a lower catalytic activity of Ni12P5 compared to Ni, or to a higher stability of the tetragonal structure of the phosphide compared to the cubic structure of metallic nickel.
It is well established that this heterogeneous reaction depends strongly on the size of catalyst particles. A size of between 10 and 20 nm appeared to be suitable for methane decomposition [57]. In our case, the particle size variation is limited since the calcination step at 550 °C leds to an increase of particle size and then to a higher stability of particles. Moreover, the average particle size is between 15 and 20 nm, this decreases the rate of increase by thermal ripening compared to that of particles smaller than 10 nm. The catalysts are therefore more stable during the methane decomposition experiments.
The TEM images of the catalysts after the calcination step show that a good dispersion of nickel nanoparticles was achieved on CAGOP (Figure 7a,b) and CAGOC (Figure 7e) ACs. The particle size distribution was relatively narrow in the two cases. TEM images of the completely deactivated CAGOP–Ni2 catalysts are also presented on Figure 7. They show the presence of carbon nano-onions which are considered responsible for the decrease of the catalytic activity of the two supports. The high level of organisation of the carbon layers (Figure 7d) is consistent with the DRX results. The reaction mechanism proposed by Amin et al. [50] involves the adsorption of methane followed by a series of dehydrogenation reactions that end with the adsorption of carbon species on the nickel particles. The adsorbed carbon diffuses through the nickel surface from the top until it reaches the support surface where carbon filaments are formed. This diffusion process occurs simultaneously with encapsulating carbon formation. This type of carbon produced by nano-onions is often observed when the carbon adsorption rate exceeds the carbon diffusion–graphite formation rate [65]. As a consequence, the nickel particles will be encapsulated by graphitic layers that inhibit the contact between Ni particles and the reactant gas causing the deactivation of the catalyst. In the case of chemically ACs, the formation of nickel phosphide inhibits the formation of this wide graphitic layer (Figure 7f).

TEM images of calcined and spent catalysts. (a)–(b) CAGOP–Ni2 fresh, (c)–(d) CAGOP–Ni2 deactivated at T = 850 °C, (e) CAGOC–Ni2 fresh, (f) CAGOC–Ni2 deactivated at T = 850 °C.
Then, the deactivation of Ni/AC catalysts may be attributed to: (1) Carbon deposit plugging the entry of micropores leading to a decrease of the carbon support reactivity; (2) carbon deposit forming onion-like structure, recovering Ni particles and reducing their availability for CH4 chemisorption; (3) Ni sintering was limited for these catalysts but an increase of the mean particle size was nevertheless observed, leading to a decrease in the nickel dispersion.
4. Conclusion
In this paper, we have studied the influence of the carbon activation process on the performances of Ni/AC catalysts for direct methane decomposition. Both catalysts, CAGOP–Ni and CAGOC–Ni, showed good catalytic activities that strongly depend on the amount of nickel loaded and on the reaction temperature. The direct reaction on the AC support has a significant influence on the reactivity and on the deactivation rate of the Ni/AC catalysts. Kinetic study was also performed revealing average reaction orders between 0.60 and 0.75. Activation energies ranged between 120 kJ/mol and 140 kJ/mol, and these values exceeded the values published for Ni-supported catalysts. This shows the major importance of the activity of the carbon surface.
Both fresh and used catalysts were characterized. As observed for similar catalysts, carbon deposit is responsible for carbon surface deactivation by micropore plugging and also for nickel particle deactivation by formation of carbon nano-onions. The behaviour of Ni on chemically AC was different from the one of physically activated one: the formation of nickel phosphide led to a lower activity compared to metallic Ni. Moreover, this AC support exhibits a lower stability with reaction time compared to the physically activated one. This behaviour could be related to the carbon support itself.
Symbol list
| FH 2,out | Outlet molar flow of hydrogen (mol/min) |
| FCH 4,in | Inlet molar flow of methane (mol/min) |
| K | Deactivation ratio between initial reaction rate and its value after 1 h |
| mcat | Catalyst mass in the bed (g) |
| rCH 4 | Specific reaction rate per gram of catalyst (mmol∕g⋅min) |
| XCH 4 | Molar fractional conversion. |
Conflicts of interest
Authors have no conflict of interest to declare.
Acknowledgments
Special appreciation to the University of Gabes-Ministry of Higher Education and Scientific Research of Tunisia for the financial support.
Vous devez vous connecter pour continuer.
S'authentifier