



 CC-BY 4.0
CC-BY 4.0
1. Introduction
To the best of our knowledge, the first study describing the synthesis of organotin trifluoromethanesulfonates dates back to the 1970s, led by Schmeißer’s group [1]. Since then, this class of compounds has attracted much interest in homogeneous catalysis and synthesis. These compounds are indeed reported to be efficient for specific metal-assisted organic reactions, acting as appropriate Lewis acid catalysts. Beneficial effects have been particularly described for the aldol reaction of Mukaiyama [2], Robinson annulation [3], acetylation of alcohols [4], transesterification of dimethyl carbonate with phenol [5], and direct synthesis of dimethyl carbonate (DMC) from methanol and carbon dioxide [6, 7]. From a synthetic point of view, organotin trifluoromethanesulfonates are generally prepared by reacting an organotin oxide with trifluoromethanesulfonic acid (CF3SO3H, TfOH) [8, 9], or alternatively, from an organotin chloride (R(4−x)SnClx with R = alkyl) in the presence of silver trifluoromethanesulfonate (AgCF3SO3) [8, 10]. Several organotin(IV) complexes have been characterized in the solid state by single crystal X-ray analysis. The CF3SO3 group can be ionic and behave as a counter anion, or be a ligand directly bound to tin. In the latter case, it can adopt various coordination modes. Examples of mono-, bi-, and tridentate as well as terminal, pseudo-terminal, and bridging CF3SO3 groups have already been reported in the literature for p-block metals and are shown in Scheme 1 [11, 12].
Molecular representations of known coordination modes involving the trifluoromethanesulfonate ligand and p-block metals (M).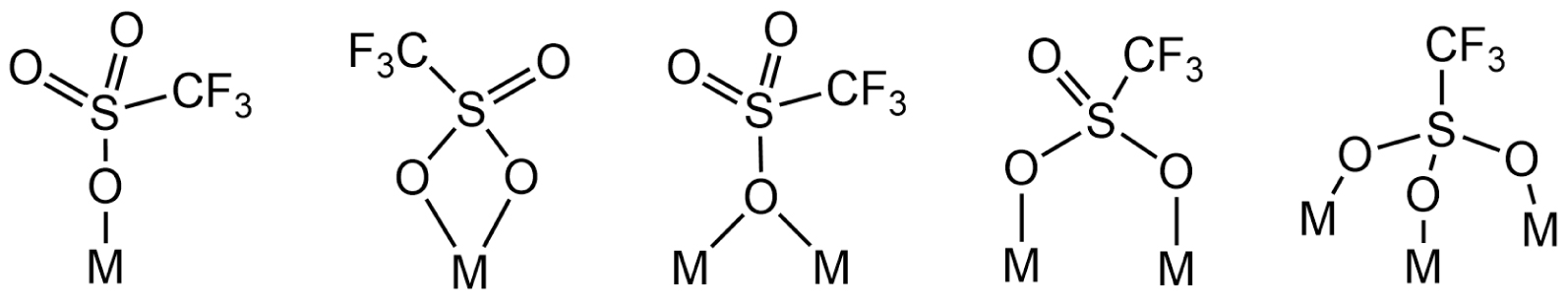
The chemistry and structural aspects of organotin trifluoromethanesulfonates were first reviewed by Beckmann in 2005 [13]. Since 2006, our group has also contributed to this field by characterizing several new specimens by X-ray crystallographic analysis: (i) ionic monobenzyltin(IV) trifluoromethanesulfonate clusters exhibiting unprecedented Sn6, Sn11, and Sn12 frameworks [14, 15], (ii) a two-dimensional organostannoxane coordination network {[n-Bu2(μ-OH)SnOSn(μ-𝜂2–O3SCF3)n-Bu2]2[n-Bu2(𝜂1-O3SCF3)SnOSn(μ-OH)n-Bu2]2} [12], which was found to be a polymorph of the tetra-n-butyldistannoxane trifluoromethanesulfonate cluster, [n-Bu2Sn2(OH)(CF3SO3)]2O (4), described initially by Otera [16, 17], (iii) a polymeric chain of dimeric hydroxo di-n-butyltin(IV) units bridged by trifluoromethanesulfonate ligands, [Sn2(CF3O3S)2(C4H9)4(OH)2]n [10]. More recently, we reported the isolation and characterization of the dihydrated di-n-butyltin(IV) trifluoromethanesulfonate salt {[n-Bu2Sn(H2O)]2O⋅n-Bu2Sn(OH)2}[CF3SO3]2 (2), which is characterized by a Sn3O3 core [18]. 2 was obtained by reacting at room temperature with the dimeric hydroxo di-n-butyl trifluoromethanesulfonate [n-Bu2Sn(μ-OH)(H2O)(CF3SO3)]2 (1) with a mixture of anthracene and phenazine.
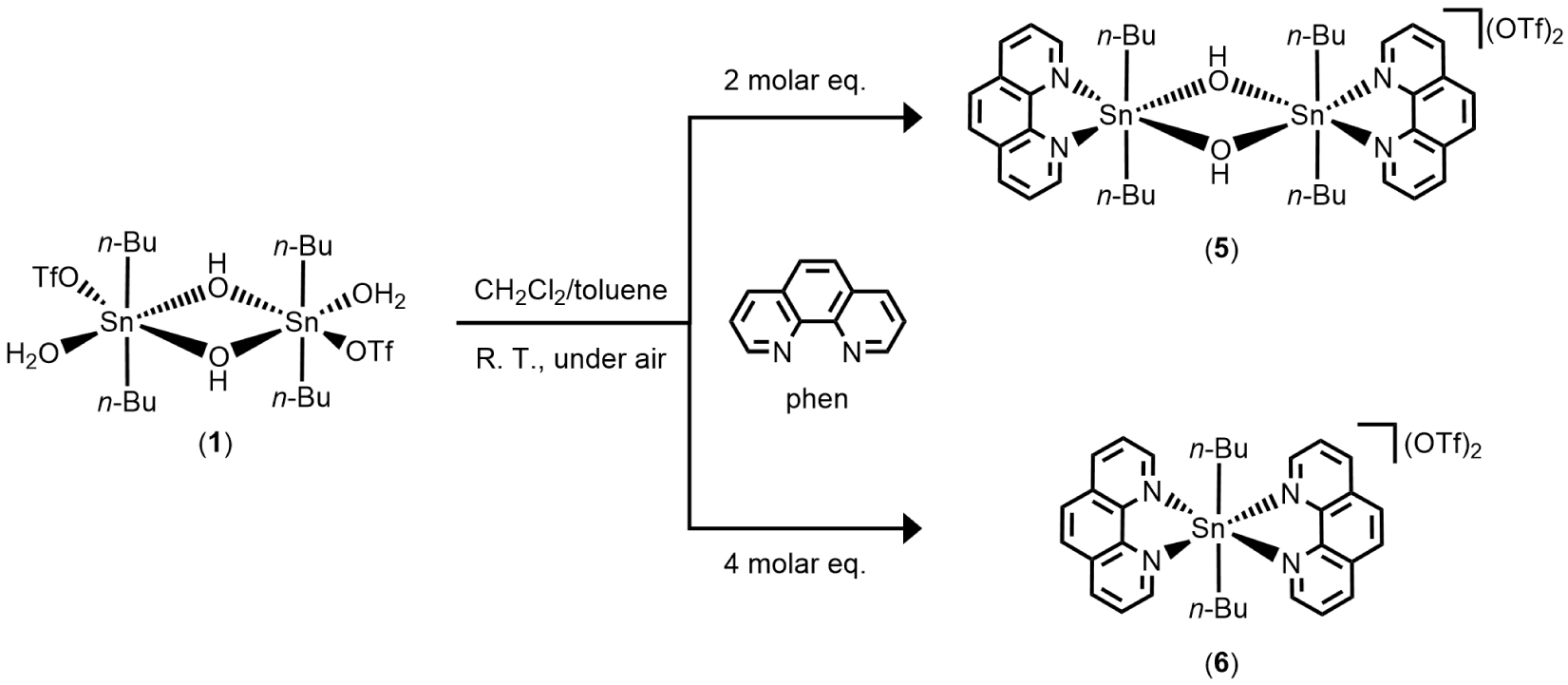
Continuing to explore the reactivity of 1 toward nitrogen-containing heterocyclic compounds, we report herein the synthesis and characterization of two novel diorganotin trifluoromethanesulfonate derivatives, [n-Bu2Sn(μ-OH)(phen)]2[CF3SO3]2 (5) and [n-Bu2Sn(phen)2][CF3SO3]2 (6), resulting from the reaction of 1 with 1,10-phenanthroline (phen), with the latter acting as bidentate ligand. Monitoring of the reaction by 119Sn{1H} NMR spectroscopy in solution (CD3CN) and as a function of the amount of phen added revealed successive in situ formation of species 2, 3, and 4. From the three molar equivalents of phen, only the resonance of 5 is visible in the spectrum. Species 2 and 4 have already been reported in the literature; however, to the best of our knowledge, 3 is a new species. Complexes 5 and 6 were fully characterized by IR, 1H, 11F, 13C, and 119Sn{1H} NMR spectroscopy, mass spectrometry, elemental analysis, and X-ray diffraction analysis. Further investigations were conducted to isolate and identify species 3. By changing the solvent conditions from acetonitrile to dichloromethane and in a 1:1 molar ratio of 1 and phen, the unprecedented mononuclear hydrated di-n-butyltin cation, [n-Bu2Sn(phen)(OH)(H2O)][CF3SO3] (7), was isolated, with the tin center N,N-chelated by a bidentate phen ligand and bearing a terminal hydroxyl group. Moreover, the reactivity of 1 was also tested in the presence of 2,9-dimethyl-1,10-phenanthroline (dmphen). From this reaction, only the phenanthrolinium triflates (dmphenHOTf and dmphen⋅dmphenHOTf), resulting from the mono-protonation of dmphen, were obtained.
2. Results and discussion
2.1. Synthesis and isolation of [n-Bu2Sn(μ-OH)(Phen)]2[CF3SO3]2 (5) and [n-Bu2Sn(Phen)2][CF3SO3]2 (6)
The air-stable dinuclear di-n-butyltin(IV) trifluoromethanesulfonate salts [n-Bu2Sn(μ-OH)(phen)]2[CF3SO3]2 (5) and [n-Bu2Sn(phen)2][CF3SO3]2 (6) were obtained at room temperature, under air atmosphere, from a mixture of dichloromethane/toluene by reacting dimeric trifluoromethanesulfonato hydroxo organotin(IV) [n-Bu2Sn(OH)(H2O)(CF3SO3)]2 (1) with two molar equivalents of 1,10-phenenthroline (C12H8N2, phen) (Scheme 2). A pinkish precipitate is first formed, leading, after recrystallization in dichloromethane/toluene, to the formation of single crystals later characterized as 6. Colorless single crystals of 5 were obtained from the filtrate of the mother liquor by slow evaporation at room temperature.
Synthetic pathway leading to 5 and 6.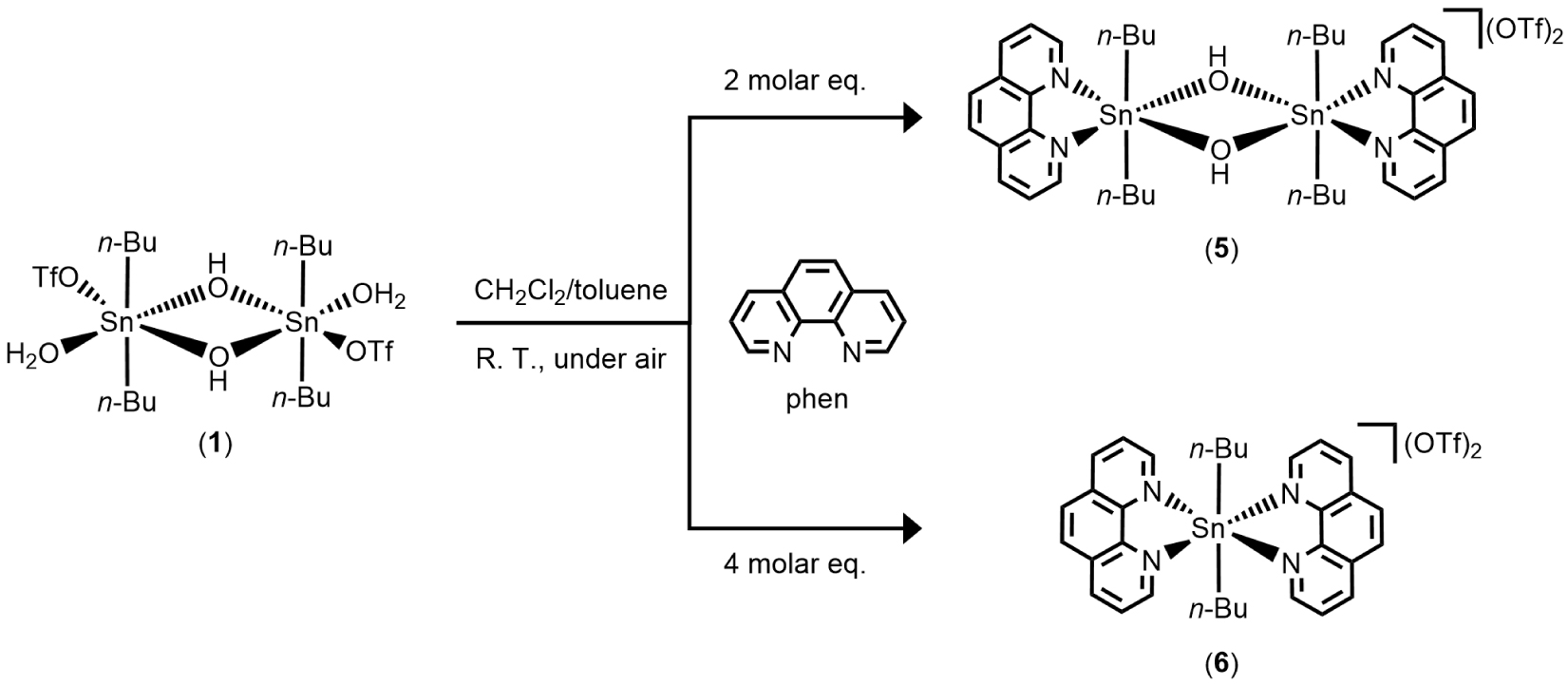
The two new organotin compounds which are ionic were characterized by elemental analysis, ESI-MS, IR and NMR spectroscopy, and single-crystal X-ray diffraction analysis. Compound 5 exhibits good solubility in organic solvents such as dichloromethane, acetone, and acetonitrile. In CD3CN, the 119Sn{1H} NMR spectrum of 5 exhibits one single resonance at 𝛿 −226 ppm (Figure S1). Such a chemical shift is in agreement with a six-coordinate tin atom substituted by two n-butyl moieties. It is interesting to note that in acetone-d6, the signal moves significantly, shifting to −150 ppm, suggesting that the tin center is pentacoordinated in this solvent environment. For comparison, the tin atoms of [n-Bu2Sn2(OH)(CF3SO3)]2O (4), which is characterized as a distannoxane, exhibit two resonances in acetone-d6 at −146 and −151 ppm [9, 12]. The 1H and 13C{1H} NMR spectra of 5 depicted in Figures S2 and S3, respectively, corroborate the structure depicted in Scheme 2, showing two sets of signals characteristic of phenanthroline ligands and n-butyl substituents. The CF3 moieties of the anions are also clearly identified in the 13C{1H} NMR spectrum, giving a typical quartet at 𝛿119.9 ppm, with a distinctive 1 JC–F coupling constant of 318 Hz. The 19F NMR spectrum exhibits one singlet at 𝛿 −78.2 ppm (Figure S4). The infrared spectrum of 5 highlights a distinctive broad absorption centered at 3321 cm−1 (Figure 1b), which is quite different from the fingerprint of 1 (Figure 1a), but can be assigned to the presence of OH groups (Figure 1b). Characteristic vibration bands of trifluoromethasulfonate ligands, in particular 𝜈(CF3) and 𝜈(SO3), are also observed for 3 in the stretching region between 1000 and 1300 cm−1 (outlined by a blue banner in Figure 1) [19, 20, 21]. The additional intense bands at 1518, 1427 and 854 cm−1 arise from the 𝜈(C=C), 𝜈(C=N) and 𝛾(C–H aromatic rings) absorptions of the phen ligands, respectively. The coordination of these ligands to tin is supported by the shift of the two bands at 1518 cm−1, and 1427 cm−1 relative to the free ligand (1503 cm−1 and 1420 cm−1) [22, 23]. An electrospray mass spectrum (positive mode) of 5 in a mixture of dichloromethane/methanol displays major mass clusters centered at m∕z = 431.11333 Da (100%), which fits closely with the [n-Bu2Sn(OH)(phen)]+ fragment (C20H27N2OSn, calc.: 431.11453 Da) (Figure S5). Finally, the micro-analytical (C, H, N, S) data also support the composition of 5 (see Section 4).
FT-IR (ATR) spectra of (a) [n-Bu2Sn(OH)(H2O)(CF3SO3)]2 (1), (b) [n-Bu2Sn(μ-OH)(phen)]2[CF3SO3]2 (5), (c) [n-Bu2Sn(phen)2][CF3SO3]2 (6), and (d) [n-Bu2Sn(phen)(OH)(H2O)][CF3SO3] (7).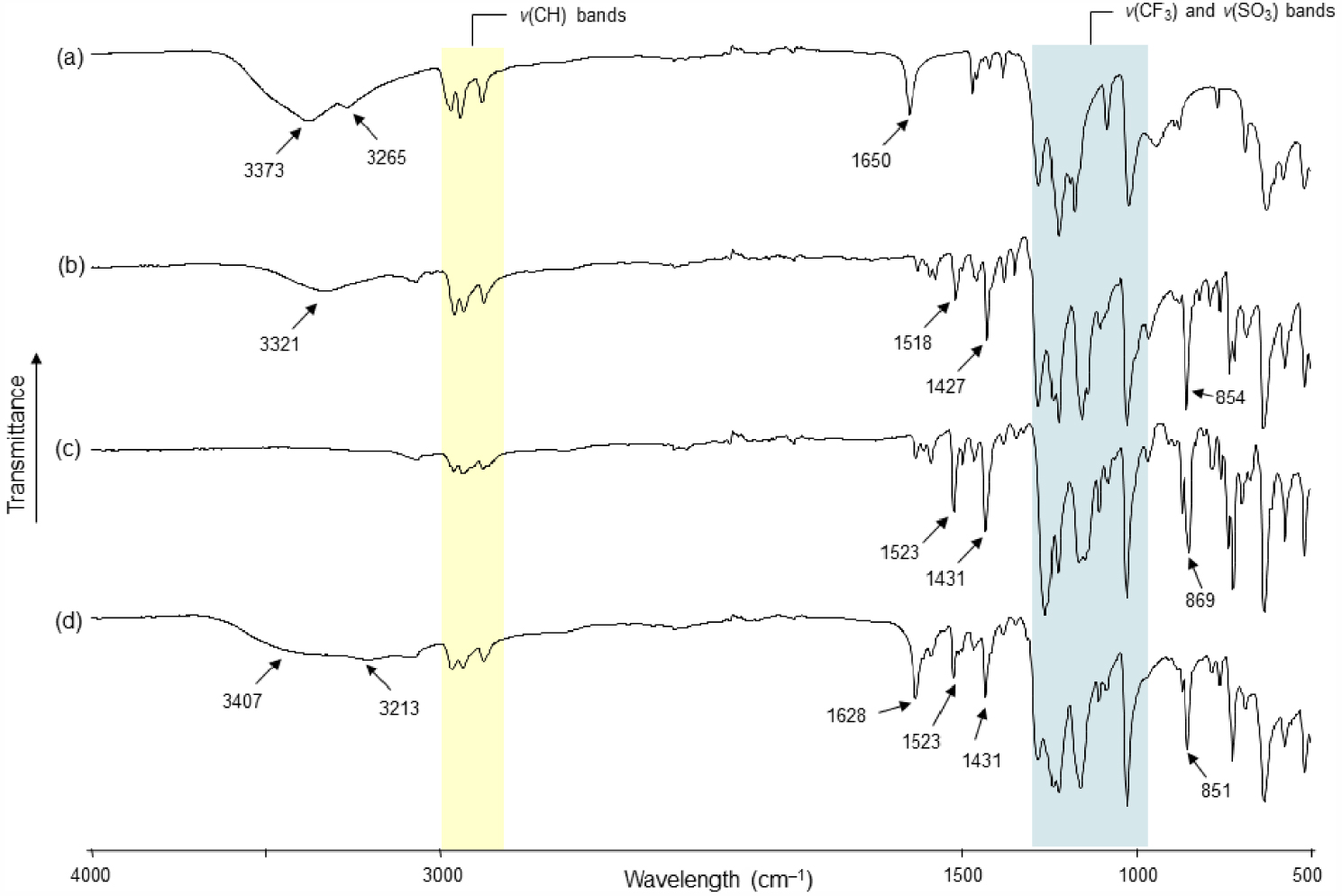
Compound 6 was collected as a precipitate from the starting reaction. However, it was found to be well soluble in acetone and dichloromethane. Suitable single-crystals for X-ray diffraction analysis were grown from a dichloromethane/toluene mixture. Compared with 1 and 5, the infrared spectrum of 6 shows no absorption bands above 3100 cm−1 excluding the presence of OH or H2O ligands. Otherwise, the characteristic absorption bands of n-butyl chain [𝜈(C–H)], CF3SO3, and phen are present and support the coordination of the phen ligand to the tin center (Figure 1c). In the aceton-d6 solution, the 119Sn{1H} NMR of 6 reveals a broad resonance at 𝛿 −231 ppm (Figure S6) [for comparison, in the same deuterated solvent, 5 exhibits a weak resonance at 𝛿 −150 ppm (Figure S7)], and the 1H NMR spectrum shows a 1:1 integration ratio between the protons of the n-butyl chains and the phen ligands (Figure S8). The 13C{1H} NMR spectrum exclusively reveals phen and n-butyl resonances and highlights the presence of anions by giving a quartet centered at 𝛿121.9 ppm, which is attributable to CF3 moieties (1 JC–F = 321 Hz) (Figure S9). A single resonance at 𝛿 −78.8 ppm is also observed in the 19F NMR spectrum (Figure S10). The electrospray mass spectrum (positive mode) of 6 in a mixture of dichloromethane/methanol displays two predominant mass clusters centred at m∕z = 445.13055 Da (100%) and 431.11559 Da (80%), which can be assigned to [n-Bu2Sn(phen)(OCH3)]+ (C21H29N2OSn, calc.: 445.13019 Da) and [n-Bu2Sn(phen)(OH)]+(C20H27N2OSn, calc.: 431.11454 Da) fragments, respectively (Figure S11).
2.2. Single-crystal X-ray diffraction analysis of 5
The solid-state structure of 5 consists of a [n-Bu2Sn(OH)(phen)] dication surrounded by two non-coordinated anions. An ORTEP view of 5 is shown in Figure 2 together with the selected bond distances and angles. The inorganic core of the cation is based on a planar four-membered distannoxane [Sn2(μ-OH)2] unit, where the tin atoms are bridged by two hydroxide ligands. The two tin atoms are bound to two n-butyl ligands [Sn–C13 = 2.131(2), Sn–C17 = 2.128(2) Å], to two oxygen atoms of bridging hydroxide ligands [Sn–O1 = 2.0828(13), Sn–O1i = 2.2334(14) Å], and are chelated by a bidentate 1,10-phenanthroline molecule forming five-membered rings. However, the Sn–N distances show significant differences [Sn–N1 = 2.4171(17), Sn–N2 = 2.6330(18) Å]. Thus, the tin centers can be considered to be hexacoordinated, adopting a markedly distorted octahedral geometry [C17–Sn–C13 = 156.93(9)°, N1–Sn–O1i = 156.85(5)°, N2–Sn–O1 = 150.33(6)°]. Furthermore, the phen ligands are parallel to each other and exhibit a twist angle of 25.55(4)° to the plane containing the Sn2O2 ring. Two trifluoromethanesulfonate anions complete the structure of 5. For both , one of their oxygen atoms is involved in hydrogen interaction with a hydroxide group of the cation [O3⋯HO = 2.791(2) Å]. Moreover, we can also suspect for each a possible weak interaction involving another oxygen atom of sulfonate functions and one tin atom of the cation [O4⋯ Sn = 3.342(2) Å], which could explain the different Sn–N distances. The existence of such long-distance Sn⋯ O interactions in the solid state has already been underlined for previous diorganotin compounds [10, 24]. In the past, Blaschette and Jones reported the isolation of [Me2{(MeSO2)2N}(phen)Sn(μ-OH)]2, a compound structurally analogous to 5 and prepared from Me2Sn[N(SO2Me)2]2 and 1,10-phenanthroline by adventitious hydrolysis [25]. Interestingly, in this compound, the (MeSO2)2N− anions operate in the same manner as the triflate anions of 5. They are non-coordinated and are involved in hydrogen bonding and long Sn⋯ O interactions. The authors then described the tin atoms as being heptacoordinated in a pentagonal bipyramidal arrangement. With regard to compound 5 and based on the 119Sn{1H} NMR chemical shift value (𝛿 −226 ppm in CD3CN), we argue in this case for the hexacoordination of the tin atoms.
Ortep drawing of 5 using a partial atom labeling scheme (30% probability thermal ellipsoids). Hydrogen atoms of phenanthroline ligands and n-butyl chains are omitted for clarity. Hydrogen bonds involving CF3SO3– and μ–OH groups are represented by red dotted lines. Selected bond lengths (Å) and angles (°): Sn–C17 2.128(2), Sn–C13 2.131(2), Sn–O1 2.0820(13), Sn–O1i 2.2343(14), Sn–N1 2.4171(17), C21–F3 1.315(3), C21–F1 1.329(3), C21–F2 1.334(3), C21–S1 1.816(3), S1–O2 1.4239(19), S1–O4 1.4331(18), S1–O3 1.4406(15); O1–Sn–C17 100.02(7), O1–Sn–C13 102.51(8), C17–Sn–C13 156.93(9), O1–Sn–O1i 72.08(6), C17–Sn–O1i 90.38(8), C13–Sn–O1i 91.85(8), O1–Sn–N1 84.98(6), C17–Sn–N1 90.58(8), C13–Sn–N1 96.24(8), O1i–Sn–N1 156.85(5), Sn–O1–Sni 107.92(6), F3–C21–F1 107.0(2), F3–C21–F2 108.8(3), F1–C21–F2 106.7(2), F3–C21–S1 112.27(19), F1–C21–S1 110.9(2), F2–C21–S1 110.95(18), O2–S1–O4 116.03(13), O2–S1–O3 114.98(13), O4–S1–O3 113.25(10), O2–S1–C21 103.25(13), O4–S1–C21 104.76(13), O3–S1–C21 102.36(10) (symmetry transformations used to generate equivalent atoms: (i) : 3/2–x, 1∕2 − y,−z).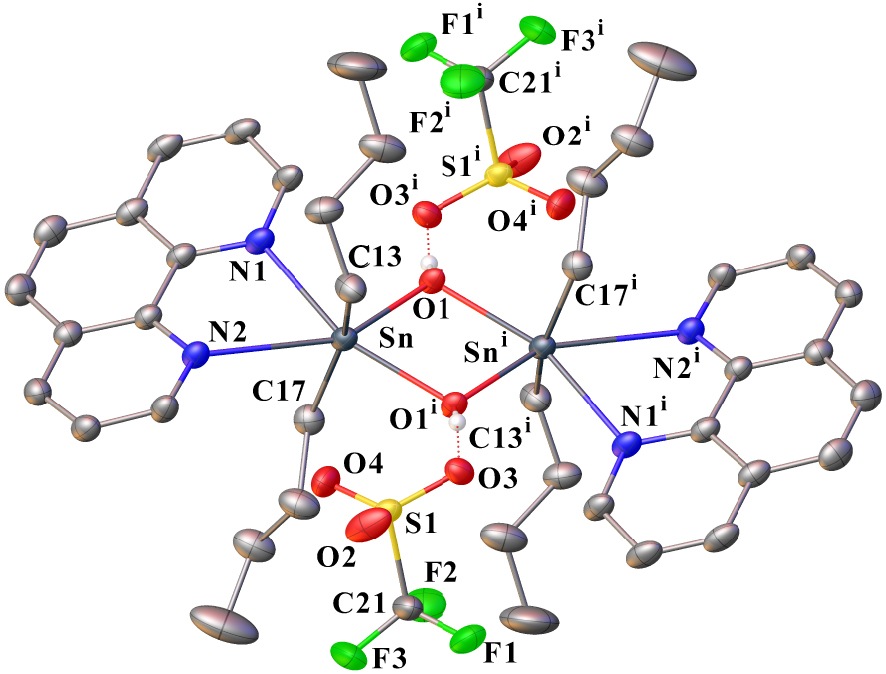
2.3. Single-crystal X-ray structural analysis of 6
The structure of salt 6 in the solid state consists of a [n-Bu2Sn(Phen)2]2+ dication surrounded by two non-coordinating anions. One toluene molecule (crystallization solvent) co-crystallizes with 6. Figure 3 shows an Ortep view of 6 with the atom numbering scheme, and Figure 4 shows a perspective view of the crystal packing in the unit cell. The molecular structure of the title compound consists of a central six-coordinated tin atom that exhibits an octahedral coordination environment comprising four nitrogen atoms from two distinct chelating phen ligands and two carbon atoms of the two n-butyl chains. The ligands are positioned in a cis-arrangement. The N1–Sn–N4, N2–Sn–C25, N3–Sn–C29 bond angles measure 154.5(3), 157.9(3), and 156.9(3)°, respectively, indicating a distortion from an ideal octahedral geometry. The two phen ligands are bidentate to tin, forming five-membered chelate rings that are nearly planar. The Sn–N distances are in the range of 2.273(8) and 2.368 Å. They are comparable to that found in [MeSn(phen)2]2+ [26], which is, to the best of our knowledge, the only example reported to date for a diorganotin complex bis-N,N-chelated by two 1,10-phenanthroline ligands.
Ortep drawing of 6 using a partial atom labeling scheme (30% probability thermal ellipsoids). Hydrogen atoms and one molecule of toluene (solvent) are omitted for clarity. Selected bond lengths (Å) and angles (°): Sn1–C29 2.162(9), Sn1–C25 2.158(9), Sn–N1 2.273(8), Sn–N2 2.369(8), Sn–N3 2.309(7), Sn–N4 2.275(8), S1–O2 1.415(8), S1–O1 1.427(8), S1–O3 1.431(9), S1–C33 1.823(11), C33–F1 1.299(12), C33–F2 1.328(14), C33–F3 1.312(13), C25–Sn1–C29 116.9(4), N1–Sn1–N2 71.9(3), N1–Sn1–N3 87.7(3), N1–Sn1–N4 154.5(3), C29–Sn1–N1 98.0(3), C25–Sn1–N1 97.0(4), N3–Sn1–N2 76.3(3), N4–Sn1–N2 87.5(3), C29–Sn1–N2 84.12(3), C25–Sn1–N2 157.9(3), N4–Sn1–N3 72.6(3), C29–Sn1–N3 156.9(3), C25–Sn1–N3 84.3(3), C29–Sn1–N4 94.6(3), C25–Sn1–N4 96.9(4).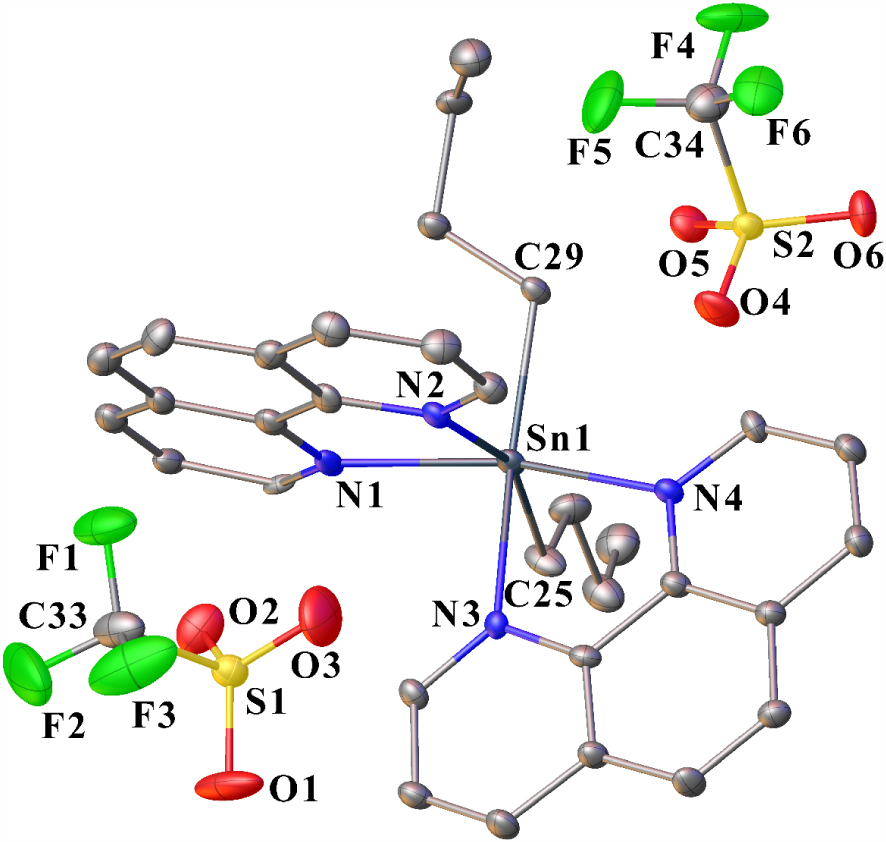
Projection of the crystal structure approximately along the b axis, showing the orientation of the phen ligands between two neighboring cations of 6. The solvent toluene molecules co-crystallizing with 6 and hydrogen protons are omitted (Mercury representation [27], color code: Sn = violet, N = blue, C = gray, O = red, S = yellow, F = green).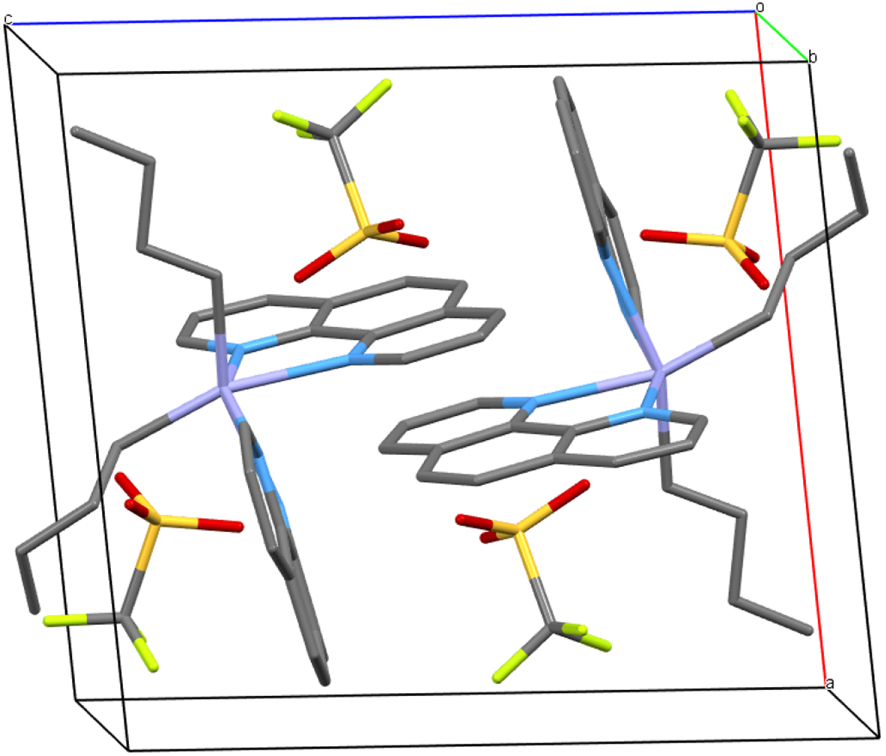
119Sn{1H} NMR monitoring of the evolution of a solution of 1 in CD3CN ([0.08 M], 298 K) as a function of successive additions of 1,10-phenanthroline (phen): (a) starting solution of 1, (b) after addition of 0.5 molar equivalent, (c) of 0.75 molar equivalent, (d) of 1 molar equivalent, (e) of 2 molar equivalent, and (f) of 3 molar equivalent.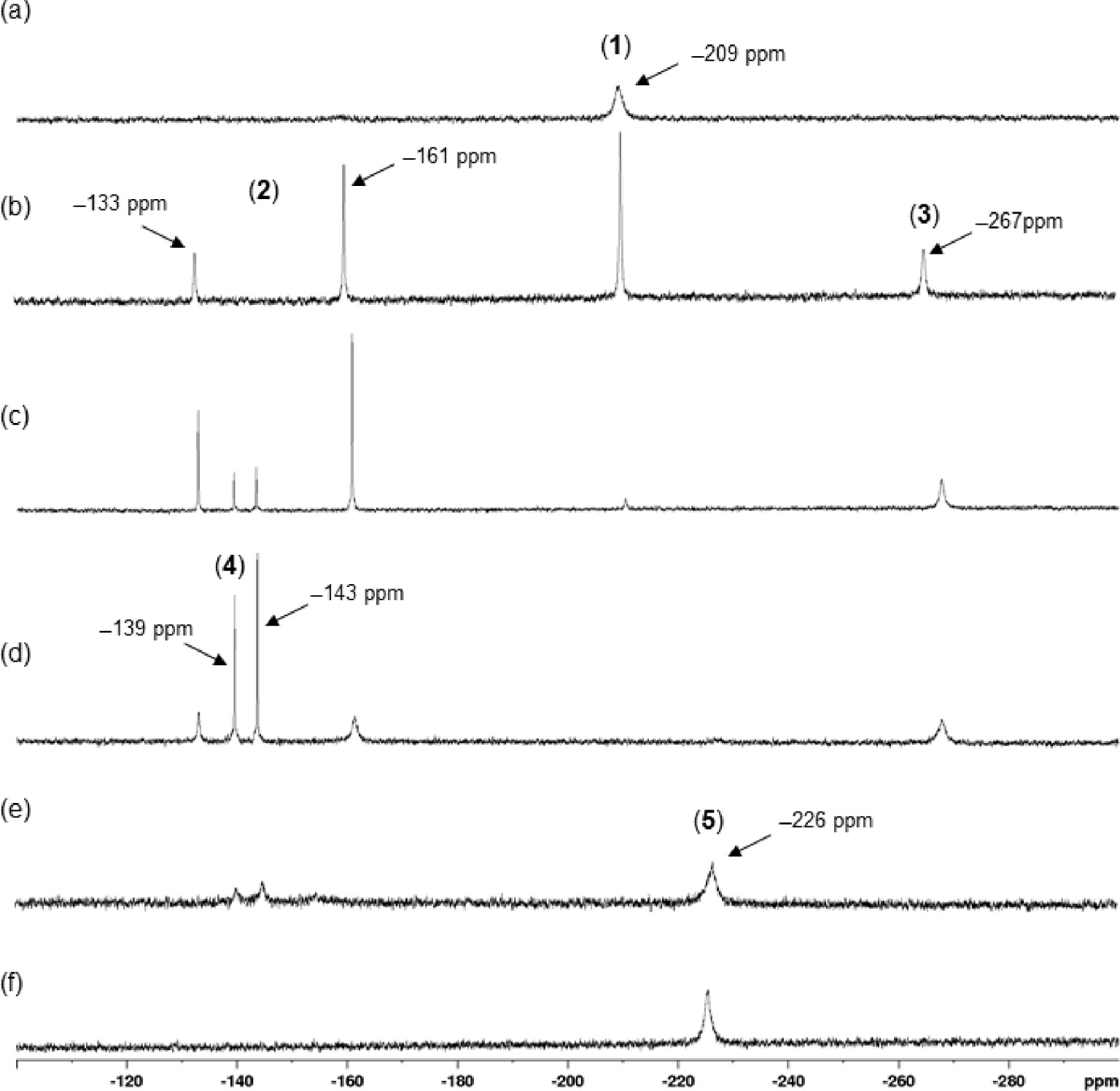
From a supramolecular point of view, the molecules of 6 are organized in pairs via offset π–π interactions involving the aromatic rings of each phen ligand (Figure 4). The interplanar and centroid-to-centroid distances between the two parallel phen molecules are 3.393 and 4.732 Å, respectively. In general, for such interactions, the interplanar distance is assumed to be in the range of 3.3–3.8 Å [28]. In the arrangement of 6, the rings are severely offset with a slippage angle (angle between the normal to the planes and the centroid vector centroid vector) of 44.19°, which corresponds to a slip distance of 3.298 Å.
2.4. Solution 119Sn{1H} NMR monitoring
The reactivity of 1 toward successive additions of 1,10-phenanthroline was followed by 119Sn{1H} spectroscopy in CD3CN directly in an NMR tube, as shown in Figure 5. In CD3CN, compound 1 shows a broad signal at 𝛿 −209 ppm in agreement with the value reported previously in the literature [8] (Figure 5a). The addition of 0.5 molar equivalents of phen leads to the appearance of three new signals at 𝛿 −133, −161, and −267 ppm in addition to the signal of 1, which is still present at 𝛿 −209 ppm (Figure 5b). The two de-shielded chemical shifts exhibiting a 1:2 intensity ratio correspond to the signature of the ionic complex 2, already known and characterized by us as the unusual trinuclear complex {[n-Bu2Sn(H2O)]2O⋅n-Bu2Sn(OH)2} [CF3SO3]2 (2) (Scheme 3a) [16], while the signal at 𝛿 −267 ppm can be attributed to a new species 3 whose composition and structure will be discussed later. Further addition of phen (up to 1 molar equivalent) lead to a progressive disappearance of the previously described resonances in favor of the emergence, and then the predominance, of a new pair of signals located at −139 and −143 ppm (Figure 5b and c). They reflect the formation of 3-hydroxy-1-(triflato)tetra-n-butyldistannoxane, [n-Bu2Sn2(OH)(CF3SO3)]2O (4), whose solid-state structure was initially established by Otera et al. (Scheme 3b) [9]. For our part, we had previously shown the possible transformation of 2 to 4 in the presence of phenazine [16]. It is likely that a similar reaction can occur in the presence of 1,10-phenanthroline. When the addition of phen reaches two molar equivalents, the residual signals of 4 are still visible, but a broad resonance, prevailing, appears at 𝛿 −226 ppm (Figure 5e). This corresponds to the formation of complex 5. In the presence of three molar equivalents, this compound is the only species detected in solution (Figure 5f). However, a white precipitate is also observed at the bottom of the NMR tube, which corresponds to complex 6 insoluble in CD3CN. The values of the 119Sn{1H} NMR chemical shifts of the different organotin species implicated in this study are summarized in Table 1.
Molecular representations of 2 (a) and 4 (b).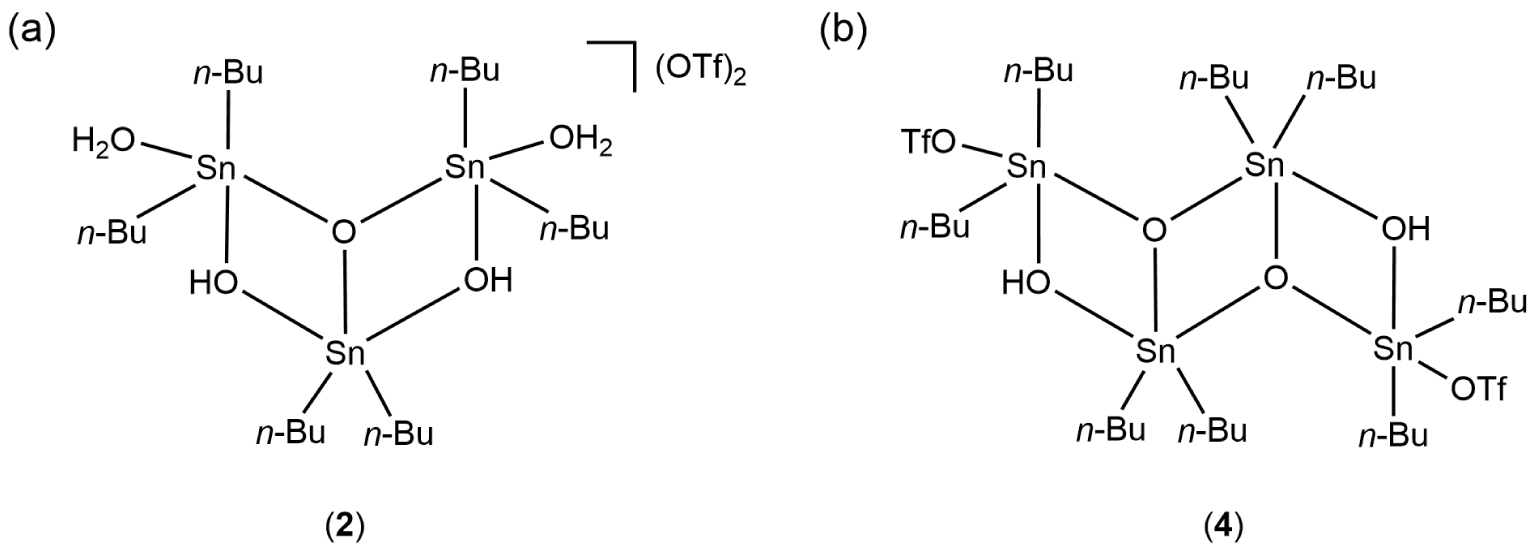
119{1H] NMR chemical shift data of di-n-butyltin(IV) trifluoromethanesulfonates
| Compounds | 𝛿 (ppm)a | ||
|---|---|---|---|
| In CD2Cl2 | In acetone-d6 | in CD3CN | |
| 1 | −145 | −209 | |
| 2 | −127, −149 [18] | −133, −161 | |
| 3 | −288 | −267 | |
| 4 | −135, −144 [18] | −146, −151 [12] | −139, −143 |
| 5 | Not visible | −150 | −226 |
| 6 | Not visible | −231 | Insoluble |
| 7 | −260 | ||
a At T = 298 K.
2.5. Isolation and characterization of species 3
During the monitoring of the reaction by 119Sn{1H} NMR, all signals were attributed to identified organotin species, except for the single broad resonance at 𝛿 −267 ppm in CD3CN (Figure S12), which was assigned to compound 3. We did not find any bibliographic data associated with this species; therefore, it appeared to be new and its composition remains to be clarified. We first sought to isolate it by reproducing the experimental conditions of the NMR measurement depicted in Figure 5d in glassware conditions, i.e., by adding 1 molar equivalent of phen to a solution of 1 in a mixture of acetonitrile/toluene (15 mL/10 mL). After three successive separations by crystallization at 4 °C (2 and 4 were collected as crystalline products) in a CH3CN/toluene mixture, it was possible to isolate only 3. After complete evaporation of the solvents, 3 consists of a colorless pasty solid. Infrared analysis, shown in Figure S13, reveals several characteristic bands: (i) 𝜈(C–H) absorption bands between 2800 and 3000 cm−1, (ii) 𝜈(C=C), 𝜈(C=N) and 𝛿(C–H) fingerprints of the phen ligand [22, 23], and (iii) stretching vibration bands of trifluoromethasulfonate ligands [𝜈(CF3) and 𝜈(SO3)] between 1000 and 1300 cm−1 [19, 20, 21]. A 119Sn{1H} NMR spectrum of a solution of 3 in CD2Cl2 shows a singlet resonance at 𝛿 −288 ppm. The 13C{1H} NMR spectrum of 3 in CD2Cl2 (Figure S14) highlights two sets of signals attributed to the presence of n-butyl and phenanthroline ligands. A quartet at 𝛿 121.2 ppm (1 JC–F = 320 Hz) reveals the presence of CF3 moieties, which is corroborated by the 19F NMR spectrum, which exhibits one singlet at 𝛿 −78.4 ppm (Figure S15). The 1H spectrum establishes a phen/n-butyl ligand ratio of 1:2 (Figure S16). The electrospray mass spectrum (positive mode) of 3 in dichloromethane/acetonitrile solution displays three intense mass clusters (Figure S17). The first one centered at m∕z = 563.0613 Da (z = 1, 67%) matches with high accuracy to a mononuclear di-n-butyltin fragment bearing a positive charge that can be assigned to [n-Bu2Sn(phen)(CF3SO3)]+ (C21H26O3N2F3SSn), calc. = 563.0628 Da), whereas the cluster centered at m∕z = 1275.07507 Da (z = 1, 100%) is consistent with the monocationic dinuclear framework of the empirical formula C43H52O9N4F9S3Sn2 (calc. = 1275.07912 Da). The intermediate cluster at m∕z = 893.0887 Da (67%) is compatible with [M+H–phen–n-Bu2Sn]+ (C34H35O6N4F6S2Sn, calc. = 893.0819 Da). Simulations of these ESI-MS mass clusters are depicted in Figures S18–S20. On the basis of these data, we suggest that for species 3, the structure reproduced on Figure 6 could correspond to two [n-Bu2Sn(phen)(CF3SO3)] entities bounded by a bridging trifluoromethanesulfonate ligand, [{n-Bu2Sn(phen)(CF3SO3)}2(μ-CF3SO3)][CF3SO3]. The overall positive charge of 3 is compensated by an anion. To the best of our knowledge, such a structure has not yet been described in the literature. However, there are some reports confirming in the solid-state the existence of –CF3SO3 groups bridging di-n-butyltin derivatives. In general, this results in the propagation of polymeric networks: [Sn2(CF3O3S)2(C4H9)4(OH)2]n [10], [9], [12]. In 3, the presence of phen ligands occupying two coordination sites on each tin atom probably prevents polymerization. In the context of the related chemistry of organotin(IV) alkanesulphonates, Shankar et al. recently reported the solid-state structures of di-n-butyltin complexes with bridging and terminal –OSO2R ligands (R = Me, Et), quite comparable to the structure suspected for 3 [29].
Possible molecular representation of 3 (OTf = CF3SO3).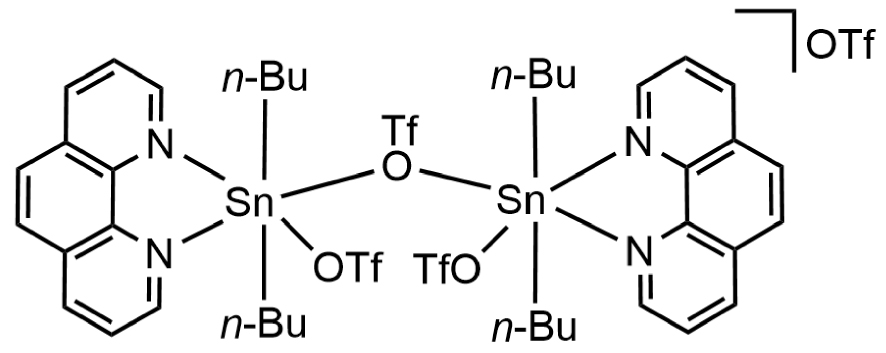
The elemental analysis calculated for [{n-Bu2Sn(phen)(CF3SO3)}2(μ-CF3SO3)][CF3SO3] (C44H52F12N4O12S4Sn2, 1422.57 g⋅mol−1) is also in accordance with the formula suggested for 3: Calculated: C, 37.17; H, 3.68; N, 3.94; S, 9.02 Found: C, 36.43; H, 3.74; N, 4.13; S, 8.16%. Furthermore, as highlighted by the NMR measurements reported in Figures 5d and 5e, we verified that the addition of phen to a solution of 3 in CDCN does indeed result in the appearance of the characteristic signal of 5 at 𝛿 −227 ppm. However, to date, it was not possible to confirm the proposed structure by single crystal X-ray diffraction analysis because the compound does not crystallize properly in acetonitrile.
2.6. Reactivity in dichloromethane
In contrast, when the reaction between 1 and phen (in 1:1 molar ratio) is conducted in dichloromethane instead of acetonitrile, colourless single crystals are grown at −20 °C from the filtrate of the reaction. They are unstable and melt very quickly as the temperature increases. This is recurrent and was observed in several batches. The crystallographic analysis could be performed despite the poor quality of crystals. However, several difficulties were encountered during the treatment of the crystallographic data: (i) the determination of the space group was delicate and finally defined as C2/c, (ii) several ligands were affected by disorders, (iii) a solvent-accessible voids remained. The resolution of the structure remains imperfect, but we believe that the structural data provided are informative in relation to the intermediate species leading to 5 and 6. Thus, the structure of compound 7 (Figure 7) established the presence of a cationic mononuclear compound based on a tin atom carrying two n-butyl chains [Sn–C13 = 2.136(7), Sn–C17 = 2.123(7) Å] in trans-position with respect to each other and chelated by a bidentate phenanthroline ligand [Sn–N1 = 2.208(11), Sn–N2 = 2.342(8) Å]. The Sn–N bond lengths are comparable with those measured for 6. Two oxygen atoms, O1 and O2, complete the coordination sphere of the tin atom, which describes a distorted octahedral geometry [N1–Sn–O1 = 157.1(3), N2–Sn–O2 = 162.2(3), C17–Sn–C13 = 156.89(9)]. The O1 and O2 atoms are assigned to OH2 and OH ligands, respectively, but unfortunately, their hydrogen atoms could not be located. Their assignments were nevertheless possible based on the Sn–O interatomic distances, which are significantly different: Sn1–O1 = 2.512(8), as opposed to Sn–O2 = 2.142(5) Å. Distances greater than 2.3 Å have already been observed for several di-n-butyltin derivatives with Sn–OH2 bonds: 2.365 Å in [n-Bu2Sn(H2O)(μ-OH)]2[CF3SO3] [5], 2.409 Å in [n-Bu2Sn(μ-OH)(H2O)(CF3SO3)]2 [4], 2.511 Å in {[n-Bu2Sn(H2O)]2O⋅n-Bu2Sn(OH)2}[CF3SO3]2 [16]. Sn–OH bonds are generally shorter: between 2.062 and 2.150 Å in [n-Bu2Sn(H2O)(μ-OH)]2[CF3SO3] [5], 2.120 Å in {[n-Bu2Sn(H2O)]2O⋅n–Bu2Sn(OH)2}[CF3SO3]2 [16]. These examples support the attribution of O1 and O2. The positive charge of [n-Bu2Sn(phen)(OH)(H2O)]+ is compensated by one non-coordinated anion. To the best of our knowledge, such a structure describing a monomeric complex is unprecedented. Diorganotin hydroxides are difficult to purify and characterize [30]. In the solid state, they are mainly described as dimers, hydroxyl groups acting as bridging ligands between two tin atoms, and also as polymers. Thus, the number of complexes with an OH group singly coordinated to tin is very limited [31, 32, 33]. In the case of 7, although the resolution of its structure is not optimal, we assume that the coordination of the phen ligand favours the existence of a monomeric form. To the best of our knowledge, another example of tin-hydroxide stabilized by a phen ligand was previously reported by Aghabozorg et al., characterized by X-ray crystallographic structure as being [Sn(pydc)(phen)(OH)2]⋅3H2O (pydc = pyridine-2,6-dicarboxyate) [34]. The two terminal OH groups occupy the apical positions of a pentagonal bipyramid. The IR spectrum recorded from the crystals of 7 corroborates the presence of H2O and OH ligands by showing two shoulders centered at 3407 and 3213 cm−1 and a sharp band at 1628 cm−1 assigned to 𝛿(H2O) elongation. The characteristic absorption bands of CF3SO3 and phen are also well visible (Figure 1d). Globally, the IR fingerprint of 7 shows strong similarities to that of 5.
Ortep drawing of 7 using the atom labeling scheme (30% probability thermal ellipsoids). The hydrogen atoms of O1 and O2 could not be precisely located. Selected bond lengths (Å) and angles (°): Sn–N1 2.208(11), Sn–N2 2.342(8), Sn–O1 2.512(8), Sn–O2 2.142(5), Sn–C13 2.136(17), Sn–C17 2.123(7), S–O3 1.466(7), S–O4 1.403(8), S–O5 1.408(6), C21–F1 1.311(13), C21–F2 1.309(12), C21–F3 1.330(11), C21–S 1.794(11); O1–Sn–O2 113.8(3), O2–Sn–O1 89.1(3), O2–Sn–N2 162.2(3), N1–Sn–O1 157.1(3), N1–Sn–N2 73.1(4), N2–Sn–O1 84.1(3), C13–Sn–C17 161.9(3), C13–Sn–O1 79.9(3), C13–Sn–O2 91.9(2), C13–Sn–N1 100.1(4), C13–Sn–N2 90.7(3), C17–Sn–O1 82.2(3), C17–Sn–O2 97.4(2), C17–Sn–N1 95.6(4), C17–Sn–N2 85.2(3), O3–S–C21 103.7(5), O4–S–C21 104.5(5), O4–S–O3 113.9(5), O4–S–O5 117.0(5), O5–S–C21 104.2(4), O5–S–O3 111.7(4). F1–C21–F3 109.1(11), F1–C21–S 110.5(7), F2–C21–F1 106.5(9), F2–C21–F3 106.6(9), F2–C21–S 113.1(9), F3–C21–S 110.8(7).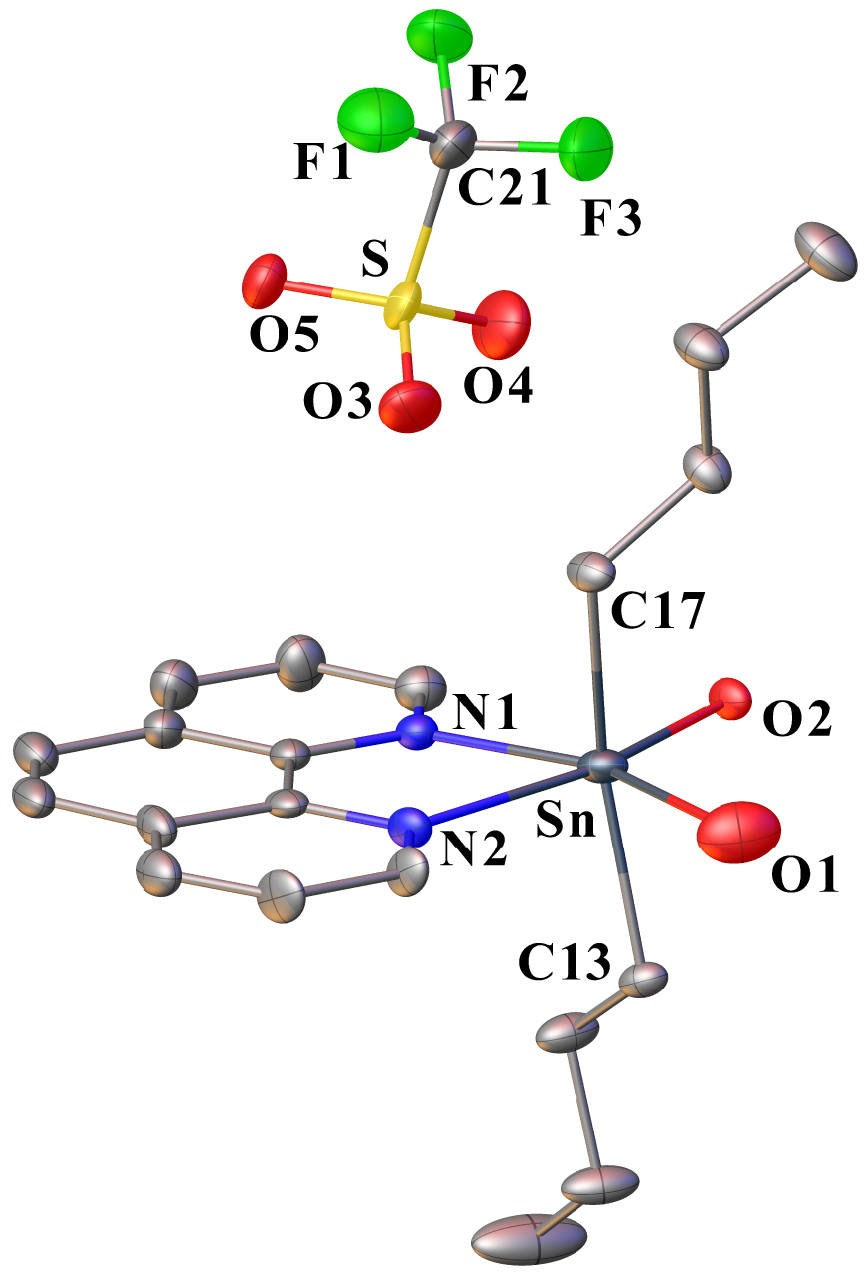
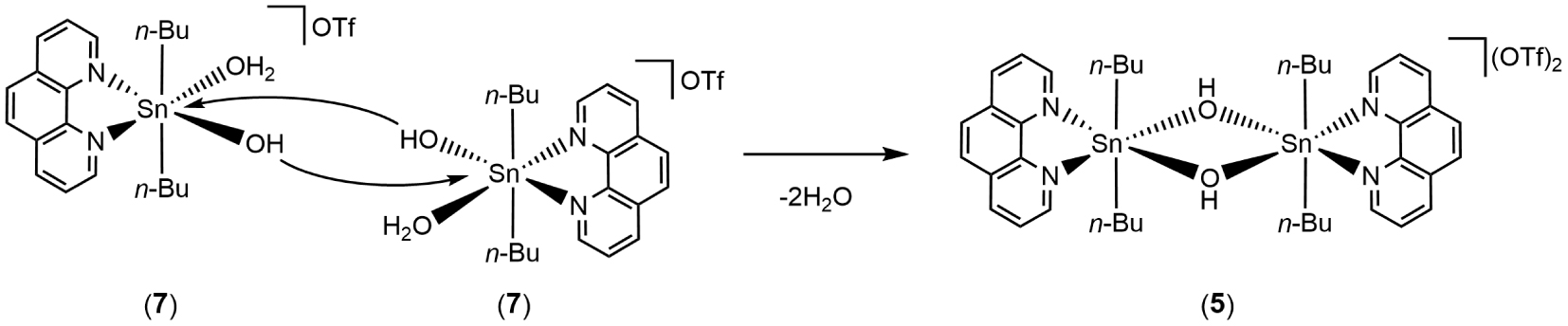
Compound 7 can be viewed as a reaction intermediate resulting from the reactivity of 1 toward 1,10-phenanthroline under sub-stoichiometric phen conditions (Equation (1)). This can then lead to the formation of 5 by dimerization, driven by the nucleophilicity of the OH group, resulting in the expulsion of aqua ligands (Scheme 4). This mechanism is similar to that proposed earlier by Chandrasekhar et al. for the dimerization of {[n-Bu2Sn(OH2)4]2+[2,5-Me2C6H3SO]2 into {[n-Bu2Sn(μ-OH)(O3SC6H3-2,5-Me2)]2}n [35].
Plausible mechanism leading to the formation of 5 from 7.
| (1) |
From a structural point of view, 7 can be related to the salt {[n-Bu2Sn(OH2)(phen)(O3SC6H3-2,5-Me2)]+[2,5-Me2C6H3SO3]−} also described by Chandrasekhar et al., and isolated by reacting {[n-Bu2Sn(OH2)4]2+[2,5-Me2C6H3SO]2 with 1,10-phen [35]. The two compounds exhibit strong similarities. They consist of mononuclear hydrated diorganotin cations chelated by a phen ligand. In the Chandrasekhar cation, the coordination of the tin atom is completed by a sulfonate ligand, whereas for 7, we claim the presence of a terminal OH group. It is interesting to note that in solution in CD3CN, the 119Sn{1H} NMR spectroscopic analysis of the crystals of 7 shows the presence of a mixture of four species (Figure S21), which highlights the instability of the mononuclear cation 7 also in solution. It is nevertheless possible to identify the characteristic resonances of compounds 2 (−133, −161 ppm), 4 (−139, −143 ppm), and 5 (−226 ppm). The most shielded signal at −260 ppm is thus attributed to 7. Based on the relative integration, the two main species are 5 and 7, which is consistent with the dimerization reaction suggested in Scheme 4. However, the presence of species 2 and 4, which are in the minority according to a 119Sn{1H} NMR spectrum and do not bear phen ligands, from 7, is still unexplained.
Thus, the solid-state structure of 7 resulting from the reaction in dichloromethane differs markedly from the results obtained in acetonitrile, leading to the hypothesis of compound 3. This implies a determining role of the solvent used on the nature of the trifluoromethanesulfonate intermediate species formed. This has already been demonstrated experimentally in the past by Otera et al., who showed that when n-Bu2SnO reacts with triflic acid in CH2Cl2 conditions, the compound 1, characterized as a dimeric cation is preferentially formed [8], whereas in acetonitrile, a polymeric structure prevails, alternating anhydrous and hydrated moieties of [n-Bu2Sn(OH)(OTf)]2 and [n-Bu2Sn(OH)(OTf)(H2O)]2, respectively [5]. In the future, we plan to use this modularity to explore the performance of the compounds described in this study for tin-assisted organic reactions, especially since 1,10-phenanthroline derivatives are known to be efficient and stable ligands in homogeneous catalysis [36].
2.7. Isolation and solid-state structure of [C12H9N2][CF3SO3] (phenHOTf)
In addition to 7, single crystals of 1,10-phenanthrolinium trifluoromethanesulfonate, [C12H9N2][CF3SO3] (phenHOTf), were obtained after a few days from the filtrate of the mother-liquor. The phenHOTf salt consists of a monoprotonated 1,10-phenanthrolinium cation interacting with a surrounding trifluoromethanesulfonate anion through N–H⋯ O hydrogen bonding [N1⋯ O3 = 2.804(2) Å, N1–H⋯ O3 = 147.61(11)°]. An Ortep representation is shown in Figure 8. To date, a large number of structures of 1,10-phenanthrolinium salts [C12H9N2][X] have been resolved by X-ray crystallography, such as X = Cl− [37], [38], [39], [40]. The structure of phenHOTf is a new example. The crystal stacking of phenHOTf (Figure 9) shows that the phenanthrolinium cations are grouped in pairs via offset π–π interactions characterized by an interplanar distance of 3.398 Å, a centroid–centroid distance of 4.878 Å, and a slippage distance of 3.50 Å (slippage angle = 45.85°).
Ortep drawing of phenHOTf using the atom labeling scheme (30% probability thermal ellipsoids). The N–H⋯ O interaction is indicated by a red dotted line. Selected bond lengths (Å) and angles (°): C1–N1 1.333(2), C5–N1 1.358(2), C5–C6 1.438(3), C6–N2 1.359(2), C12–N2 1.326(3), O1–S 1.4367(14), O2–S 1.4417(14), O3–S 1.4505(14), C13–S 1.825(2), C13–F1 1.336(2), C13–F2 1.335(2), C13–F3 1.335(2); C1–N1–C5 123.04(17), C12–N2–C6 116.54(18), O1–S–C13 103.50(9), O1–S–O2 115.68(8), O1–S–O3 114.80(8), O2–S–C13 103.31(9), O2–S–O3 114.88(8), O3–S–C13 102.07(9), F1–C13–F3 107.57(16), F1–C13–S 111.34(13), F2–C13–F1 107.49(16), F2–C13–F3 107.62(15), F2–C13–S 111.31(14), F3 C13 S 111.31(14).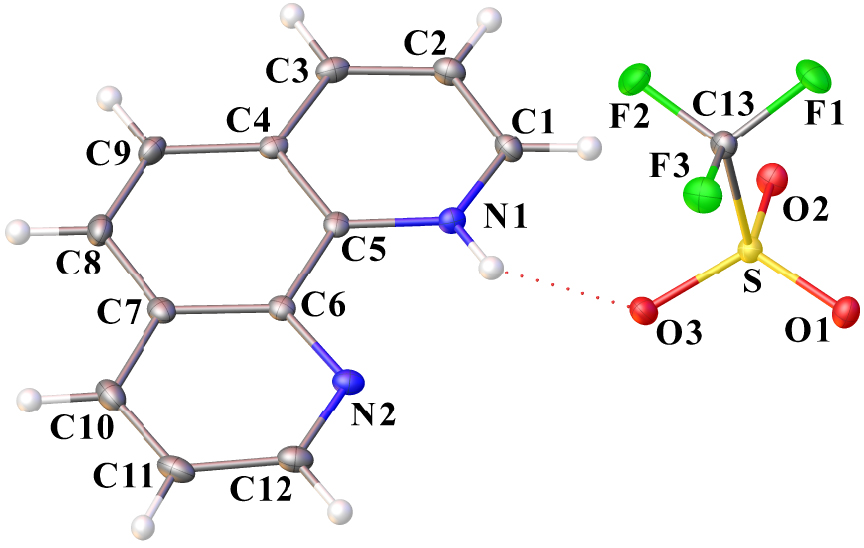
Crystal packing of phenHOTf along the a-axis. (Mercury representation [27], color code: N = blue, C = gray, H = white, O = red, S = yellow, F = green.)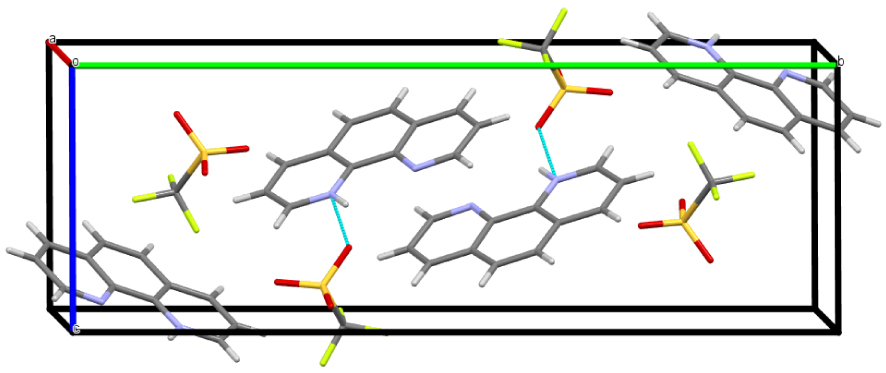
2.8. Reactivity of 1 to 2,9-dimethyl-1,10-phenanthroline
Subsequently, the reactivity of 1 was extended to the disubstituted 1,10-phenanthrolines. Under the reaction conditions (room temperature, dichloromethane/toluene mixture), we found that 4,7-diphenyl-1,10-phenanthroline (bathophenanthroline) did not react, fully recovering the starting compounds. However, in the presence of 2,9-dimethyl-1,10-phenanthroline (dmphen), two types of colorless crystals were successively obtained from the mother liquor. Monitoring of the reaction by 119Sn{1H} NMR in CD2Cl2 revealed the transformation of 1 into distannoxanes 2 and 4. However, we did not detect the formation of organotin species coordinated by dmphen ligands, as in the case of compounds 5, 6 and 7. This is probably due to steric hindrance caused by the presence of methyl substituents. The first type of crystals collected as long needles was characterized by X-ray diffraction analysis as being the organic salt [C14H13N2][CF3SO3] (dmphenHOTf). An Ortep representation is shown in Figure 10. The monoprotonation of dmphen led to the formation of the cation [C14H13N2]+. The positive charge is balanced by a triflate anion, which is also involved in hydrogen bonding to the cation via an N–H⋯ O interaction [N1⋯ O1 = 2.910 Å, N1–H1⋯ O1 = 157.63(14)°]. The crystal packing view of dmphenHOTf shows a folded sheet organization (Figure 11), stacked along the a-axis via π–π interactions between the aryl rings of [C14H13N2]+ with an interplanar distance of 3.449 Å, a centroid–centroid distance of 3.626 Å, and a slippage distance of 1.119 Å (slip angle of 17.98°). Recently, Assefa and Gore unintentionally obtained the same compound by adding 2,9-dimethyl-1,10-phenanthroline drop wise to a Eu(CF3O3S)3 solution [41]. In addition, several structures of 2,9-dimethyl-1,10-phenanthrolinium salts [C14H13N2][X] have already been solved. This is the case for [42], Cl− [43], [44], [45].
Ortep drawing of dmphenHOTf using the atom labeling scheme (30% probability thermal ellipsoids). The N–H⋯ O interaction is indicated by a red dotted line. Selected bond lengths (Å) and angles (°): N1–C5 1.371(3), N1–C1 1.336(3), N2–C6 1.355(3), N2–C12 1.327(3), S–O1 1.4472(17), S–O2 1.4306(19), S–O3 1.4359(17), S–C15 1.832(3), F1–C15 1.329(3), F2–C15 1.334(3), F3–C15 1.336(3); O1–S–C15 102.31(11), O3–S–O1 115.26(11), O3–S–C15 103.14(12), O2–S–O1 115.68(12), O2–S–O3 114.96(11), O2–S–C15 102.71(14), F2–C15–S 111.2(2), F2–C15–F3 107.5(2), F3–C15–S 111.46(18), F1–C15–S 111.25(18), F1–C15–F2 108.1(2), F1–C15–F3 107.2(2).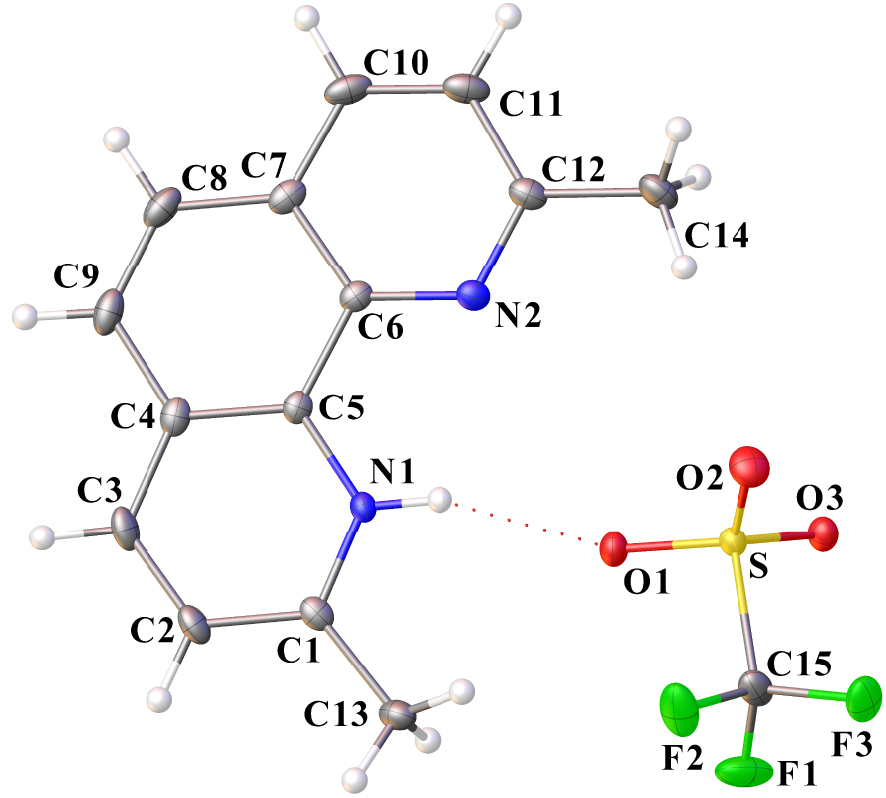
Organization of dmphenHOTf in the crystal lattice along the a-axis. Hydrogen atoms are omitted for clarity (Mercury representation [27], color code: N = blue, C = gray, O = red, S = yellow, F = green).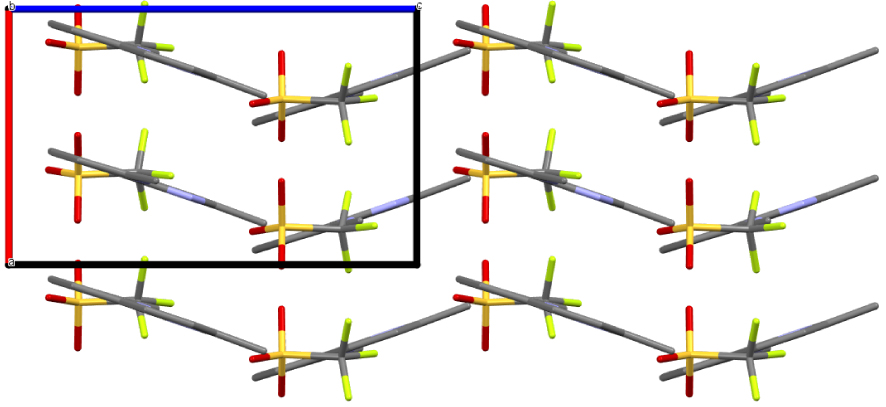
A second type of colorless crystals, exhibiting a different shape (prism), was collected from the filtrate of the solution from which dmphenHOTf crystals were initially obtained. Their composition consists of a monoprotonated dmphen molecule (dmphenH) co-crystallizing with a free dmphen molecule to form dmphenHOTf⋅dmphen. The two components interact through an N–H⋯ N hydrogen bond [N1⋯ N3 = 2.927(3) Å]. In fact, the hydrogen atom is split between the two nitrogen atoms [N1–H⋯ N3 = 152.78(14)°, N3–H⋯ N1 = 159.92(14)°]. Interestingly, the steric hindrance of the methyl substituents leads to a positioning close to orthogonality between the two heterocycles. Their arrangement can be described as a head–tail assembly. A dihedral angle of 77.74(3)° was determined between the two planes containing dmphenH and dmphe. The overall positive charge is compensated by the presence of a triflate anion. An Ortep view of dmphenHOTf⋅dmphen is shown in Figure 12. The crystal packing view, depicted in Figure 13, also shows that the dmphen rings are in π–π aromatic interaction and are organized in pairs. However, two distinctive stacking patterns can be observed for dmphen and dmphenH, respectively, characterized by interplanar distances of 3.631 and 3.353 Å, centroid–centroid distances of 3.762 Å and 4.445 Å, and slip distances of 0.984 Å (slip angle of 15.16°) and 2.918 Å (slip angle of 41.03°), respectively.
Ortep drawing of dmphenHOTf⋅dmphen using the atom labeling scheme (30% probability thermal ellipsoids). The N–H⋯ N interaction is indicated by a blue dotted line. Selected bond lengths (Å) and angles (°): C1–N1 1.336(3), C5–N1 1.366(3), C6–N2 1.366(3), C12–N2 1.324(3), C15–N3 1.335(3), C19–N3 1.367(3), C20–N4 1.357(3), C26–N4 1.330(3), C29–F1 1.334(3), C29–F2 1.333(3), C29–F3 1.340(3), C29–S 1.832(3), O1–S 1.4364(18), O2–S 1.4423(19), O3–S 1.4426(18); F2–C29–F3 107.3(2), F2–C29–F1 107.4(2), F2–C29–S 111.80(18), F3–C29–S 110.85(17), F1–C29–F3 107.1(2), F1–C29–S 112.11(17), O2–S–C29 102.19(12), O2–S–O3 114.84(11), O3–S–C29 103.17(11), O1–S–C29 103.78(11), O1–S–O2 115.20(12), O1–S–O3 115.13(12).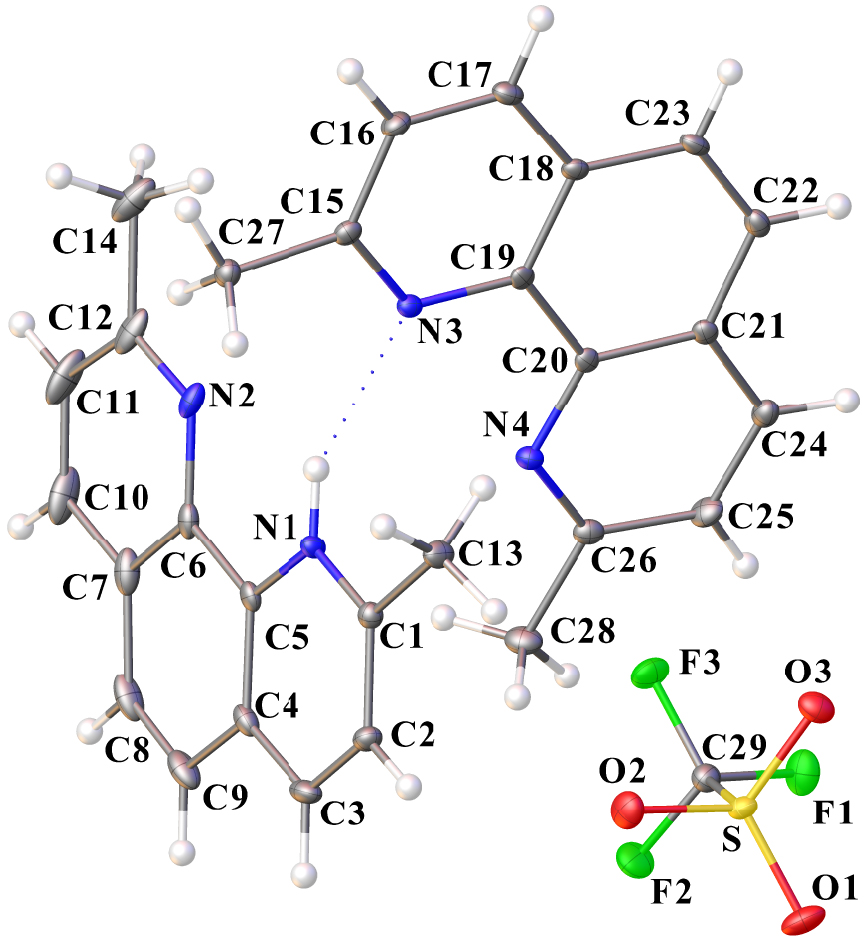
Crystal packing of dmphenHOTf⋅dmphen along the b-axis. Hydrogen atoms are omitted for clarity (Mercury representation [27], color code: N = blue, C = gray, O = red, S = yellow, F = green).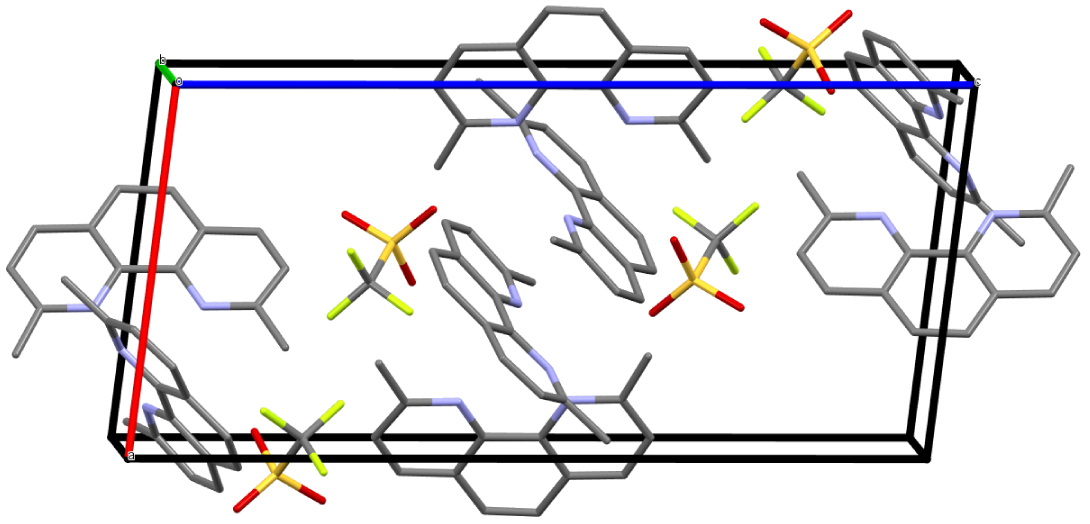
In the past, we have shown the possibility of accessing phenazinium and acridinium trifluoromethanesulfonate salts by reacting compound 1, at room temperature, in the presence of phenazine (phz) [12] and acridine (acr) [46], respectively. The isolation of original architectures based on molecular stacks driven by hydrogen and π–π interactions, has underlined the predisposition of N-heterocyclic molecules as suitable building-blocks. In our opinion, this approach, via the assistance of an organotin compound, could be seen as an innovative method of crystal engineering. It is successfully applied here to 1,10-phenanthroline and 2,9-dimethyl-1,10-phenanthroline, leading to new phenanthrolinium trifluoromethanesulfonate salts.
3. Conclusion and perspectives
In conclusion, the study of the reactivity between 1,10-phenanthroline (phen) and the complex [n-Bu2Sn(μ-OH)(H2O)(CF3SO3)]2 (1) led to the complete characterization of two new di-n-butyltin(IV) trifluoromethanesulfonates, 5 and 6, N,N-bis-chelated with phen ligands. Furthermore, using 119Sn(1H) NMR spectroscopy as an investigation probe under deuterated acetonitrile conditions, we were also able to highlight the formation of additional tin trifluoromethanesulfonate intermediates. Two, 2 and 4, exhibiting distannoxane-like frameworks but without phen-coordinated ligands, were clearly identified by comparison with previous work while investigations were conducted to clarify the unknown 119Sn(1H) NMR fingerprint attributed to 3. Crystallization attempts are underway to corroborate the suggested dinuclear structure. We also observed that the identity of the solvent used for the reaction had a notable impact on the intermediates formed. In the presence of dichloromethane, the solid-state structure of a mononuclear hydrated di-n-butyltin hydroxide, stabilized by a phen ligand and assigned to 7, was revealed. To the best of our knowledge, there are few comparable examples to date. Thus, new structural and spectroscopic insights into organotin(IV) trifluoromethanesulfonates have been obtained, thereby opening the way for further investigations. In the future, we plan to explore the catalytic properties of these compounds.
4. Experimental section
4.1. Materials and instrumentation
Organic solvents, dichloromethane (Carlo Erba, 99.5% purity), toluene (Acros, 99.99%), acetonitrile (99.9% purity), were refluxed over appropriate dessicants, distilled, and saturated with argon prior to use. Chemicals were purchased from Aldrich, Acros Organics, and Fluka and used without further purification. The starting compound [n-Bu2Sn(OH)(H2O)(CF3SO3)]2 (1) was synthesized from n-Bu2SnO (Acros, 98% purity) and trifluoromethanesulfonic acid (Fluka, 98% purity) in acetonitrile, according to a published method [8]. The 1H, 19F, 119Sn{1H}, and 13C{1H} NMR experiments were recorded on Bruker Avance 300 and 500 MHz spectrometers and calibrated with Me4Si, trifluoromethylbenzene, or Me4Sn as an internal standard. Chemical shift 𝛿 values are given in ppm. FT-IR spectra were recorded on a Bruker Alfa spectrometer equipped with a Specac Golden Gate™ ATR device. ESI-MS spectra were obtained on a Bruker micro Q-TOF instrument using acetonitrile, dichloromethane and methanol mobile phases. Elemental analyses (C, H, N, S) were performed at the Institut de Chimie Moléculaire de l’Université de Bourgogne, Dijon.
4.2. Preparation of [n-Bu2Sn(OH)(phen)]2[CF3SO3]2 (5) and [n-Bu2Sn(Phen)2][CF3SO3] (6) from [n-Bu2Sn(OH)(H2O)(CF3SO3)]2 (1) and 1,10-phenanthroline
Two molar equivalents of 1,10-phenanthroline (C8H8N2, Sigma-Aldrich, 99% purity) (0.130 g, 0.72 mmol) were added to a colourless solution of [n-Bu2Sn(OH)(H2O)(CF3SO3)]2 (1) (0.300 g, 0.360 mmol) in a mixture of dichloromethane/toluene (15 mL/10 mL). The reaction medium is stirred for 3 h at room temperature leading to a clear pinkish solution. A precipitate (0.160 g) was collected after one week of evaporation at room temperature. This corresponds to the formation of compound 6, which was then recrystallized as fine needles in dichloromethane/toluene. Compound 5 was obtained a few days later from the reaction filtrate as colorless, parallelepipedal crystals (0.050 g). The use of four equivalents of 1,10-phenanthroline gave exclusive access to compound 6.
5: 1H NMR (300 MHz, CD2Cl2, 301 K): 𝛿0.5–2.1 (m, 36H, Ant), 3.78 (br, 2H), 8.02 (m, 4H), 8.11 (s, 4H), 8.66 (m, 4H), 9.37 (m, 4H); 13C{1H} NMR (75 MHz, CD2Cl2, 300 K): 𝛿13.4, 26.5, 27.6, 120.8 (q, 1 JCF = 320 Hz), 125.7, 127.8, 130.1, 139.9, 141.7, 149.9; 19F NMR (282 MHz, CD2C12, 301 K): 𝛿 −78.5 (s, ); 119Sn{1H} (186 MHz, CD3CN, 298 K): 𝛿 −226; 119Sn{1H} (186 MHz, acetone-d6, 298 K): 𝛿 −150; ESI-HRMS (+): m∕z 431.1133 Da (100%) [M–OH–phen–n-Bu2Sn]+ (C20H27N2OSn, calc.: 431.1145 Da); FT-IR (ATR, cm−1): 3321, 3081, 3066, 2956, 2929, 2871, 1626, 1575, 1589, 1543, 1518, 1427, 1379, 1282, 1237, 1223, 1155, 1027, 854, 731, 717, 683, 633, 573, 515. Anal. Calc. For C42H54F6N4O8S2Sn2 (1158.44): C, 43.55; H, 4.70; N, 4.84; S, 5.54. Found: C, 43.42; H, 5.45; N, 4.90; S, 4.32%.
6: 1H NMR (500 MHz, acetone-d6, 298 K): 0.65 (t, J = 7.3 Hz, 6H), 1.20 (m, 4H), 1.34 (m, 4H), 2.10 (m, 4H), 8.26 (m, 4H), 8.50 (s, 4H), 9.18 (m, 8H); 13C{1H} NMR (125 MHz, acetone-d6, 298 K): 𝛿13.1, 26.5, 27.8, 27.81, 121.9 (q, 1 JCF = 321 Hz), 127.20, 128.7, 131.0, 138.5, 142.9, 149.0; 19F NMR (470 MHz, acetone-d6, 298 K): 𝛿 −78.8 (s, ); 119Sn{1H} (186 MHz, acetone-d6, 298 K): 𝛿 −231; ESI-HRMS (+): m/z 445.1305 Da (100%) [M–phen+OCH3]+ (C21H29N2OSn, calc.: 445.1301 Da), m∕z 431.11559 Da (80%) [M–phen+OH]+ (C20H27N2OSn, calc.: 431.11454 Da); FT-IR (ATR, cm−1): 3104, 3070, 2960, 2925, 2870, 2860, 1630, 1589, 1543, 1523, 1431, 1259, 1223, 1153, 1027, 868, 720, 633, 573, 516. Anal. Calc. For C34H34F6N4O6S2Sn (891.49): C, 45.81; H, 3.84; N, 6.28; S, 7.19 Found: C, 45.56; H, 3.38; N, 6.30; S, 7.29%.
4.3. Isolation and characterization of 3
The protocol used the same conditions as those used in the 119Sn{1H} NMR experiment shown in Figure 5d. One molar equivalent of 1,10-phenanthroline (0.065 g, 0.360 mmol) was added to a colourless solution of [n-Bu2Sn(OH)(H2O)(CF3SO3)]2 (1) (0.300 g, 0.360 mmol) in a mixture of acetonitrile/toluene (15 mL/10 mL). The solution was progressively enriched in compound 3 by eliminating species 2, 3, and 4 by successive crystallizations, and the filtrate was stored each time at −20 °C. 3 was finally obtained after three cycles (119Sn{1H} NMR monitoring) and after the total evaporation of the solvents leading to a colorless pasty solid.
3: 1H NMR (499 MHz, CD3CN, 298 K): 0.61 (t, J = 7.0 Hz, 6H), 1.09 (m, 4H), 1.17 (m, 4H), 2.08 (m, 4H), 8.36 (m, 2H), 8.41 (s, 2H), 9.13 (m, 2H), 9.53 (m, 2H); 13C{1H} NMR (125 MHz, CD3Cl2, 298 K): 13.1, 25.9, 27.3, 33.9, 120.55 (1 JCF = 319 Hz), 127.2, 128.6, 130.9, 139.4, 143.0, 150.3; 19F NMR (470 MHz, CH2Cl2, 298 K): 𝛿 −78.4 (s, CFSO3); 119Sn{1H} (149 MHz, CD3CN, 298 K): 𝛿 −266, 119Sn{1H} (186 MHz, CD2Cl2, 298 K): 𝛿 −288; ESI MS (+): m∕z 1275.07507 Da (100%) [M]+ (C43H52O9N4F9S3Sn2, calc.: 1275.07912 Da), m∕z 893.0887 Da (67%) [M+H–phen–n-Bu2Sn]+ (C34H35O6N4F6S2Sn, calc.: 893.09201 Da), m∕z 563.06134 Da (z = 1, 67%) [n-Bu2Sn(phen)(CF3SO3)]+ (C21H26O3N2F3SSn, calc.: 563.06328 Da); FT-IR (ATR, cm−1): 3091, 3071, 2960, 2931, 2871, 1631, 1607, 1586, 1525, 1264, 1200, 1162, 1016, 866, 723, 691, 631, 573, 513, 426. Anal. Calc. For C44H52F12N4O12S4Sn2 (1422.57): C, 37.15; H, 3.68; N, 3.94; S, 9.02 Found: C, 36.43; H, 3.74; N, 4.13; S, 8.16%.
4.4. Isolation and characterization of phenHOTf
Small crystals of phenHOTf were obtained from the filtrate of the mother-liquor from which compound 7 had been isolated.
phenHOTf: 1H NMR (500 MHz, CD2Cl2, 298 K): 𝛿8.03 (dd, 1H, J = 8.17, 5.02 Hz), 8.08 (s, 1H), 8.16 (brs, 2H), 8.71 (d, 1H, J = 8.12 Hz), 8.79 (d, 1H, J = 7.30 Hz), 9.36 (d, 1H, J = 4.35 Hz), 9.64 (brs, 1H); 19F NMR (470 MHz, CD2Cl2, 298 K): 𝛿 −78.57 (s, CFSO3); 13C{1H} NMR (125 MHz, CD2Cl2, 298 K): 120.4 (1 JCF = 320 Hz), 125.6, 126.6, 127.5, 129.8, 130.3, 137.8, 141.4, 147.8, 150.5; FT-IR (ATR, cm−1): 3100, 3070, 3043, 2960, 2930, 2872, 1598, 1542, 1525, 1497, 1471, 1438, 1283, 1223, 1152, 10256, 847, 719, 632, 571, 514, 463.
4.5. Isolation and characterization of dmphenHOTf and dmphenHOTf⋅dmphen
Four molar equivalents of 2,9-dimethyl-1,10-phenanthroline (C10H12N2, Sigma-Aldrich, 99% purity) (0.200 g, 0.96 mmol) were added to a colourless solution of [n-Bu2Sn(OH)(H2O)(CF3SO3)]2 (1) (0.200 g, 0.24 mmol) in a dichloromethane/toluene mixture (10 mL/5 mL). The reaction medium is stirred for 3 h at room temperature in ambient air. In the following days, dmphenHOTf crystals first grew and then, after filtration of the mother-liquor, new crystals characterized as dmphenHOTf⋅dmphen were collected.
dmphenHOTf: 1H NMR (300 MHz, CD2Cl2, 299 K): 𝛿8.65 (d, 2H, J = 8.4 Hz), 8.08 (s, 2H), 7.90 (d, 2H, J = 8.4 Hz), 7.66 (br, 1H), 3.18 (s, 6H, CH3); 19F NMR (470 MHz, CD2Cl2, 298 K): 𝛿 −78.93 (s, CFSO3); 13C{1H} NMR (125 MHz, CD2Cl2, 298 K): 23.3, 121.2 (1 JCF = 320 Hz), 126.8, 126.9, 128.2, 137.4, 141.5, 159.9; FT-IR (ATR, cm−1): 3174, 3111, 3046, 3016, 2980, 1635, 1604, 1533, 1503, 1463, 1278, 1253, 1246, 1222, 1142, 1032, 813, 719, 691, 677, 631, 573, 542, 513; Anal. Calc. For C15H13F3N2O3S (358.34): C, 50.28; H, 3.66; N, 7.829; S, 8.95. Found: C, 49.96; H, 4.05; N, 7.82; S, 6.29%.
dmphenHOTf⋅dmphen: 1H NMR (500 MHz, CD2Cl2, 298 K): 𝛿8.33 (d, 4H, J = 8.30 Hz), 7.84 (s, 4H), 7.57 (d, 4H, J = 8.38 Hz), 7.41 (br, 1H), 2.59 (s, 12H, CH3); 19F NMR (470 MHz, CD2Cl2, 298 K): 𝛿–78.92 (s, CFSO3); 13C{1H} NMR (125 MHz, CD2Cl2, 298 K): 23.6, 121.3 (1 JCF = 321 Hz), 125.4, 126.4, 127.96, 139.08, 141.7, 159.1; FT-IR (ATR, cm−1): 2958, 2929, 2858, 1636, 1626, 1605, 1595, 1541, 1499, 1466, 1353, 1257, 1221, 1152, 1027, 853, 754, 735, 724, 634, 570, 546, 515, 441; Anal. Calc. For C29H25F3N4O3S (566.59): C, 61.47; H, 4.45; N, 9.89; S, 5.66. Found: C, 60.93; H, 4.80; N, 10.08; S, 5.09%.
4.6. X-ray diffraction analysis and refinement
Crystallographic data and structure refinement details for 5, 6, 7, phenHOTf, dmphenHOTf⋅ and dmphenHOTf⋅dmphen are reported and detailled in Supporting information – Crystallographic Data.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Funding
The authors are grateful to the University of Burgundy-Dijon and the CNRS for their constant financial support.
Acknowledgements
The authors would particularly like to thank Dr. Q. Bonin, Ms M.-J. Penouilh (ESI-HRMS) and Ms T. Régnier (elemental analysis), as well as the Professor Klaus Jurkschat and the second anonymous reviewer for their corrections and suggestions, which significantly improved the initial manuscript.
Supplementary data
Supporting information for this article is available on the journal’s website under https://doi.org/10.5802/crchim.260 or from the author.
Additional spectroscopic data (1H, 19F, 13C{1H}, 119Sn{1H}, FT-IR, and ESI-MS spectra; Figures S1–S21) related to complexes 5, 6, 7 are given in Supporting Information—Spectroscopic Data. Crystallographic data regarding the X-ray structures reported in this study (compounds 5, 6, 7, phenHOTf, dmphenHOTf, and dmphenHOTf⋅dmphen) are available in Supporting Information—Crystallographic Data.
CCDC 2254272 (5), 2254273 (6), 2254274 (7), 2254275 (phenHOTf), 2254276 (dmphenHOTf), and 2254277 (dmphenHOTf⋅dmphen) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.