



 CC-BY 4.0
CC-BY 4.0
1. Introduction
Drug resistance is a constantly growing issue that poses a major challenge for the development of new drugs. In the context of antibiotic discovery, the situation is alarming, with antimicrobial resistant bacteria emerging and spreading all around the world [1]. Our ability to treat common infectious diseases is now compromised as some infections are already impossible to treat with the existing therapeutic repertoire. Antibiotic resistance not only increases mortality but also has a severe impact on medical expenses and hospitalization time.
The World Health Organization (WHO) published in 2017 a list of prioritization of bacteria to guide research and development of novel antibiotics [2]. In the latest WHO report dated December 9, 2022 [3], Dr. Tedros Adhanom Ghebreyesus, WHO Director-General, stated that “Antimicrobial resistance undermines modern medicine and puts millions of lives at risk”. Given the severe menace caused by antibiotic-resistant bacteria, it is more than urgent to discover new antibacterial agents with new modes of action. In this context, the methylerythritol phosphate pathway (MEP, Scheme 1), responsible for the biosynthesis of the universal precursors of terpenoids in most bacteria, has emerged as an attractive target for drug development [4, 5, 6, 7].
The MEP pathway.
Terpenoids, also known as isoprenoids, represent the most diverse family of natural products, with over 55,000 known compounds. They are present in all living organisms and are involved in many important biological processes such as electron transport, cell wall biosynthesis, and protein prenylation [8, 9]. Terpenoids are biosynthesized by the addition of one or more molecules of isopentenyl diphosphate (IPP, 1) to its isomer dimethylallyl diphosphate (DMAPP, 2, Scheme 1) [8]. Most pathogenic bacteria, including almost all that were prioritized by the WHO [2], use the MEP pathway (Scheme 1) for the production of IPP and DMAPP, whereas the biosynthesis of these building blocks relies exclusively on the mevalonate pathway in humans and animals [10]. This metabolic difference makes enzymes of the MEP pathway interesting targets for new antibacterial drug development that are expected to have no or reduced side effects in humans. Despite the hope sparked by the discovery of the MEP pathway, only fosmidomycin, an inhibitor of 1-deoxyxylulose-5-phosphate reductoisomerase (DXR), the second enzyme of the MEP pathway (Scheme 1), reached clinical trials as an antimalarial agent (P. falciparum is dependent on the MEP pathway) in combination with clindamycin and piperaquine [11]. This finding validates the MEP pathway as an innovative target for the development of new drugs [12]. However, new molecules need to enter the therapeutic pipeline to combat deadly bacterial infections.
Here, we focus our efforts on discovering new inhibitors of IspH, also called LytB, the last enzyme of the MEP pathway.
IspH contains an oxygen-sensitive [4Fe–4S]2+ center that is essential for catalysis and converts (E)-4-hydroxy-3-methylbut-2-enyl diphosphate (HMBPP, 3) into a mixture of IPP and DMAPP (Scheme 2). Mössbauer spectroscopy [13, 14] and Nuclear Resonance Vibrational Spectroscopy studies highlighted that the [4Fe–4S]2+ cluster of substrate-free IspH is particular, as one of its four iron sites is an Fe(II) atom in an octahedral coordination geometry, linked to three inorganic sulfur atoms of the iron–sulfur cluster and three water molecules (Scheme 2) [15]. This unusual Fe(II) coordination with three labile ligands is at the origin of the instability of the [4Fe–4S]2+ cluster of IspH in the presence of oxygen. Indeed, the oxidation of this Fe(II) may trigger the decomposition of the prosthetic group. Consequently, IspH is only stable under anaerobic conditions, making this enzyme difficult to study and hence an underexplored target. No crystal structure of substrate-free IspH in its [4Fe–4S]2+ form has been reported as the apical Fe(II) might dissociate during the crystallization process. The first X-ray structure of IspH described with an intact [4Fe–4S] cluster was obtained for the E. coli homolog in complex with HMBPP (Scheme 2) [16]. Since then, several other IspH structures harboring the [4Fe–4S] center in complex with ligands have been published (for a review see [17]).
IspH-catalyzed reaction and structures of potent inhibitors.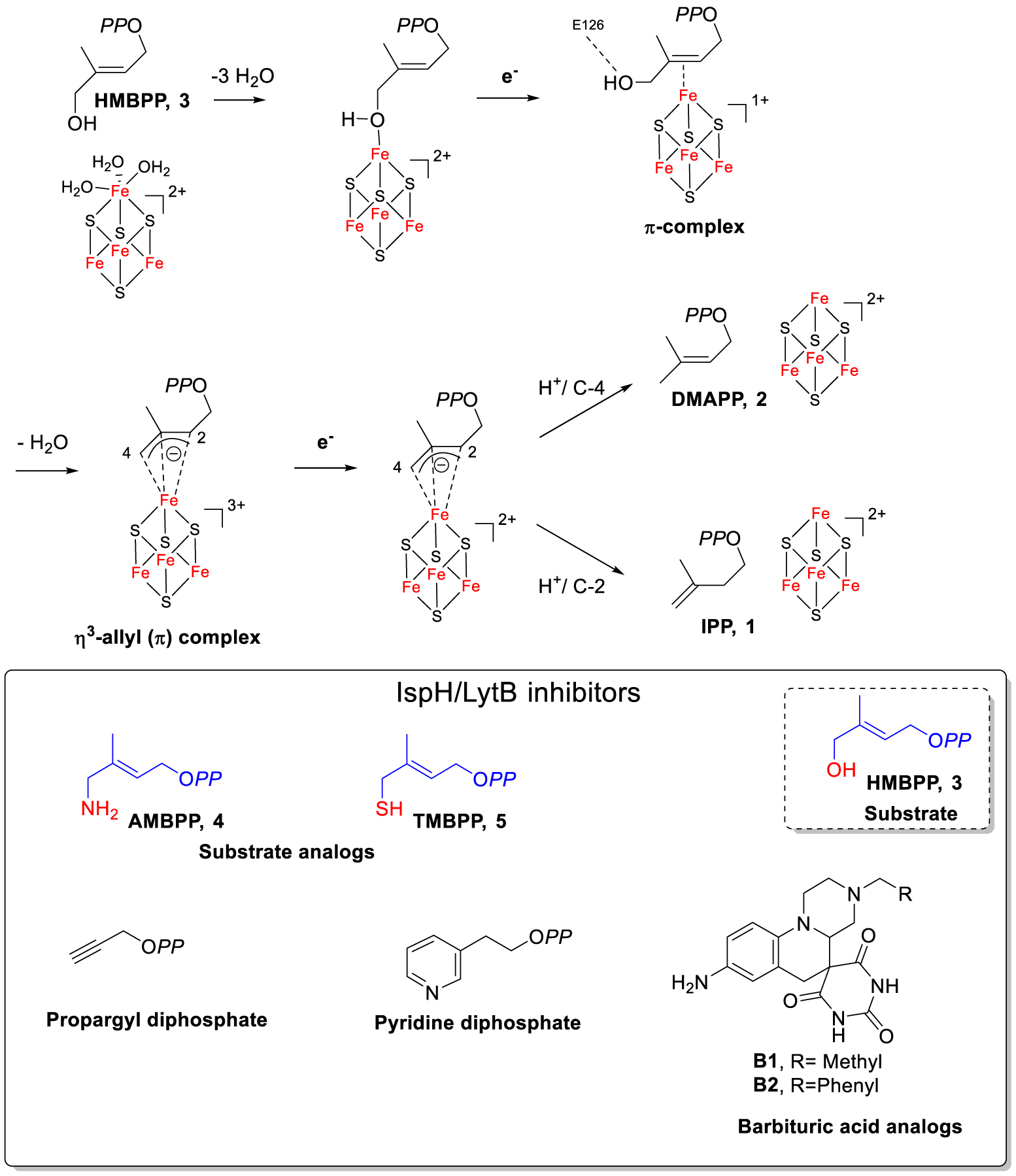
The IspH mechanism is peculiar and involves bioinorganic and bioorganometallic intermediates (for reviews see [17, 18, 19]). It formally involves removal of the hydroxyl group, transfer of two electrons from the [4Fe–4S] cluster, and protonation of an intermediate allylic anion (Scheme 2). The binding of the OH group of HMBPP to the unique fourth iron site of the [4Fe–4S]2+ cluster, leading to the change in coordination geometry of this iron from octahedral to tetrahedral upon binding of the substrate (Scheme 2), was shown using Mössbauer spectroscopy [13] and is illustrated in the X-ray structure of the E. coli IspH–HMBPP complex [16]. After reduction of the first bioinorganic complex (Scheme 2), the OH group of the substrate undergoes a rotation to interact with E126, leading to a π-complex [20, 21, 22, 23]. EPR/ENDOR investigations led to the characterization of a η3-allyl (π) complex that forms after water elimination [24, 25]. Further reduction of the paramagnetic η3-allyl (π) complex followed by protonation at the si face of C-2 yields IPP, while protonation at C-4 yields DMAPP [26].
We and others further exploited the acquired knowledge of the IspH mechanism to design inhibitors [17]. In this context, we have already reported two molecular tools that were HMBPP analogs (Scheme 2) in which the OH group of HMBPP was replaced by an amino (AMBPP, 4) or a thiol group (TMBPP, 5). We had expected AMBPP and TMBPP to tightly bind to the IspH [4Fe–4S] cluster but not being capable of undergoing the elimination step. Enzymatic studies have revealed that these molecules are very potent inhibitors of IspH with Ki values in the nanomolar range: TMBPP is a tight-binding inhibitor (IC50 = 210 nM, Ki = 20 nM, E. coli IspH) and AMBPP is a slow-binding inhibitor (IC50 = 150 nM, Ki = 54 nM, E. coli IspH) [27]. These molecules remain the best IspH inhibitors known to date [17]. The mode of binding of these inhibitors to the apical iron of IspH [4Fe–4S] via the thiol or the amino function has been further confirmed [28, 29, 30]. In addition to these two inhibitors, two other potent inhibitors of A. aeolicus IspH were reported (Scheme 2): a propargyl diphosphate (IC50 = 6.7 μM) and a pyridine diphosphate (IC50 = 9.1 μM) [18]. However, all these inhibitors are diphosphate derivatives and therefore have substantial liabilities with respect to poor transport across bacterial membranes and inactivation upon hydrolysis by secreted phosphatases. Barbituric acid analogs, very different in structure compared to the substrate, were recently developed. They were found to be less potent IspH inhibitors (B1: IC50 = 22 μM, P. aeruginosa IspH; B2: IC50 = 23 μM, E. coli IspH, Scheme 2) [31].
All these results comforted us in our approach to optimize these substrate-based inhibitors. In this context, we report here the synthesis of analogs of AMBPP 4, in which the diphosphate moiety was replaced by simple mimics, and the results of their biological evaluation on IspH.
2. Results and discussion
When preparing molecules harboring a diphosphate, this functional group is usually introduced at the end of the synthesis as its presence leads to a molecule almost insoluble in most common organic solvents, which limits further chemical transformations [27, 28, 29, 30, 31, 32, 33, 34]. Moreover, the purification of diphosphorylated molecules is often tedious. Diphosphate entities are also prone to hydrolysis catalyzed by phosphatases excreted by bacteria, which would result in the inactivation of the diphosphate-containing inhibitors.
To avoid these issues, we investigated the replacement of the diphosphate moiety of the AMBPP inhibitor with more stable mimics. In this context, sulfonate, phosphonate or phosphinophosphonate were chosen. Sulfonate is used as isostere of phosphate as it has a tetrahedral shape similar to that of the phosphate group but is more acidic. The methylene phosphonate moiety is an isostere of phosphate with its phosphorus–carbon bond more stable towards hydrolysis compared to the phosphorus–oxygen bond of phosphate. These two moieties are shorter than diphosphate [35]. Finally, the phosphinophosphonate group is an isostere of the diphosphate group but less sensitive to hydrolysis. The structures of the corresponding AMBPP analogs that we investigated are displayed in Figure 1.
AMBPP and its phosphonate, sulfonate, and phosphinophosphonate analogs.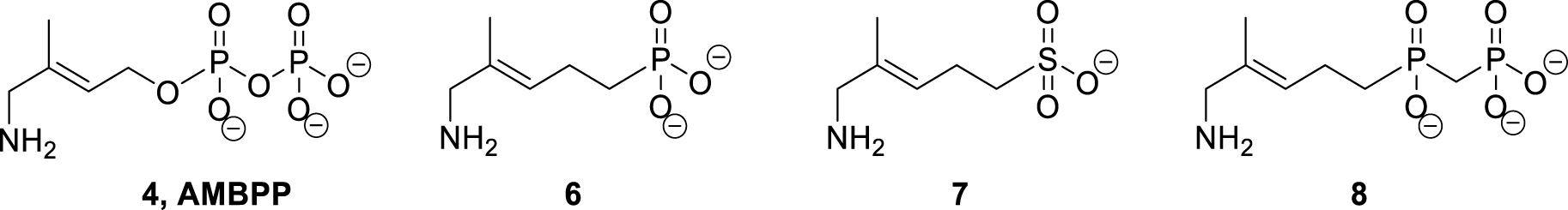
2.1. Preliminary docking experiments
Preliminary in silico docking and scoring experiments were performed using phosphonate 6, sulfonate 7, and phosphinophosphonate 8. Due to the difference in pKa values of these different entities and the fact that the activity of IspH is determined at pH = 8, docking experiments were performed at pH = 7 and at pH = 8.
Experiments were carried out starting from the X-ray structure of IspH in complex with AMBPP (PDB: 3ZGL) [30]. Interestingly, the docking scores of the sulfonate analog 7 (D.S. = −6.93; −6.92, Figure 2) are slightly better than the docking scores of the parent molecule AMBPP (D.S. = −6.75; −5.33, Figure 2). Replacement of the diphosphate by a simple phosphonate or a phosphinophosphonate leads to still acceptable docking scores (Figure 2). The corresponding docking poses revealed that compounds 6, 7, and 8 docked in the active site and placed their sulfonate, phosphonate or phosphinophosphonate group in the diphosphate binding pocket of AMBPP (Figure 2). The amino groups in these AMBPP analogs were also found close to the apical iron.
Docking experiments were performed using the X-ray structure of the E. coli IspH:AMBPP complex (PDB: 3ZGL). Upper panel: structural formula and docking score (D.S) at pH = 7 (left) and pH = 8 (right). Lower panel: docking poses of the compound in the IspH active site. Docked molecules are depicted with green carbon atoms. Docked AMBPP is superposed onto its corresponding crystallographic structure (gray carbon atoms). Iron and sulfur atoms are represented in orange and yellow, respectively.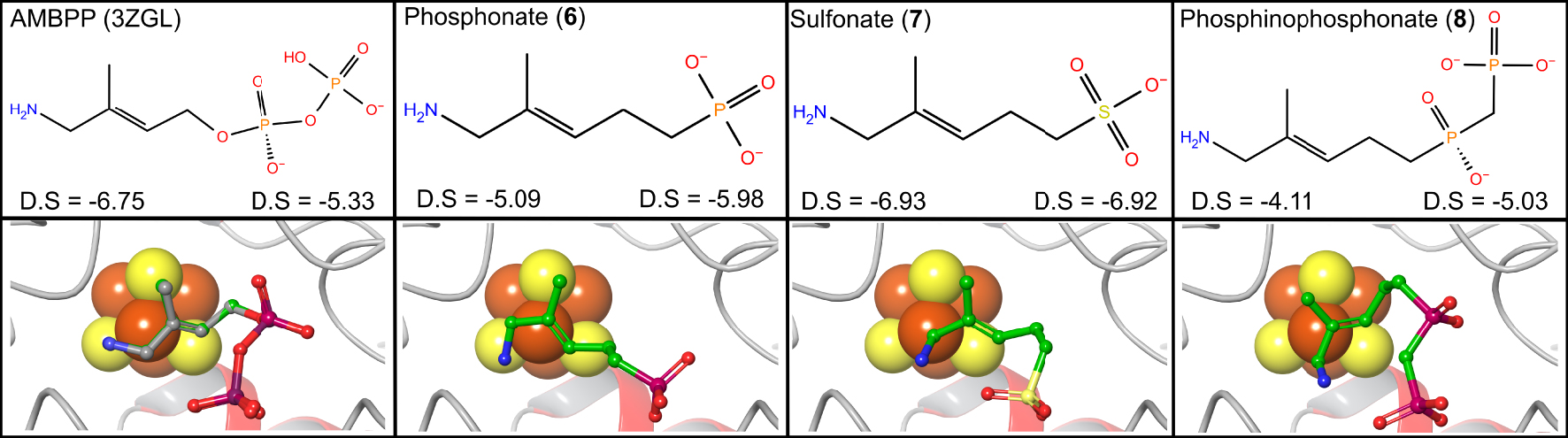
Encouraged by these indicators, compounds 6, 7, and 8 were prepared.
2.2. Chemistry
Target compounds were synthesized starting from dimethylallyl bromide 9. The syntheses of 6 and 8 are outlined in Scheme 3.
Synthesis of phosphonate 6 and phosphinophosphonate 8 analogs.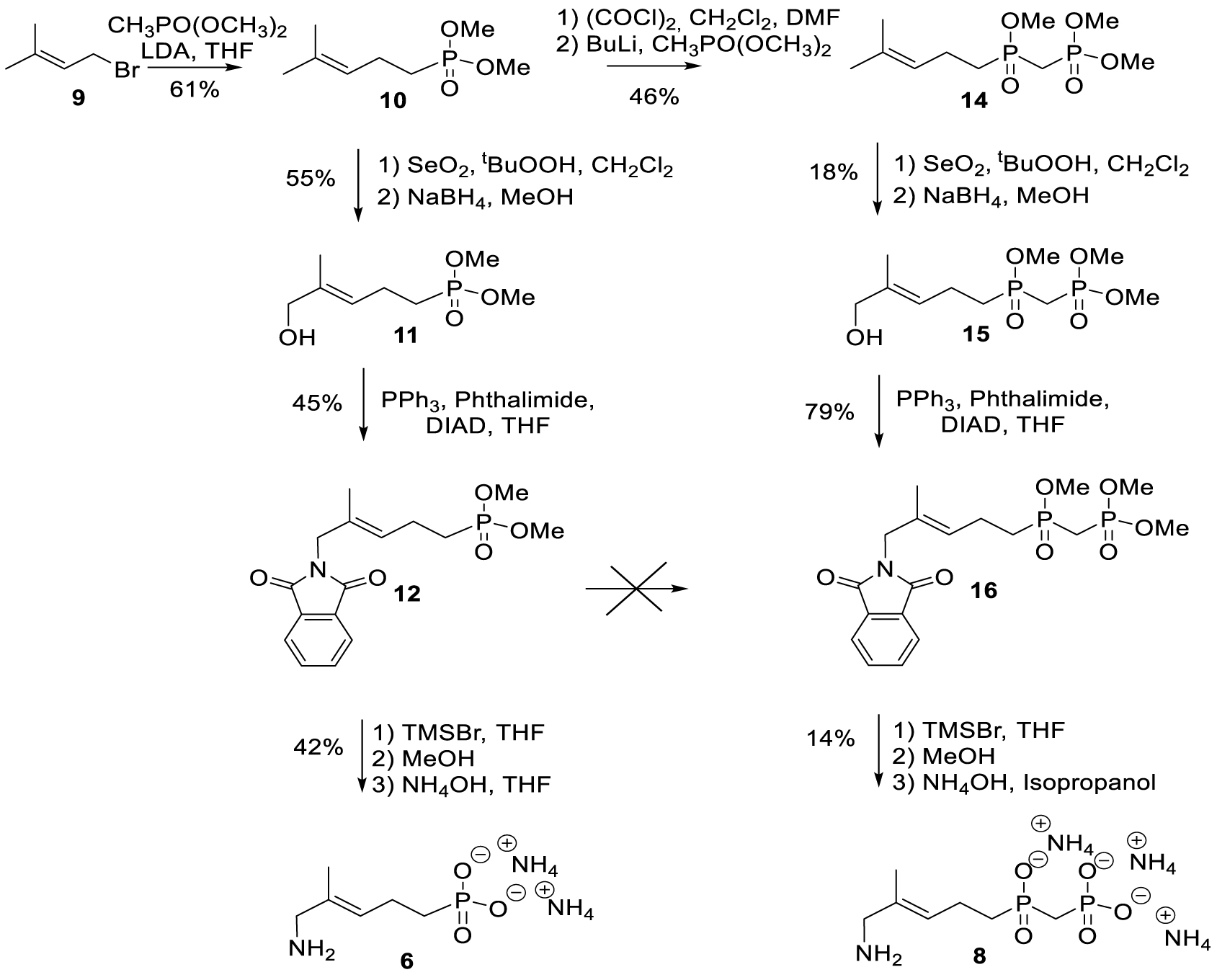
2.2.1. Synthesis of (E)-(5-amino-4-methylpent-3-en-1-yl) phosphonate 6
Dimethylphosphonate 10 was synthesized by nucleophilic substitution of the bromine atom in dimethylallyl bromide 9 with deprotonated dimethyl methylphosphonate following the procedure reported by Wiemer [36]. Phosphonate 10 was selectively oxidized with selenium dioxide to yield the corresponding E-configurated aldehyde, which was subsequently reduced by NaBH4 to alcohol 11 [37, 38]. Displacement of the hydroxyl group in 11 with phthalimide under Mitsunobu conditions yielded 12. Methyl phosphoesters and phthalimide were deprotected by treatment with bromotrimethylsilane and aqueous ammonia, respectively, to provide the corresponding amine 6.
2.2.2. Synthesis of (E)-(5-amino-4-methylpent-3-en-1-yl) phosphinophosphonate 8
Phosphinophosphonate 16 could not be obtained by sequential activation of phosphonate 12 by oxalyl chloride to the corresponding phosphonic acid chloride and treatment of the latter by deprotonated dimethyl methylphosphonate. As an alternative, we used the procedure published by Wiemer and coworkers for the formation of 14 [39]. Subsequent allylic oxidation with selenium dioxide yielded 15. Phosphinophosphonate 8 was then obtained following the same strategy used for phosphonate 6.
The synthesis of sulfonate 7 is outlined in Scheme 4.
Synthesis of sulfonate analog 7.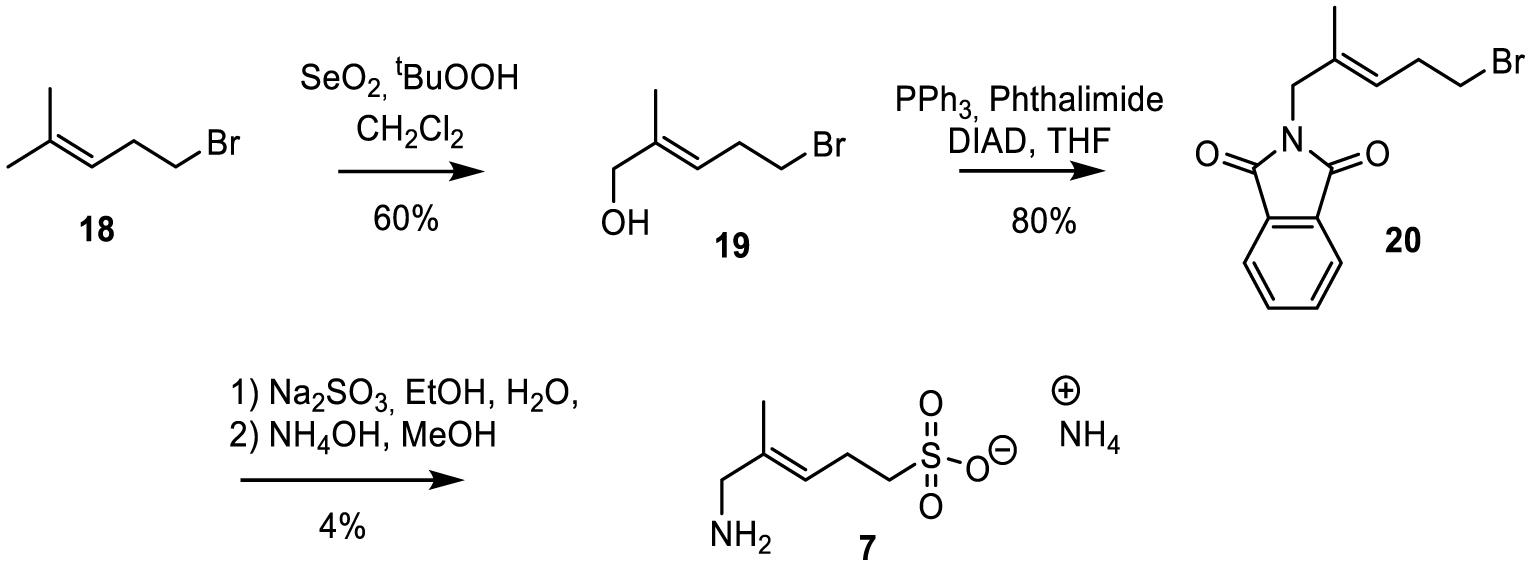
2.2.3. Synthesis of (E)-5-amino-4-methylpent-3-ene-1-sulfonate 7
The sulfonate analog 7 was prepared in three steps. Commercially available alkyl bromide 18 was selectively oxidized to alcohol 19, as reported by Gaich and Mulzer [40]. A Mitsunobu reaction using phthalimide as the nucleophile allowed the synthesis of compound 20. Bromide was substituted by treatment with sodium sulfite, and hydrolysis of the phthalimide by ammonia yielded 7.
Biological experiments were further carried out to test the ability of 6, 7, 8 to act as IspH inhibitors.
2.3. Biological evaluation
E. coli IspH contains an oxygen-sensitive [4Fe–4S]2+ cluster that is essential for catalysis and therefore needs to be handled in a glove box under a strictly inert (N2) atmosphere. IspH converts HMBPP into a mixture of IPP and DMAPP in the presence of an external reduction system. In E. coli, the natural flavodoxin (FldA)/flavodoxin reductase (FpR1)/NADPH system plays this role [41, 27]. Enzyme activity was therefore determined by monitoring NADPH consumption. The progress curve (Figure 3, navy blue) showed a sharp drop in the NADPH concentration in the first 7 min that was due to IspH catalysis under multiple turnover conditions. The resulting E. coli IspH activity was 990 nmol⋅min−1⋅mg−1, in agreement with previous reports [13, 23, 27]. It should be noticed that the low-slope region observed after 7 min is due to spontaneous NADPH degradation at pH = 8.
IspH enzymatic assays. Decrease in the absorbance of NADPH at 340 nm in the presence or absence of 6, 7, 8, or AMBPP. Conditions: NADPH (2.2 mM), FldA (30 μM), FpR1 (17 μM), IspH (0.5 μM) in 50 mM Tris HCl buffer pH = 8 at 37 °C; HMBPP alone (dark blue) or in the presence of 1 mM of 6 (yellow), 7 (green), 8 (blue), or 25 μM AMBPP 4 (orange) that were preincubated with IspH for 15 min at 37 °C before initiating the reaction by addition of HMBPP (150 μM). Samples were prepared in a glove box, and additions were performed using a gastight syringe. A control sample (light gray) was prepared under the same conditions but replacing HMBPP by buffer.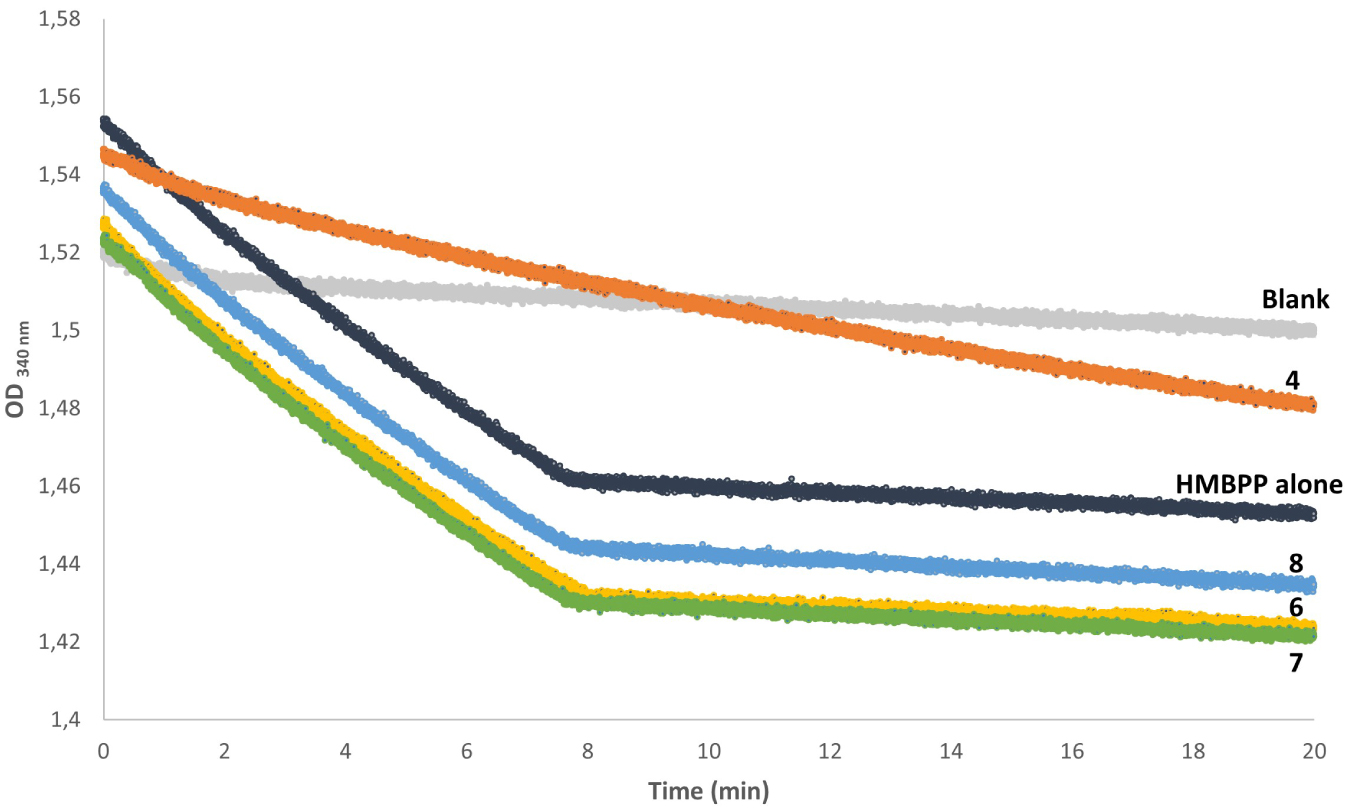
We previously reported that AMBPP 4 is a slow-binding inhibitor of IspH under the conditions used in this assay and hypothesized that the slow-binding step might be due to the formation of the nonprotonated amine required for binding to the apical iron of the [4Fe–4S]2+ cluster [27]. As 6, 7, and 8 also contain the amine function, this slow-binding behavior would also be expected for these compounds. As a consequence, we tested the ability of 6, 7, and 8 to promote IspH inhibition by first preincubating IspH with each of these compounds for 15 min, in order to favor the formation of the enzyme–inhibitor complex, and then initiate the IspH-catalyzed reaction by the addition of the HMBPP substrate. Progress curves recorded using 6, 7, or 8 at a concentration of 1 mM displayed the same initial slope as the curve recorded for IspH with HMBPP alone, indicating the same steady-state rates (Figure 3, yellow, green, blue). In contrast, the progress curve recorded under the same conditions for AMBPP at a concentration as low as 25 μM showed a drastic decrease in the IspH reaction rate, indicative of an E. coli IspH activity of 263 nmol⋅min−1⋅mg−1 that corresponds to 73% enzyme inhibition (Figure 3, orange). Together, these results reveal that replacement of the diphosphate in AMBPP with a phosphonate, a sulfonate, or a phosphinophosphonate, compromise the inhibition potential of the resulting derivatives 6, 7, and 8 towards IspH.
3. Conclusion
Three novel analogs of AMBPP, in which the diphosphate group was replaced by a sulfonate or a methylene phosphonate or a phosphinophosphonate, were synthesized and characterized by NMR spectroscopy and mass spectrometry. In contrast to the parent molecule AMBPP, which is one of the best two inhibitors known to date for E. coli IspH, a metalloenzyme containing an oxygen sensitive [4Fe–4S] cluster involved in the MEP pathway, these new molecules did not affect IspH activity. These results illustrate the essentiality of the diphosphate group of AMBPP, so far. The lack of inhibition potential of the sulfonate or methylene phosphonate analogs is most probably due to the size of these phosphate mimics that are shorter than diphosphate and might not completely fill the diphosphate binding pocket of IspH. In contrast, phosphinophosphonate has the same size as AMBPP and could undergo the same interactions as AMBPP with the surrounding amino acids of IspH. However, phosphinophosphonates are known to be less acidic than diphosphates. As a consequence, the phosphinophosphonate might retain a proton that might weaken some interactions within the active site.
Based on the knowledge gained from this study, new inhibitors derived from AMBPP or other promising IspH inhibitors need to be elaborated. Such optimization could consist in the use of other diphosphate isosteres such as difluoromethylphosphonates, difluoromethanediphosphonates or via structure-based fragment selection to find new scaffolds binding to the diphosphate pocket of IspH that do not rely on phosphate chemistry.
4. Experimental section
4.1. Molecular docking
In silico docking experiments were carried out using the Schrödinger suite 2020-4 (Schrödinger LLC, New York, NY, USA). The X-ray structure of IspH in complex with AMBPP (PDB: 3ZGL) was used for the studies. The protein structure was processed as previously described [30]. To generate the docking grid, we used AMBPP as the reference ligand and the protein model without water molecules. The binding region was defined by a square box centered on the inhibitor. Sizes that largely exceeded the volume of the binding site were used for both the enclosing (10 Å × 10 Å × 10 Å) and bounding box (20 Å × 20 Å × 20 Å). LigPrep was used for energy minimization, to generate the 3D structures of the compounds, and to produce the tautomers and the ionization states at pH = 7 and pH = 8. The docking study was performed using Glide’s extra precision mode [42, 43]. No constraints (such as hydrogen bond or atom position) were applied to guide the binding. Results of the in silico docking experiments were sorted according to the Glide docking score.
4.2. Syntheses
All reactions in nonaqueous solvents were conducted under an argon atmosphere with a magnetic stir bar. All reagents and solvents were purchased from commercial sources and used without further purification. Anhydrous CH2Cl2 and tetrahydrofuran (THF) were purchased (99.85%, water < 50 ppm). All other solvents were of HPLC grade. Reactions were monitored by thin layer chromatography (TLC) with silica gel 60-F254 plates. Flash column chromatography was performed using silica gel (0.04–0.063 mm, 230–400 mesh) under pressure. Yields refer to chromatographically and spectroscopically pure compounds.
NMR spectra were recorded on a 300- or 500-MHz spectrometer. All NMR spectra were measured in CDCl3 or D2O solutions and referenced, respectively, to the residual CHCl3 signal (1H, δ = 7.26 ppm; 13C, δ = 77.16 ppm) or H2O (δ = 4.79 ppm). For 31P NMR spectroscopy, 85% phosphoric acid in D2O was used as external reference (δ = −0.85 ppm). Chemical shifts and coupling constants are reported in ppm and Hz, respectively. High-resolution mass spectra were obtained using ESI-TOF.
4.2.1. Synthesis of (E)-(5-amino-4-methylpent-3-en-1-yl) phosphonate 6
∙ Synthesis of dimethyl (4-methylpent-3-en-1-yl) phosphonate 10
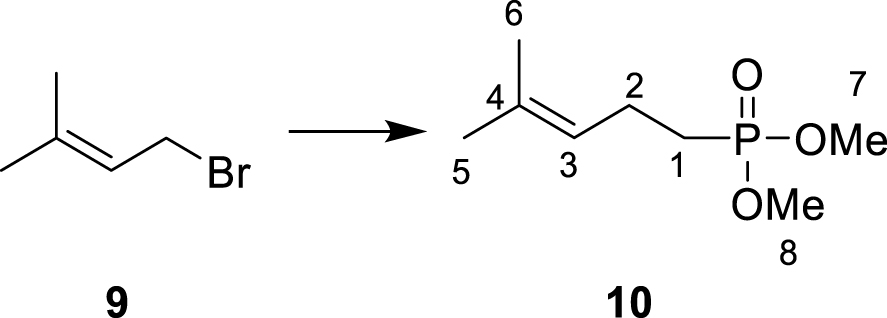
Dimethyl methylphosphonate (2.1 mL, 19.4 mmol, 1 eq.) was added dropwise at −78 °C to a stirred solution of LDA (2 M in THF, 10 mL, 20 mmol, 1 eq.) in dry THF (80 mL). The reaction mixture was stirred at −78 °C for 15 min before 1-bromo-3-methylbut-2-ene 9 (2.3 mL, 19.9 mmol, 1 eq.) was added dropwise. The reaction mixture was stirred at −78 °C for 30 min and then left to stand at 20 °C overnight. A saturated aqueous solution of NH4Cl was then added, and the different layers were separated. The aqueous layer was extracted using diethyl ether. The combined organic layers were dried over Na2SO4, filtered, and the solvent was removed under reduced pressure. The crude product was purified by column chromatography on silica gel (ethyl acetate/petroleum ether, 8:2) yielding dimethyl (4-methylpent-3-en-1-yl) phosphonate 10 as an oil (2.26 g, 11.8 mmol, 61%, Rf (ethyl acetate/cyclohexane, 9:1) = 0.25). 1H NMR (500 MHz, CDCl3): δ(ppm) = 1.61 (3H, s, H-6), 1.68 (3H, s, H-5), 1.70–1.82 (2H, m, H-1), 2.21–2.33 (2H, m, H-2), 3.73 (6H, d, J = 12.0 Hz, H-7 + H-8), 5.10 (1H, tq, J = 7.2 Hz, J = 1.5 Hz, H-3). 13C NMR (125 MHz, CDCl3): δ(ppm) = 17.8 (C-6), 21.1 (d, J = 5.0 Hz, C-2), 25.1 (d, J = 137.0 Hz, C-1), 25.8 (C-5), 52.4 (d, J = 6.6 Hz, C-7 + C-8), 123.1 (d, J = 17.5 Hz, C-3), 133.1 (d, J = 2.5 Hz, C-4). 31P NMR (121 MHz, CDCl3): δ(ppm) = 34.5.
∙ Synthesis of dimethyl (E)-(5-hydroxy-4- methylpent-3-en-1-yl) phosphonate 11
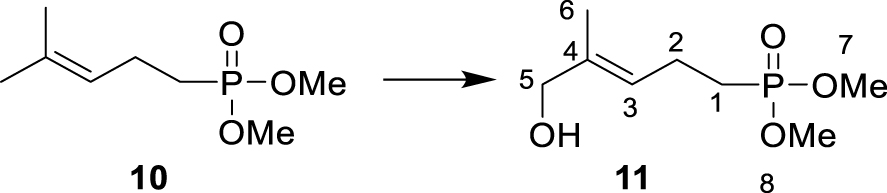
Dimethyl (4-methylpent-3-en-1-yl) phosphonate 10 (500 mg, 2.60 mmol, 1 eq.), SeO2 (220 mg, 1.95 mmol, 0.75 eq.) and tert-butyl hydroperoxide (70% in water, 1.4 mL, 10.4 mmol, 4 eq.) were dissolved in CH2Cl2 (15 mL). The reaction mixture was stirred at 20 °C for 16 h before the reaction was quenched by the addition of a saturated aqueous solution of NaCl. The different layers were separated, and the aqueous layer was extracted with CH2Cl2. The combined organic layers were washed with an aqueous solution of Na2S2O3, dried over Na2SO4, filtered, and the solvent was removed under reduced pressure. The resulting crude product was dissolved in methanol (7.5 mL), and NaBH4 (200 mg, 5.2 mmol, 2 eq.) was added portionwise. The reaction mixture was stirred at 20 °C for 2 h before being quenched by the addition of a saturated aqueous solution of NH4Cl. The resulting mixture was extracted with diethyl ether, and the combined organic layers were dried over Na2SO4, filtered, and the solvent was removed under reduced pressure. The crude product was purified by column chromatography on silica gel (MeOH/DCM, 4:96) yielding dimethyl (E)-(5-hydroxy-4-methylpent-3-en-1-yl) phosphonate 11 as an oil (298 mg, 1.43 mmol, 55%, Rf (ethyl acetate/cyclohexane, 9:1) = 0.11). 1H NMR (300 MHz, CDCl3): δ(ppm) = 1.68 (3H, s, H-6), 1.77–1.84 (2H, m, H-1), 2.31–2.38 (2H, m, H-2), 3.74 (6H, d, J = 10.0 Hz, H-7 + H-8), 4.00 (2H, s, H-5), 5.42 (1H, tq, J = 7.0 Hz, J = 1.5 Hz, H-3). 13C NMR (125 MHz, CDCl3): δ(ppm) = 13.8 (C-6), 20.8 (d, J = 5.0 Hz, C-2), 24.8 (s, J = 139.0 Hz, C-1), 52.5 (d, J = 6.28 Hz, C-7 + C-8), 68.6 (C-5), 124.2 (d, J = 16.25 Hz, C-3), 136.4 (d, J = 1.25 Hz, C-4). 31P NMR (121 MHz, CDCl3): δ(ppm) = 34.29.
∙ Synthesis of dimethyl (E)-(5-(1,3- dioxoisoindolin-2-yl)-4-methylpent-3-en-1-yl) phosphonate 12

Dimethyl (E)-(5-hydroxy-4-methylpent-3-en-1-yl) phosphonate 11 (398 mg, 1.9 mmol, 1.1 eq.), PPh3 (515 mg, 2.0 mmol, 1.1 eq.) and phthalimide (250 mg, 1.7 mmol, 1 eq.) were dissolved in dry THF (12 mL). DIAD (430 μL, 2.0 mmol, 1.1 eq.) was added dropwise at 0 °C, and the reaction mixture was stirred at 0 °C for 30 min and then left to stand at 20 °C overnight. The reaction was quenched by the addition of MeOH (0.5 mL), and the solvent was evaporated under vacuum. The crude product was purified by column chromatography on silica gel (ethyl acetate) yielding 12 as a white solid (260 mg, 0.77 mmol, 45%, Rf (MeOH/DCM, 5:95) = 0.54). 1H NMR (300 MHz, CDCl3): δ(ppm) = 1.66 (3H, s, H-6), 1.70–1.81 (2H, m, H-1), 2.24–2.36 (2H, m, H-2), 3.70 (6H, d J = 9.0 Hz, H-7 + H-8), 4.18 (2H, s, H-5), 5.31 (1H, tq, J = 8.7 Hz, J = 1.5 Hz, H-3), 7.70–7.74 (2H, m, H-11 or H-12), 7.81–7.86 (2H, m, H-11 or H-12). 13C NMR (125 MHz, CDCl3): δ(ppm) = 14.8 (C-6), 20.9 (d, J = 4.5 Hz, C-2), 24.5 (d, J = 139.0 Hz, C-1), 44.8 (C-5), 52.4 (d, J = 6.5 Hz, C-7 + C-8), 123.5 (C-10), 125.7 (d, J = 16.9 Hz, C-3), 130.9 (d, J = 1.6 Hz, C-4), 132.1 (C-11 or C-12), 134.2 (C-11 or C-12), 168.3 (C-9). 31P NMR (121 MHz, CDCl3): δ(ppm) = 33.94.
∙ Synthesis of (E)-(5-amino-4-methylpent-3-en-1-yl) phosphonate 6

Me3SiBr (180 μL, 1.68 mmol, 9 eq.) was added dropwise to a stirred solution of dimethyl (E)-(5-(1,3-dioxoisoindolin-2-yl)-4-methylpent-3-en-1-yl) phosphonate 12 (60 mg, 0.178 mmol, 1 eq.) in dry CH2Cl2 (1.5 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 30 min and at 20 °C for 1 h. Methanol (1 mL) was added, and the reaction was further stirred for 1 h. The solvents were then evaporated under reduced pressure, and the resulting oil was dissolved in THF (4 mL). NH4OH (1 mL) was added, and the reaction mixture was stirred overnight. Solvents were evaporated under reduced pressure, and the resulting mixture was purified by column chromatography on silica gel (Isopropanol/H2O/NH4OH, 7:1:2 to 5:3:2) yielding 6 as a white solid, which was dissolved in water, lyophilized, and further dried under high vacuum (13 mg, 0.074 mmol, 42%, Rf (Isopropanol/H2O/NH4OH, 6:2:2) = 0.20). 1H NMR (300 MHz, D2O): δ(ppm) = 1.61–1.72 (2H, m, H-1), 1.75 (3H, s, H-6), 2.26–2.38 (2H, m, H-2), 3.53 (2H, s, H-5), 5.62 (1H, tq, J = 7.5 Hz, J = 1.5 Hz, H-3). 13C NMR (125 MHz, D2O): δ(ppm) = 13.5 (C-6), 21.6 (d, J = 3.75 Hz, C-2), 27.2 (d, J = 132.5 Hz, C-1), 46.3 (C-5), 127.5 (C-4), 130.9 (d, J = 15.0 Hz, C-3). 31P NMR (121 MHz, D2O): δ(ppm) = 25.32. High-resolution MS (ES-) m/z: [M–H]- (C6H13NO3P) calculated 178.0638, found 178.0650.
4.2.2. Synthesis of (E)-(((5-amino-4-methylpent-3-en-1-yl) oxidophosphoryl) methyl) phosphonate 8
∙ Synthesis of dimethyl ((methoxy(4-methylpent-3-en-1-yl) phosphoryl) methyl) phosphonate 14

Oxalyl chloride (3 mL, 35 mmol, 3 eq.) was added dropwise to a stirred solution of dimethyl (4-methylpent-3-en-1-yl) phosphonate 10 (2.05 g, 11.5 mmol, 1 eq.) and dry DMF (0.10 mL, cat.) in dry CH2Cl2 (60 mL) at 0 °C. The solution was stirred overnight at 20 °C. Solvents were then removed under reduced pressure, yielding a chlorinated compound, which was used without further purification. Dimethyl methylphosphonate (3.5 mL, 33 mmol, 2.9 eq.) was added dropwise to a stirred solution of BuLi (2.5 M in hexane, 12.5 mL, 31 mmol, 2.7 eq.) at −78 °C. The mixture was further stirred for 30 min before a solution of the chlorinated compound in CH2Cl2 (10 mL) was added dropwise. The reaction mixture was allowed to reach 20 °C while being stirred overnight. A saturated aqueous solution of NH4Cl was then added, and the different layers were separated. The aqueous layer was extracted using CH2Cl2. The combined organic layers were dried over Na2SO4, filtered, and the solvent was removed under reduced pressure. The resulting mixture was purified by column chromatography on silica gel (EtOH/PE, 2:8) yielding 14 (1.50 g, 5.28 mmol, 46%, Rf (MeOH/DCM, 5:95) = 0.17) as yellowish oil. 1H NMR (300 MHz, CDCl3): δ(ppm) = 1.63 (3H, s, H-5), 1.68 (3H, s, H-6), 1.92–2.02 (2H, m, H-1), 2.25–2.35 (2H, m, H-8), 2.33–2.46 (2H, m, H-2), 3.76 (3H, d, J = 11.1 Hz, H-7), 3.81 (6H, d, J = 11.4 Hz, H-9 + H-10), 5.13 (1H, tq, J = 7.2 Hz, J = 1.5 Hz, H-3). 13C NMR (125, CDCl3): δ(ppm) = 17.8 (C-6), 20.4 (d, J = 4.12 Hz, C-2), 25.8 (C-5), 26.4 (dd, J = 134.2 Hz, J = 75.5 Hz, C-8), 29.5 (d, J = 96.6 Hz, C-1), 51.6 (d, J = 6.9 Hz, C-7), 53.2 (t, J = 6.7 Hz, C-9 + C-10), 122.9 (d, J = 15.6 Hz, C-3) and 133.4 (d, J = 1.4 Hz, C-4). 31P NMR (121 MHz, CDCl3): δ(ppm) = 22.76 (d, J = 4.4 Hz), 48.34 (d, J = 4.5 Hz).
∙ Synthesis of dimethyl (E)-(((5-hydroxy-4- methylpent-3-en-1-yl) (methoxy)phosphoryl) methyl) phosphonate 15
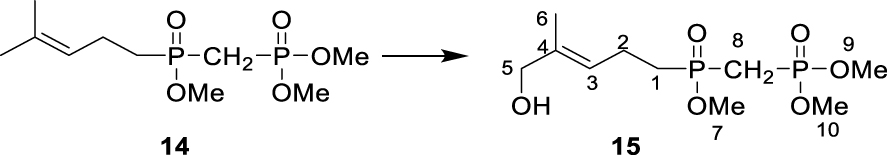
Dimethyl ((methoxy(4-methylpent-3-en-1-yl) phosphoryl) methyl) phosphonate 14 (911 mg, 3.2 mmol, 1 eq), SeO2 (215 mg, 1.9 mmol, 0.6 eq.) and tert-butyl hydroperoxide (70% in water, 2.15 mL, 15.7 mmol, 5 eq.) were dissolved in CH2Cl2 (20 mL). The reaction mixture was stirred at 20 °C for 16 h before being quenched by the addition of a saturated aqueous solution of NaCl. The different layers were separated, and the aqueous layer was extracted with CH2Cl2. The combined organic layers were washed with an aqueous solution of Na2S2O3, dried over Na2SO4, filtered, and the solvent was removed under reduced pressure. The resulting crude product was dissolved in methanol, and NaBH4 (150 mg, 3.9 mmol, 1.2 eq.) was added at 0 °C. The reaction mixture was stirred at room temperature for 2 h before being quenched by the addition of a saturated aqueous solution of NH4Cl (2 mL). The solvents were removed under reduced pressure, and the resulting mixture was purified over column chromatography on silica gel (MeOH/CH2Cl2, 8:92) yielding 15 as an oil (170 mg, 0.56 mmol, 18%, Rf (MeOH/DCM, 8:92) = 0.16). 1H NMR (500 MHz, CDCl3): δ(ppm) = 1.69 (3H, s, H-6), 1.73 (bs, 1H, OH), 2.00–2.06 (2H, m, H-1), 2.35–2.45 (4H, m, H-2 + H-8), 3.77 (3H, d, J = 11.0 Hz, H-7), 3.81 (6H, dd, J = 11.0 Hz, J = 2.0 Hz, H-9 + H-10), 4.00 (2H, s, H-5), 5.45 (1H, tq, J = 7.5 Hz, J = 1.5 Hz, H-3). 13C NMR (125, CDCl3): δ(ppm) = 13.7 (C-6), 19.9 (d, J = 4.2 Hz, C-2), 26.4 (dd, J = 134.5 Hz, J = 75.8 Hz, C-8), 29.0 (d, J = 97.6 Hz, C-1), 51.5 (d, J = 6.7 Hz, C-7), 53.1 (d, J = 6.5 Hz, C-9 + C-10), 68.4 (C-5), 123.8 (d, J = 14.1 Hz, C-3), 136.6 (d, J = 1.0 Hz, C-4). 31P NMR (121 MHz, CDCl3): δ(ppm) = 23.07 (d, J = 4.2 Hz), 48.26 (d, J = 4.0 Hz).
∙ Synthesis of dimethyl (E)-(((5-(1,3- dioxoisoindolin-2-yl)-4-methylpent-3-en-1-yl) (methoxy)phosphoryl) methyl) phosphonate 16
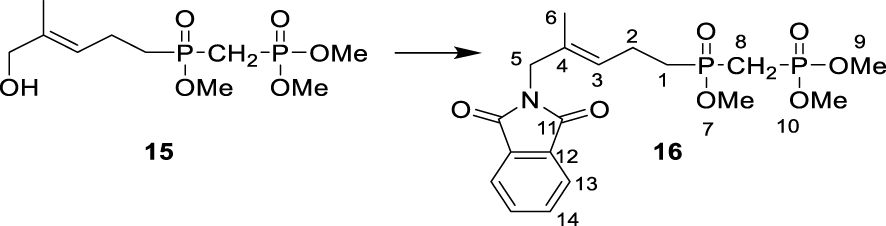
Dimethyl (E)-(((5-hydroxy-4-methylpent-3-en-1-yl) (methoxy)phosphoryl)methyl)phosphonate 15 (170 mg, 0.57 mmol, 1.2 eq.), PPh3 (155 mg, 0.59 mmol, 1.2 eq.) and phthalimide (70 mg, 0.48 mmol, 1 eq.) were dissolved in dry THF. DIAD (130 μL, 0.64 mmol, 1.3 eq.) was added dropwise at 0 °C, and the reaction mixture was stirred at 0 °C for 30 min and left to stand at 20 °C for 2 h. The reaction was quenched by the addition of MeOH (500 μL), and the solvent was removed under reduced pressure. The crude product was purified by column chromatography on silica gel (MeOH/CH2Cl2, 8:92). A second column chromatography on silica gel (ethyl acetate) yielded 16 as a white oil (162 mg, 0.38 mmol, 79%, Rf (ethyl acetate) = 0.43). 1H NMR (500 MHz, CDCl3): δ(ppm) = 1.67 (3H, s, H-6), 1.93–2.00 (2H, m, H-1), 2.30–2.42 (4H, m, H-2 + H-8), 3.73 (3H, d, J = 11 Hz, H-7), 3.78 (6H, d, J = 11 Hz, H-9 + H-10), 4.17 (2H, s, H-5), 5.35 (1H, tq, J = 7.5 Hz, J = 1.5 Hz, H-3), 7.70–7.72 (2H, m, H-13), 7.83–7.84 (2H, m, H-14). 13C NMR (125 MHz, CDCl3): δ(ppm) = 14.8 (C-6), 20.1 (d, J = 16 Hz, C-2), 26.4 (dd, J = 134.1 Hz, J = 76.1 Hz, C-8), 29.0 (d, J = 97.4, C-1), 44.7 (C-5), 51.6 (d, J = 6.3 Hz, C-7), 53.2 (dd, J = 6.5 Hz, J = 3.1 Hz, C-9 + C-10), 123.4 (C-13), 125.5 (d, J = 15.5 Hz, C-3), 131.2 (d, J = 1.4 Hz, C-4), 132.1 (C-14), 134.1 (C-12), 168.3 (C-11). 31P NMR (121 MHz, CDCl3): δ(ppm) = 22.96 (d, J = 4.0 Hz), 47.97 (d, J = 4.0 Hz).
∙ (E)-(((5-amino-4-methylpent-3-en-1-yl) oxidophosphoryl) methyl) phosphonate 8
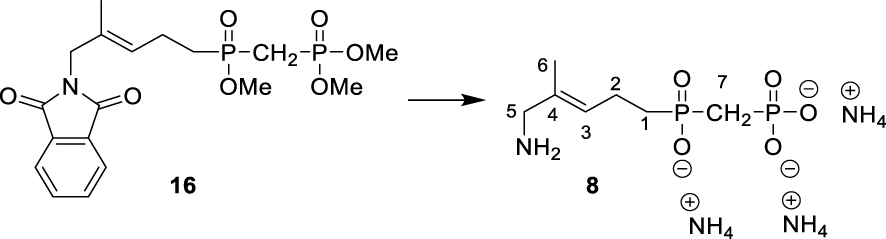
Bromotrimethylsilane (340 μL, 3.2 mmol, 9.4 eq.) was added dropwise to a solution of dimethyl (E)-(((5-(1,3-dioxoisoindolin-2-yl)-4-methylpent-3-en-1-yl) (methoxy)phosphoryl) methyl) phosphonate 16 (146 mg, 0.34 mmol, 1 eq.) in dry CH2Cl2 (3 mL) while being stirred at 0 °C. The reaction mixture was further stirred at 0 °C for 30 min and then at 20 °C for 1 h. Methanol was added, and the mixture was further stirred for 1 h. Solvents were then removed under reduced pressure, and the resulting oil was dissolved in acetone. A mixture of isopropanol, water and ammonia (6/2/2) was added, and the mixture was stirred overnight. After evaporation of the solvents, the crude product was purified by column chromatography on silica gel (IPA/H2O/NH4OH, 6:2:2 to 5:3:2) yielding 8 as a white solid, which was dissolved in water, lyophilized, and further dried under high vacuum yielding 17 (12 mg, 0.046 mmol, 14%, Rf (IPA/H2O/NH4OH, 6:4:2) = 0.25). 1H NMR (300 MHz, D2O): δ(ppm) = 1.69 (3H, s, H-6), 1.70–1.76 (2H, m, H-1), 1.98–2.06 (2H, m, H-7), 2.23–2.30 (2H, m, H-2), 3.46 (2H, s, H-5), 5.54 (1H, t, J = 7.0 Hz, H-3). 13C NMR (125 MHz, D2O): δ(ppm) = 13.6 (C-6), 20.4 (d, J = 3.6 Hz, C-2), 29.9 (d, J = 95.0 Hz, C-1), 30.7 (dd, J = 119.8 Hz, J = 75.9 Hz, C-7), 46.2 (C-5), 127.5 (C-4), 130.8 (d, J = 14.4 Hz, C-3). 31P NMR (121 MHz, D2O): δ(ppm) = 15.40 (d, J = 5.5 Hz), 38.13 (d, J = 5.5 Hz). High-resolution MS (ES-) m/z: [M–H]- (C7H16NO5P2) calculated 256.0509, found 256.0526.
4.2.3. Synthesis of (E)-5-amino-4-methylpent-3-ene-1-sulfonate 7
∙ (E)-5-bromo-2-methylpent-2-en-1-ol 18
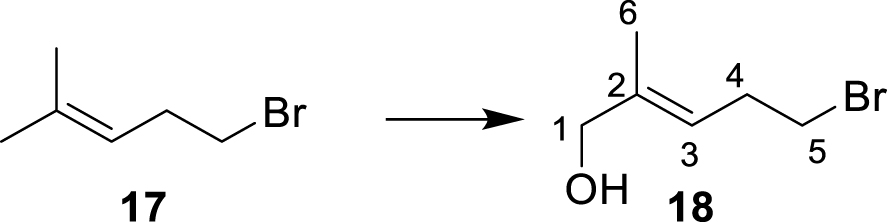
Tert-butyl hydroperoxide (70% in water, 1.7 mL, 12.6 mmol, 2 eq.) was added to a suspension of SeO2 (340 mg, 3.01 mmol, 0.5 eq.) stirred at 0 °C. The reaction mixture was stirred at 0 °C for 5 min and at 20 °C for 30 min. 5-Bromo-2-methylpent-2-ene 17 (820 μL, 6.13 mmol, 1 eq.) was added dropwise at 0 °C. The reaction mixture was stirred overnight while slowly being allowed to warm up to 20 °C. It was then diluted with diethyl ether and washed twice with an aqueous solution of KOH (1M) then brine. The organic layer was dried over Na2SO4, filtered, and the solvent was removed under reduced pressure. The crude product was purified by column chromatography on silica gel (ethyl acetate/petroleum ether, 2:8) yielding 18 (660 mg, 3.69 mmol, 60%). 1H NMR (500 MHz, CDCl3): δ(ppm) = 1.69 (3H, s, H-6), 2.63 (2H, dt, J = 7.0 Hz, 6.5 Hz, H-4), 3.39 (2H, t, J = 7.0 Hz, H-5), 4.03 (2H, s, H-1), 5.44 (1H, m, H-3). 13C NMR (125 MHz, CDCl3): δ(ppm) = 14.0 (C-6), 31.3 (C-4), 32.6 (C-5), 68.5 (C-1), 122.1 (C-3), 138.1 (C-2).
∙ (E)-2-(5-bromo-2-methylpent-2-en-1-yl) isoindoline-1,3-dione 19
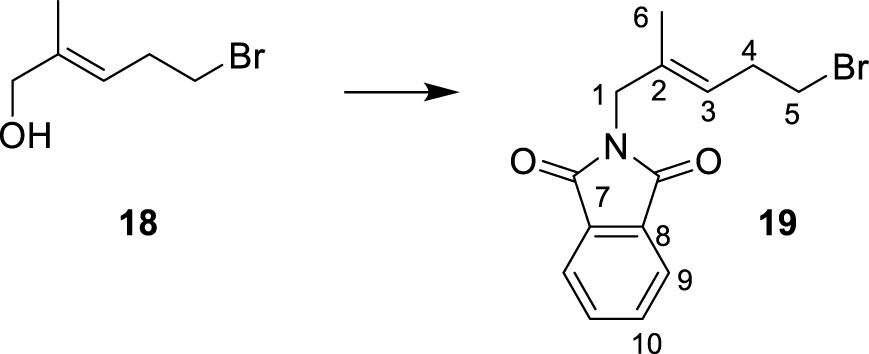
DIAD (400 μL, 2.0 mmol, 1.2 eq.) was added to a solution of phthalimide (300 mg, 2.0 mmol, 1.2 eq.), PPh3 (530 mg, 2.0 mmol, 1.2 eq.) and 18 (300 mg, 1.68 mmol, 1 eq.) in dry THF while stirring at 0 °C. The reaction mixture was stirred for 3 h and simultaneously allowed to warm up to 20 °C. The reaction was quenched by the addition of MeOH (1 mL), and the solvent was removed under reduced pressure. The crude product was purified by column chromatography on silica gel (ethyl acetate/petroleum ether 1:9) yielding 19 as an oil (415 mg, 1.35 mmol, 80%, Rf (ethyl acetate/cyclohexane, 20:80) = 0.60). 1H NMR (300 MHz, CDCl3): δ(ppm) = 1.68 (3H, s, H-6), 2.59 (2H, dt, J = 7.2 Hz, 6.3 Hz, H-4), 3.33 (2H, t, J = 7.2 Hz, H-5), 4.22 (2H, s, H-1), 5.37 (1H, m, H-3), 7.69–7.76 (2H, m, H-9), 7.83–7.89 (2H, m, H-10). 13C NMR (125 MHz, CDCl3): δ(ppm) = 15.0 (C-6), 31.5 (C-4), 32.0 (C-5), 44.8 (C-1), 123.5 (C-9), 124.1 (C-3), 132.1 (C-8) 132.7 (C-2), 134.2 (C-10), 168.3 (C-7).
∙ (E)-5-amino-4-methylpent-3-ene-1-sulfonate 7
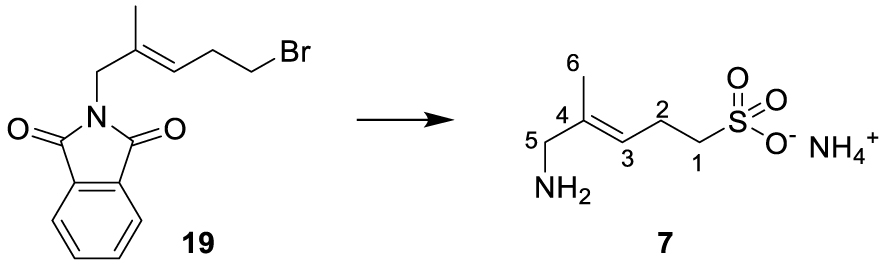
A solution of sodium sulfite (360 mg, 2.86 mmol, 2.2 eq.) in water (5 mL) was added to a stirred solution of 19 (400 mg, 1.30 mmol, 1 eq.) in EtOH (5 mL). The reaction mixture was further stirred under reflux overnight. The suspension was filtered, and the filtrate was evaporated under reduced pressure. Nonpolar products were discarded by filtration on silica gel (MeOH/CH2Cl2/NEt3, 2/8/0.2), and the most polar product was recovered and then dissolved in MeOH (2 mL) without further purification. NH4OH (2 mL) was added, and the reaction mixture was stirred overnight. Solvents were removed under reduced pressure. Consecutive column chromatography (isopropanol/H2O/NH4OH, 8:0.8:2 then isopropanol/H2O, 9:1) yielded 7 as a white solid, which was dissolved in water, lyophilized, and further dried under high vacuum (10 mg, 0.056 mmol, 4%, Rf (IPA/H2O, 8:2) = 0.27). 1H NMR (300 MHz, D2O): δ(ppm) = 1.76 (3H, s, H-6), 2.54 (2H, dt, J = 7.2 Hz, J = 6.2 Hz, H-2), 2.98 (2H, t, J = 7.8 Hz, H-1), 3.55 (2H, s, H-5), 5.60 (1H, m, H-3). 13C NMR (125 MHz, D2O): δ(ppm) = 14.3 (C-6), 23.5 (C-2), 46.8 (C-1), 50.6 (C-5), 128.4 (C-3), 129.9 (C-4). High-resolution MS (ES-) m/z: [M–H]- (C6H12NO3S) calculated 178.0543, found 178.0541.
4.3. Biological experiments
4.3.1. IspH production
Production and purification of E. coli IspH were performed as previously described [30]. Protein concentration was measured using the Bradford method with bovine serum albumin as a standard [44]. Iron was quantified according to Fish [45] and sulfide as described by Beinert [46]. UV/visible spectrum, iron and sulfur content of E. coli IspH were similar to those previously reported [13], in accordance with the presence of [4Fe–4S]2+ cluster.
4.3.2. Enzymatic assays
Enzyme activity was determined by monitoring NADPH consumption in the presence of optimized concentrations of the reducing system under anaerobic conditions. A HMBPP solution (final concentration 150 μM) was added through a gas-tight syringe to a 0.1 cm light path cuvette prepared in an anaerobic glove box and containing NADPH (2.2 mM), FldA (30 μM), FpR1 (17 μM), IspH (0.5 μM), and either 6 or 7 or 8 or AMBPP in 50 mM Tris–HCl pH = 8 that had previously been incubated for 15 min at 37 °C. The reaction was monitored spectrophotometrically at 340 nm with a Cary 100 UV/visible spectrophotometer (Varian) maintained at 37 °C using a thermostat equipped with a Peltier element.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Funding
This work was funded by the Fondation Jean-Marie Lehn, the European Union’s Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement No. 860816 and the “Université franco-allemande”. This work of the Interdisciplinary Thematic Institute InnoVec, as part of the ITI program of the University of Strasbourg, CNRS and Inserm, was supported by IdEx Unistra (ANR-10-IDEX-0002) and by SFRI-STRAT’US project (ANR-20-SFRI-0012) under the framework of the French Investments for the Future Program.
Acknowledgments
We appreciate the help from the staff of the computing facility provided by the Commissariat à l’Energie Atomique et aux énergies renouvelables (CEA/DSV/GIPSI), Saclay, France, and the Centre de Calcul Recherche et Technologie (CEA/CCRT), Bruyères-le-Châtel, France.