

1 Introduction
In the past, several examples of organotin(IV) trifluoromethanesulfonate complexes have been synthesized and structurally characterized by single-crystal X-ray diffraction analysis. The most recent review in this research area was published by J. Beckmann in 2005 [1]. From a synthetic point of view, organotin(IV) trifluoromethanesulfonates are obtained either by reacting organotin oxide with trifluoromethanesulfonic acid (HO3SCF3, HOTf) [2], or alternatively organotin chloride (R(4−x)SnClx) with silver(I) trifluoromethanesulfonate (AgO3SCF3, Ag(OTf)) [3]. The resulting solid-state reports highlight a large structural versatility for F3CSO3− which can be ionic or non-ionic, describing then various coordination modes to tin atoms: mono-, bi- and tridentate as well as terminal, pseudo-terminal, and bridging [4]. Furthermore, organotin(IV) trifluoromethanesulfonate complexes can be used as pre-catalysts or catalysts for several organic reactions, such as the Mukaiyama aldol reaction [5], the Robinson annulation [6], the acetylation of alcohols [7], the transesterification of dimethyl carbonate with phenol [8], and the direct synthesis of dimethyl carbonate from carbon dioxide and methanol [9].
Parallel to our investigations on carbon dioxide activation by organotin(IV) compounds [10], we previously reported the reaction of phenazine (Phz) and di-n-butyltin(IV) trifluoromethanesulfonato complexes affording phenazinium trifluoromethanesulfonate salts. In the solid state hydrogen bonding and π-π stacking interactions lead to the self-assembly of supramolecular edifices [4b,11]. Herein we report the application of this unusual crystal engineering method with acridine (Acr), and three new supramolecular architectures. They are based on acridinium stacks promoted by the dimeric hydroxo di-n-butylstannane trifluoromethanesulfonato complex [n-Bu2Sn(OH)(H2O)(CF3SO3)]2 (1) (Scheme 1).
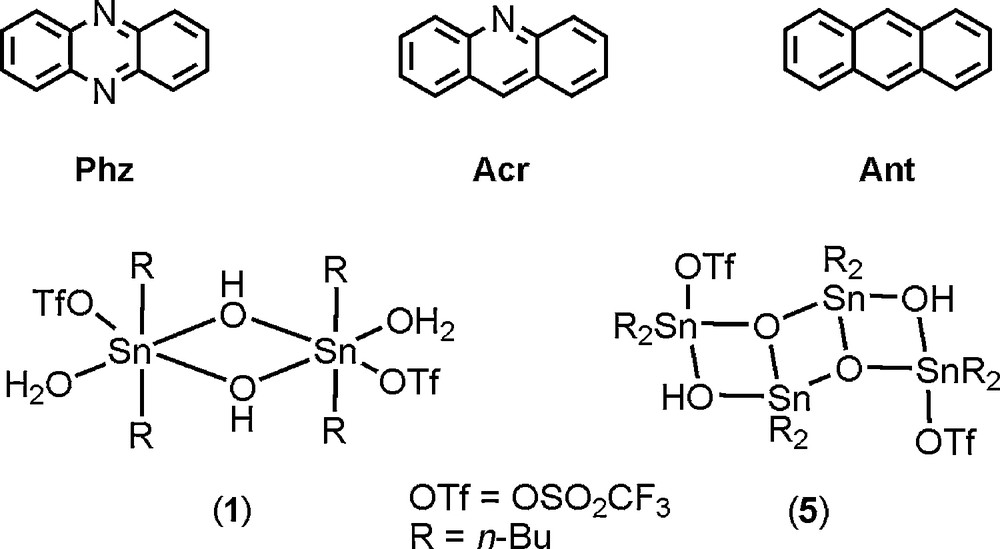
Organic reactants and organotin(IV) compounds relating to this study.
2 Experimental
2.1 Materials and instrumentation
Organic solvents, dichloromethane (Carlo Erba, 99.5% purity), toluene (Acros, 99.99%) and n-pentane (Carlo Erba, 99% purity) were refluxed over appropriate dessicants, distilled, and saturated with argon prior to use. Chemicals were purchased from Acros Organics and Fluka, and used without further purification. The starting compound [n-Bu2Sn(OH)(H2O)(CF3SO3)]2 (1) was synthesized from n-Bu2SnO (Acros, 98% purity) and trifluoromethanesulfonic acid (Fluka, 98% purity) according to a published method [2a]. The 1H, 19F, 119Sn{1H}, and 13C{1H} NMR experiments were run at 298 K on a Bruker Avance 300 spectrometer (at the Plateforme d’Analyses Chimiques et de Synthèse Moléculaire de l’Université de Bourgogne – PACSMUB), at 300.131, 282.404, 111.910, and 75.475 MHz, respectively, and calibrated with Me4Si, trifluoromethylbenzene or Me4Sn as an internal standard. Chemical shift δ values are given in ppm. IR spectra were recorded on a Bruker Vector 22 equipped with a Specac Golden Gate™ ATR device. Melting points were determined in capillaries on an Electrothermal 9100 apparatus. Elemental analyses (C, H, N) were performed at the Institut de Chimie Moléculaire de l’Université de Bourgogne, Dijon.
2.2 Isolation of [C13H10N·H2O][CF3SO3] (2) and [C13H10N][CF3SO3] (3)
Three molar equivalents of acridine (C13H9N, Acros, 98% purity) (0.237 g, 1.322 mmol) were added to a colourless solution of [n-Bu2Sn(OH)(H2O)(CF3SO3)]2 (1) (0.360 g, 0.441 mmol) in dichloromethane (15 mL) at room temperature. After stirring for two hours, fresh toluene (10 mL), used as co-solvent for crystallization, was then introduced to the resulting orange solution. After few days at room temperature, colourless single-crystals (squares and rods) were first obtained and attributed according to their IR fingerprints to oxo-cluster 5 which crystallized in fact in two polymorphic forms, 5a (square) and 5b (rod). After separation, two new types of coloured crystals, yellow needles and yellow squares, were grown from the mother-liquor by slow evaporation. The crystals were then filtered off, washed with portions of n-pentane, dried in air at room temperature, collected separately and finally characterized as salt 2 and 3, respectively.
[C13H10N·H2O][CF3SO3] (2) (mp 202 °C). 1H NMR (CD2Cl2): δ 15.94 (brs, 1H, AcrH), 9.59 (s, 1H, AcrH), 8.70 (d, J = 8.9 Hz, 2H, AcrH), 8.38 (d, J = 8.5 Hz, 2H, AcrH), 8.30 (m, 2H, AcrH), 7.94 (m, 2H, AcrH), 1.56 (brs, 2H, H2O). 19F NMR (CD2Cl2): δ –78.97 (s, CF3SO3−). 13C{1H} NMR (CD2Cl2): δ 148.5, 140.4, 138.4, 129.7, 128.8, 126.5, 121.0 (q, 1JC-F = 319 Hz, CF3SO3−), 120.8. Anal. Calc. For C14H12F3NO4S (347.31): C, 48.41; H, 3.48; N, 4.03. Found: C, 48.30; H, 3.29; N, 4.30%. Main IR bands (ν, cm−1): 3387, 3088, 3048, 2522, 2052, 1643, 1486, 1467, 1402, 1382, 1321, 1246, 1224, 1166, 1152, 1143, 1025, 902, 862, 825, 778, 743, 675.
[C13H10N][CF3SO3] (3) (mp 208 °C). 1H NMR (CD2Cl2): δ 15.82 (brs, 1H, AcrH), 9.62 (s, 1H, AcrH), 8.65 (d, J = 8.9 Hz, 2H, AcrH), 8.38 (d, J = 8.5 Hz, 2H, AcrH), 8.27 (m, 2H, AcrH), 7.92 (m, 2H, AcrH), 19F NMR (CD2Cl2): δ –78.94 (s, CF3SO3−).13C{1H} NMR (CD2Cl2): δ 148.5, 140.4, 138.4, 129.7, 128.8, 126.5, 121.0 (q, 1JC-F = 319 Hz, CF3SO3−), 120.8. Anal. Calc. For C14H10F3NO3S (329.29): C, 51.06; H, 3.06; N, 4.25. Found: C, 51.18; H, 2.96; N, 4.27%. Main IR bands (ν, cm−1): 3090, 3048, 3011, 2916, 2888, 2837, 2777, 1647, 1626, 1587, 1481, 1464, 1425, 1401, 1284, 1221, 1156, 1026, 776, 742, 674, 632, 601, 572, 515.
2.3 Isolation of {([C13H10N][CF3SO3])2·C14H10} (4)
Three molar equivalents of anthracene (C14H10, Acros, 99% purity) (0.466 g, 2.607 mmol) and acridine (C13H9N, Acros, 98% purity) (0.467 g, 2.607 mmol) were successively added to a colourless solution of [n-Bu2Sn(OH)(H2O)(CF3SO3)]2 (1) (0.709 g, 0.869 mmol) in dichloromethane (25 mL) at room temperature. After stirring for two hours, fresh toluene (15 mL), used as co-solvent for crystallization, was then introduced to the resulting dark red solution. After few days at room temperature, colourless square single-crystals were first obtained, corresponding to organotin(IV) complex 5a. After separation, dark red square single crystals were grown from the mother liquor by slow evaporation. The crystals were filtered off, washed with portions of n-pentane, dried in air at room temperature, and finally characterized as salt {([C13H10N][CF3SO3])2·C14H10} 4 (mp 178 °C). 1H NMR (CD2Cl2): δ 15.88 (brs, 2H, AcrH), 9.59 (s, 2H, AcrH), 8.69 (d, J = 8.9 Hz, 4H, AcrH), 8.47 (s, 2H, Ant), 8.37 (d, J = 8.5 Hz, 4H, AcrH), 8.29 (m, 4H, AcrH), 8.05 (m, 4H, Ant), 7.94 (m, 4H, AcrH), 7.50 (m, 4H, Ant). 19F NMR (CD2Cl2): δ –78.94 (s, CF3SO3−). 13C{1H} NMR (CD2Cl2): δ 148.5 (AcrH), 140.4 (AcrH), 138.4 (AcrH), 132.0 (Ant), 129.7 (AcrH), 128.8 (AcrH), 128.4 (Ant), 126.5 (AcrH), 126.4 (Ant), 125.7 (Ant), 121.0 (q, 1JC-F = 319 Hz, CF3SO3−), 120.7 (AcrH). Anal. Calc. For C42H30F6N2O6S2 (836.82): C, 60.28; H, 3.61; N, 3.35. Found: C, 59.90; H, 3.16; N, 3.52%. Main IR bands (ν, cm−1): 3302, 3257, 3208, 3148, 3103, 3059, 3024, 2989, 2948, 2872, 1650, 1476, 1427, 1257, 1224, 1154, 1028, 873, 790, 757, 727, 618, 574, 516.
2.4 X-Ray diffraction analysis
Diffraction data were collected from suitable crystals on a Bruker Nonius ApexII CCD (Mo Kα radiation, λ = 0.71073 Å). The structures were solved using Charge Flipping Algorithm Methods (SUPERFLIP) [12] and refined with full-matrix least-squares methods based on F2 (SHELX-97) [13] with the aid of the WINGX program (14]. All non-hydrogen atoms were refined with anisotropic thermal parameters and hydrogen atoms were included in their calculated positions and refined with a riding model. Programs used for the representation of the molecular and crystal structures: ORTEP [15], DIAMOND [16], and MERCURY [17]. Crystallographic data and structures refinement details or 2, 3 and 4 are summarized in Table 1.
Crystal and structure refinement data for 2, 3 and 4.
| Compounds | 2 | 3 | 4 |
| Empirical formula | C13H10N·H2O, CF3SO3 | C13H10N, CF3SO3 | C13H10N·0.5 C14H10, CF3SO3 |
| Formula weight (g.mol−1) | 347.31 | 329.29 | 418.40 |
| Temperature (K) | 115(2) | 115(2) | 115(2) |
| Crystal system | Orthorhombic | Triclinic | Triclinic |
| Space group | Pnma | ||
| a (Å) | 11.6289(7) | 7.0899(3) | 9.0072(4) |
| b (Å) | 6.6998(5) | 9.6716(4) | 10.1453(4) |
| c (Å) | 18.6130(11) | 10.5407(4) | 11.2361(5) |
| α (°) | 74.269(2) | 69.110(2) | |
| β (°) | 78.855(2) | 81.158(2) | |
| γ (°) | 81.869(2) | 69.894(2) | |
| Volume (Å3) | 1450.16(16) | 679.55(5) | 900.24(7) |
| Z | 4 | 2 | 2 |
| ρcalc (g.cm−3) | 1.591 | 1.609 | 1.544 |
| μ (mm−1) | 0.277 | 0.286 | 0.234 |
| F(000) | 712 | 336 | 430 |
| Crystal size (mm3) | 0.25 × 0.05 × 0.05 | 0.22 × 0.13 × 0.10 | 0.42 × 0.08 × 0.04 |
| sin(θ)/λ max (Å−1) | 0.65 | 0.65 | 0.65 |
| Index ranges | h: −14; 9 | h: −9; 9 | h: −11; 11 |
| k: −7; 8 | k: −12; 12 | k: −13; 13 | |
| l: −22; 24 | l: −13; 12 | l: −14; 14 | |
| Reflections collected | 5343 | 5501 | 12073 |
| Rint | 0.0502 | 0.0159 | 0.0582 |
| Reflections with I ≥ 2σ(I) | 1360 | 2852 | 3466 |
| Data/restraints/parameters | 1740/0/133 | 3074/0/199 | 4074/0/262 |
| Final R indices [I ≥ 2σ(I)] | R1a = 0.0487 | R1a = 0.0388 | R1a = 0.0471 |
| wR2b = 0.0911 | wR2b = 0.0995 | wR2b = 0.1016 | |
| R indices (all data) | R1a = 0.0703 | R1a = 0.0420 | R1a = 0.0581 |
| wR2b = 0.1013 | wR2b = 0.1018 | wR2b = 0.1085 | |
| Goodness-of-fitc on F2 | 1.143 | 1.104 | 1.062 |
| Largest difference peak and hole (e Ǻ−3) | 0.307 and –0.320 | 0.407 and −0.310 | 0.348 and −0.462 |
| CCDC deposition no. | 891927 | 891928 | 891929 |
a R1 = Σ(||Fo|–|Fc||)/Σ|Fo|.
b wR2 = [Σw(Fo2–Fc2)2/Σ[w(Fo2)2]1/2 where w = 1/[σ2(Fo2 + (0.0107P)2 + 1.9231P] for 2, w = 1/[σ2(Fo2 + (0.0347P)2 + 0.5841P] for 3, w = 1/[σ2(Fo2 + (0.0253P)2 + 1.0201P] for 4, where P = (Max(Fo2.0) + 2Fc2)/3.
c S = [Σw(Fo2–Fc2)2/(n–p)]1/2 (n = number of reflections, p = number of parameters).
Crystallographic data for the structures reported in this paper have been deposited at the Cambridge Crystallographic Data Centre, CCDC, No. CCDC 891927 for 2, No. CCDC 891928 for 3, and No. CCDC 891929 for 4. Copies of the data may be obtained free of charge from the Director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: int Code +44 1223 336 033; e-mail: deposit@ccdc.cam.ac.uk or http://www.ccdc.cam.ac.uk).
3 Results and discussion
Synthetic pathways of compounds 2-4 are summarized in Scheme 2.
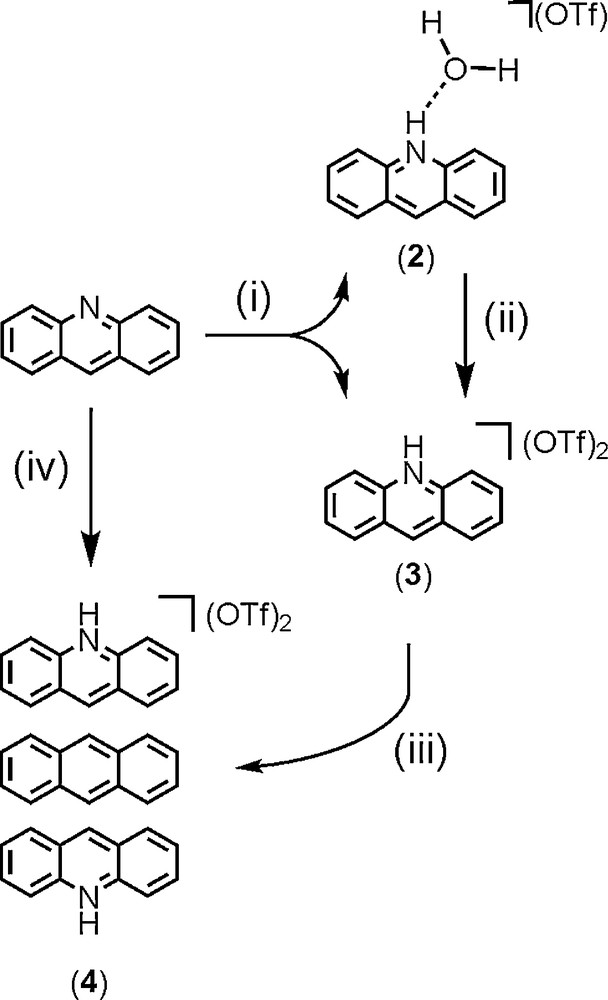
Reagents and conditions: (i) Complex 1, CH2Cl2, room temperature (RT), crystallisation from CH2Cl2/toluene; (ii) a) molecular sieves, CH2Cl2/toluene, b) solventless conditions, 100 °C, under vacuum; (iii) Ant, CH2Cl2/toluene, RT; (iv) Ant, complex 1, CH2Cl2, RT, crystallisation from CH2Cl2/toluene.
3.1 Isolation and crystal structures of acridinium salts 2 and 3
The strategy recently developed for the design of phenazinium-based stacks [4b,11] was applied to acridine. Thus, the reaction of the dimeric hydroxo di-n-butylstannane trifluoromethanesulfonato complex [n-Bu2Sn(μ-OH)(H2O)((1-O3SCF3)]2 (1) with acridine (C13H9N, Acr) (1:3 molar ratio) in dichloromethane at room temperature led to colourless crystals of the tetra-n-butyldistannoxane trifluoromethanesulfonato cluster (5). Compound 5 could be isolated in two polymorphic forms 5a and 5b which crystallized in two distinct crystal systems (triclinic- and monoclinic-P21/n, respectively), and with specific crystal habits (square and rod) that easily facilitate their visual identification [4b]. According to the infrared fingerprints recorded on crystals, and which are also characteristic for each polymorph (shown in the Supporting Information, Fig. S1), we can unambiguously conclude that both structures of 5 are formed successively during the reaction. Moreover, the monitoring of the reaction by 119Sn{1H} spectroscopy showed, after addition of three molar equivalents of acridine to a CD2Cl2 solution of 1, the predominant formation of the pair of the characteristic resonances of the distannoxane species 5a at δ = –135 and −143 ppm (Fig. S2). Upon crystallization of 5a and 5b, and separation by filtration, two new types of yellow crystals were grown together from the mother liquor. They were collected as needles and plates, and were identified as the acridinium trifluoromethanesulfonate salts [C13H10N]+[CF3SO3]–, 2 and 3, respectively. From a mechanistic point of view, the removal of protons from 1 by acridine molecules leads to the in situ formation of acridinium cations (AcrH), and promotes the rearrangement of organotin species into tetranuclear oxo-cluster 5. This subsequently led to the release of CF3SO3−, which ultimately compensates the positive charge of AcrH.
The difference between the acridinium salts 2 and 3 comes exclusively from the presence of a water molecule in the crystal lattice of one of the two salts. Indeed, the infrared spectrum recorded from 2 exhibits the characteristic vibration bands of the trifluoromethasulfonate anions (in the stretching region between 1000 and 1300 cm−1 [18], and also observed for 3), and a broad elongation band at 3387 cm−1. This band is a strong evidence of the presence of water. It is not present in the spectrum of 3. It can be attributed to the σ(OH) vibration. The 1H NMR spectra of 2 and 3 corroborate this observation (IR and NMR spectra are shown in the Supporting Information). The X-ray crystallographic analysis of 2 confirms the hydrogen bond occurring between the NH group of the acridium cation (AcrH) and one molecule of water [ Å and 179.4°]. The acridinium and water molecules are located in the same mirror plane. Furthermore, the water molecule is also in hydrogen bonding interaction with one oxygen atom of the trifluoromethanesulfonate anion [ Å and 177.3°]. An ORTEP view of 2 is shown in Fig. 1a together with selected bond distances and angles. Interestingly, and from a supramolecular point of view, the water molecule in each [C13H10N·H2O][CF3SO3] unit is also implicated in a third hydrogen-bonding interaction, with an identical distance that previously [ Å], with an oxygen atom of an adjacent trifluoromethanesulfonate anion, leading to the formation of an one-dimensional infinite polymer chain propagating along the b axis, and which consists of building blocks as repeating unit. The AcrH cations are orthogonally positioned along this chain, parallel to plane (010) and in an isotactic arrangement. As a result of the self-assembly induced by π-π interactions between anti-parallel acridinium rings, two polymeric chains are fitted into each other. It leads to a 2-D architecture which can be compared to a ladder structure. The construction of the resulting supramolecular edifice is summarized in Scheme 3 and a view of the crystal packing is shown in Fig. 1b. The interplanar and the centroid-to-centroid distances between parallel AcrH are 3.35 and 4.35 Å, respectively. The difference in these two distances indicates that acridinium rings are strongly slipped. The slip angle (angle between the normal to the planes and the centroid–centroid vector) is 39.75°, corresponding to a slippage distance of 2.79 Å. Typically for such interactions, the interplanar distance between the arene planes is commonly found around 3.3 to 3.8 Å [19]. Interestingly, the organization of the crystal packing described for 2 is comparable to ionic supramolecular architectures reported in the past by Wolf and Mei, and prepared from acridine and dicarboxylic acids [20].
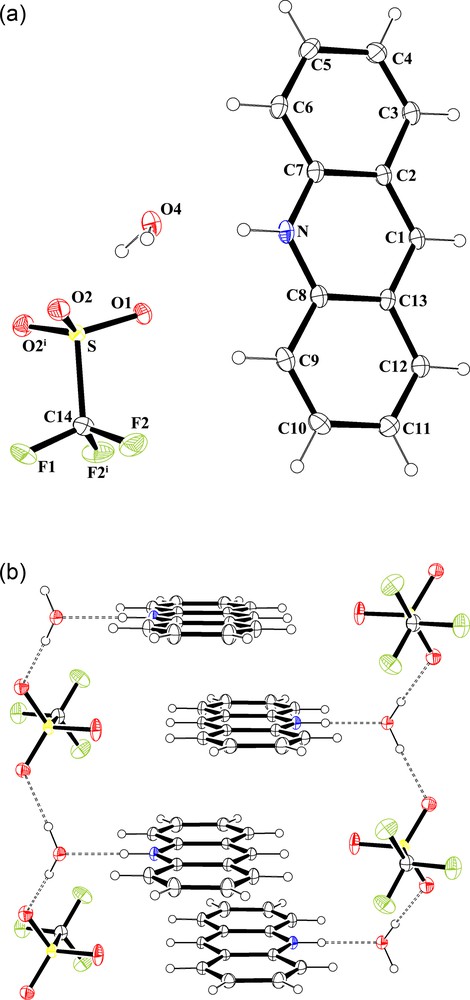
a: molecular structure of [C13H10N·H2O][CF3SO3] (2) showing the atom labelling scheme (ORTEP view). Colour code: sulphur: yellow; oxygen: red; nitrogen: blue; fluoride: green; carbon: grey; hydrogen: white (online). Selected bond lengths (A) and angles (°): NC7 1.350(4), NC8 1.360(4), C1C2 1.388(4), C1C13 1.393(4), C2C3 1.421(5), C2C7 1.427(4), C3C4 1.359(5), C4C5 1.417(5), C5C6 1.360(5), C6C7 1.409(5), C8C9 1.408(5), C8C13 1.426(4), C9C10 1.360(5), C10C11 1.432(5), C11C12 1.353(5), C12C13 1.425(5), SO1 1.435(2), SO2 1.445(2), SC14 1.825(4), F1C14 1.327(4), F2C14 1.323(3); C7NC8 123.6(2), C2C1C13 121.1(2), C1C2C3 123.3(3), C1C2C7 118.7(3), C3C2C7 118.0(3), C4C3C2 120.7(3), C3C4C5 120.3(3), C6C5C4 121.3(3), C5C6C7 119.3(3), NC7C6 120.6(3), NC7C2 119.0(3), C6C7C2 120.4(3), NC8C9 120.9(3), NC8C13 118.8(3), C9C8C13 120.4(3), C10C9C8 119.5(3), C12C11C10 120.3(3), C11C12C13 120.5(3), C1C13C12 123.0(3), C1C13C8 118.7(3), C12C13C8 118.3(3), O1SO2 115.39(9), O1SC14 103.67(16), O2SC14 103.17(10), F2C14F1 108.0(2), F2C14S 111.05(18), F1C14S 110.6(2). Symmetry transformations used to generate equivalent atoms (i): x,–y + 3/2,z; b: the crystal packing of 2 in the unit cell (DIAMOND representation). Hydrogen bonding interactions [ and ] are shown by dashed lines. Colour code: sulphur: yellow; oxygen: red; nitrogen: blue; fluoride: green; carbon: grey; hydrogen: white (online).
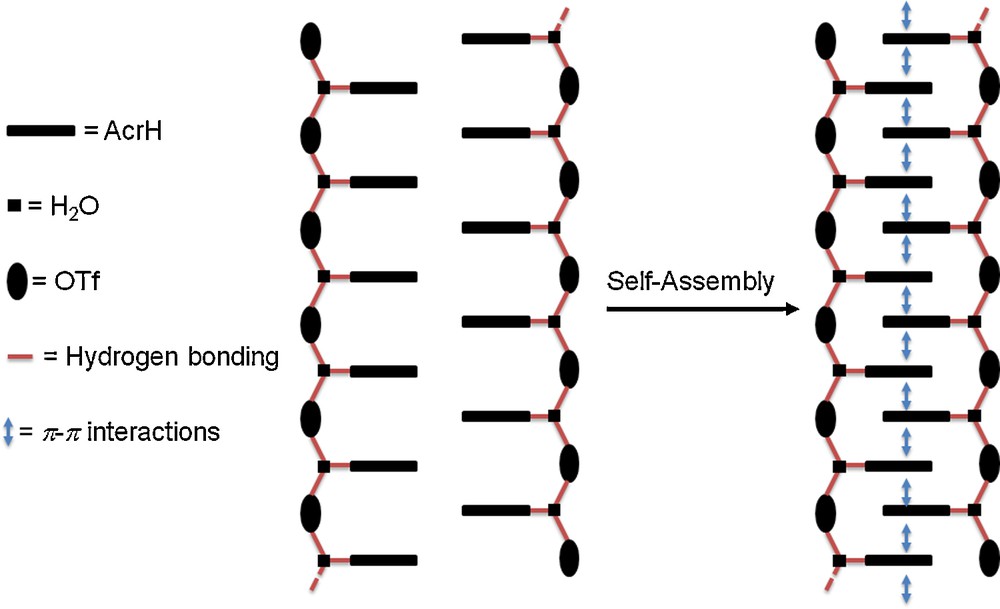
Schematic representation of the supramolecular construction of 2.
Salt 3 corresponds to the dehydrated form of 2. Initially, both salts crystallized together from the same batch but they are only distinguished by their respective shape (needles for 2 and plates for 3). However, the conversion of 2 into 3 occurs when molecular sieves were added to a dichloromethane solution of 2 or alternatively when 2 is heated at the solid state under vacuum at 100 °C for 1 hour (Scheme 2, pathway (ii)). On the basis on an X-ray crystallographic analysis, the molecular structure of 3 can be described as constituted by an acridium cation, AcrH [C13H10N]+, and by a trifluoromethanesulfonate anion [CF3SO3]− in hydrogen bonding interaction with the NH group of the cation [ Å and 169.34°]. An ORTEP representation of 3 is shown in Fig. 2a.
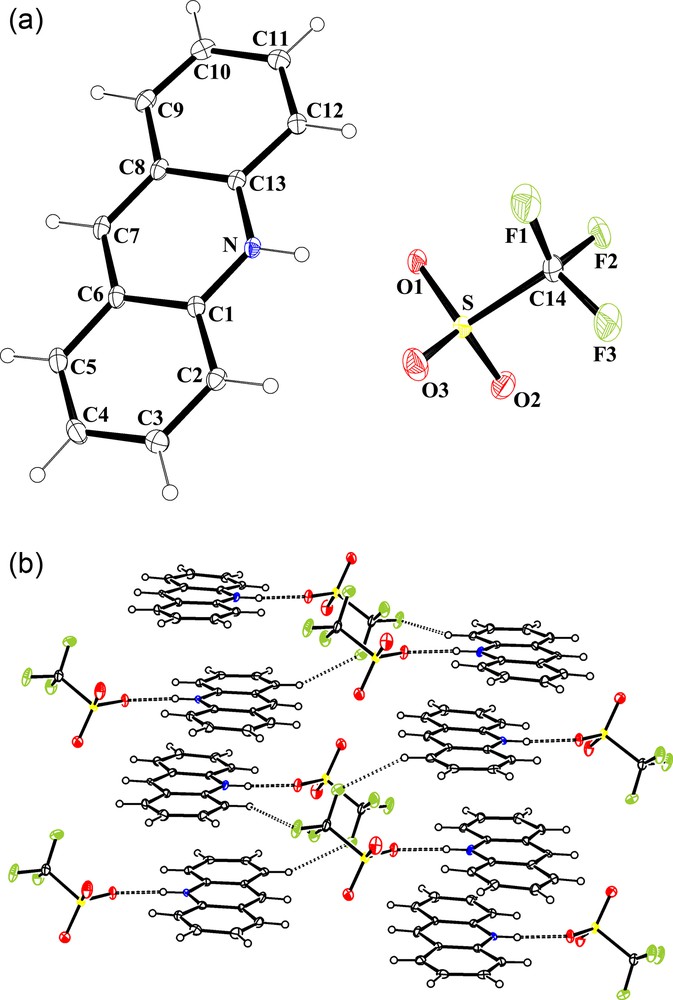
a: molecular structure of [C13H10N][CF3SO3] (3) showing the atom labelling scheme (ORTEP view). Colour code: sulphur: yellow; oxygen: red; nitrogen: blue; fluoride: green; carbon: grey; hydrogen: white (online). Selected bond lengths (A) and angles (°): C1N 1.359(2), C1C2 1.411(2), C1C6 1.427(2), C2C3 1.366(2), C3C4 1.418(2), C4C5 1.361(3), C5C6 1.430(2), C6C7 1.395(2), C7C8 1.397(2), C8C13 1.426(2), C8C9 1.430(2), C9C10 1.357(3), C10C11 1.423(2), C11C12 1.368(2), C12C13 1.415(2), C13N 1.355(2), C14F1 1.323(2), C14F3 1.330(2), C14F2 1.332(2), C14S 1.8249(18), SO2 1.4357(14), SO3 1.4389(14), SO1 1.4511(12); NC1C2 120.28(14), NC1C6 118.89(14), C2C1C6 120.83(15), C3C2C1 118.85(15), C2C3C4 121.36(16), C5C4C3 120.77(16), C4C5C6 120.03(15), C7C6C1 118.53(15), C7C6C5 123.34(15), C1C6C5 118.13(15), C6C7C8 121.28(14), C7C8C13 118.58(15), C7C8C9 123.48(14), C13C8C9 117.95(15), NC13C12 120.06(14), NC13C8 118.94(14), C13NC1 123.75(13), F1C14F3 108.84(16), F1C14F2 107.52(16), F3C14F2 107.05(14), F1C14S 111.56(12), F3C14S 110.92(13), F2C14S 110.77(13), O2SO3 115.58(10), O2SO1 114.43(9), O3SO1 113.78(8), O2SC14 103.84(8), O3SC14 104.06(9), O1SC14 103.10(8); b: the crystal packing of 3 in the unit cell (DIAMOND representation). Hydrogen bonding interactions [] and [] are shown by dashed lines. Colour code: sulphur: yellow; oxygen: red; nitrogen: blue; fluoride: green; carbon: grey; hydrogen: white (online).
From a supramolecular point of view, acridinium rings of 3 are self-assembled in antiparallel arrangement along the a-axis, and through π-π interactions leading to columnar oblique stacks in which the rings emerge tilted by 22.6° with respect to the (100) plane. Two values of interplanar distances, alternating, have been determined between AcrH: 3.218 Å [centroid-to-centroid distance = 4.584 Å, implicating a slippage of 3.26 Å (slip angle 45.35°)] and 3.349 Å [centroid-to-centroid distance = 3.698 Å, implicating a slippage of 1.56 Å (slip angle 25.03°)]. Additionally, short intramolecular contacts exist also between fluorine and oxygen atoms of trifluoromethanesulfonate anions and the nearest neighbour molecules of acridinium, through and interactions, and thus uniting the columns, to each others. A short distance (2.910(2) Å), slightly lower than the sums of the van der Waals radii (2.94), is also observed. A view of the crystal packing of 3 in the unit cell is reported in Fig. 2b.
3.2 Isolation and crystal structures of acridinium salt 4
In previous studies, we reported the incorporation of neutral polyaromatic molecules, e.g. phenazine and anthracene, between two phenazinium salts, leading to unusual sandwich-type structures [4b,11]. A similar methodology was used in the case of acridine. When an equimolar mixture of acridine and anthracene reacts with organotin(IV) complex 1, nice red-blood square crystals were grown in addition to colourless crystals corresponding to organotin(IV) cluster 5. The X-ray crystallographic analysis performed on suitable red crystals, indeed, revealed the expected sandwich-type structure and determined as {([C13H10N][CF3SO3])2·C14H10} (4) resulting from the intercalation of a neutral molecule of anthracene between two acridiniunium trifluoromethanesulfonate salt molecules via slipped parallel π-π aromatic interactions. The micro-analytical data corroborates the formulation reported for 4. The average inter-planar distance between the aromatic rings is 3.424 Å. The two rings are slipped by 1.52 Å (slip angle equal to 23.90°) and the ring-centroid to ring-centroid distances are equal to 3.74 Å. One of the oxygen atoms of the trifluoromethanesulfonate anion is engaged in an intermolecular hydrogen bonding interaction with the NH function of the acridinium cation [ Å and 169.06°]. As in the case of 3, additional short intramolecular contacts are observed between fluorine and oxygen atoms of trifluoromethanesulfonate anions and surrounding molecules of anthracene or acridinium, through and interactions. An ORTEP representation and the crystal packing of salt 4 are reported in Fig. 3a and b, respectively.
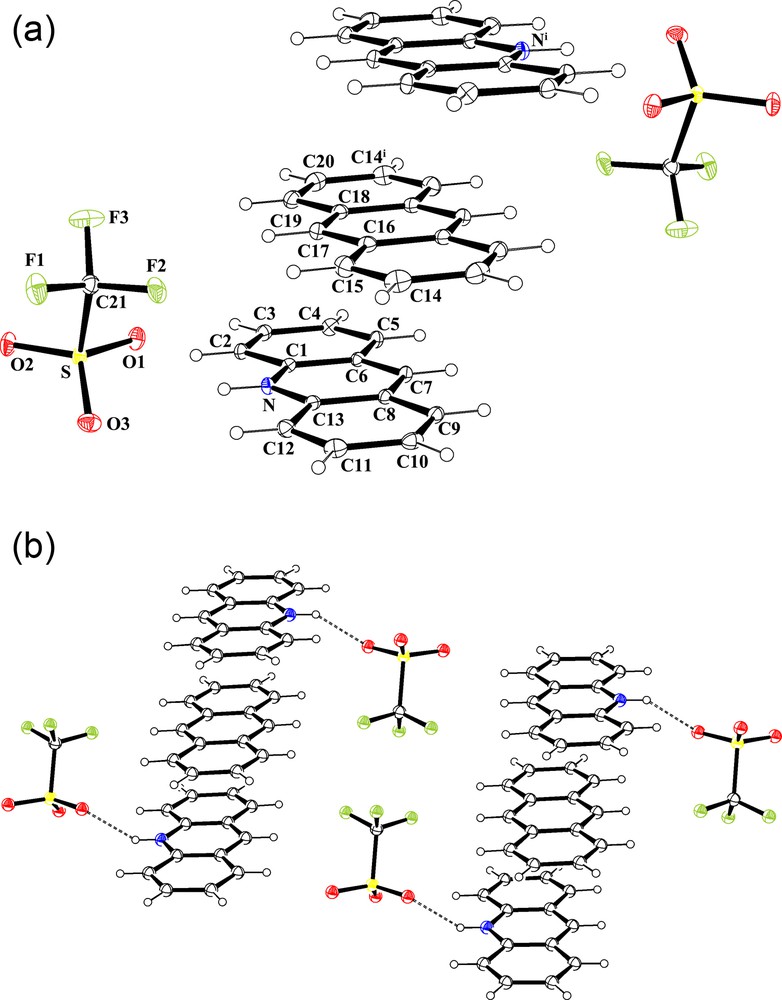
a: molecular structure of {([C13H10N][CF3SO3])2·C14H10} (4) showing the atom labelling scheme (ORTEP view). Colour code: sulphur: yellow; oxygen: red; nitrogen: blue; fluoride: green; carbon: grey; hydrogen: white (online). Selected bond lengths (A) and angles (°): C1C6 1.369(3), C1C2 1.421(3), C2C3 1.366(3), C3C4 1.428(3), C4C7 1.395(3), C4C5 1.422(3), C5N 1.354(2), C5C6 1.420(3), C7C8 1.393(3), C8C9 1.425(3), C8C11 1.429(3), C9N 1.354(2), C9C14 1.415(3), C11C12 1.361(3), C12C13 1.418(3), C13C14 1.365(3), C15C16 1.362(3), C16C17 1.430(3), C17C18 1.393(3), C18C19 1.398(3), C19C20 1.430(3), C20C21 1.361(3), C21F3 1.324(2), C21F2 1.336(2), C21F1 1.340(2), C21S 1.826(2), O1S 1.4472(16), O2S 1.4429(16), O3S 1.4386(16); C6C1C2 121.79(18), C3C2C1 120.40(18), C2C3C4 120.05(17), C7C4C5 118.87(17), C7C4C3 122.61(17), C5C4C3 118.52(17), NC5C6 120.38(17), NC5C4 118.73(17), C6C5C4 120.89(17), C1C6C5 118.35(17), C8C7C4 121.17(17), C7C8C9 118.32(17), C7C8C11 123.29(17), C9C8C11 118.39(18), NC9C14 120.30(17), NC9C8 119.19(17), C14C9C8 120.50(18), C9NC5 123.71(16), C12C11C8 120.20(18), C11C12C13 120.35(19), C14C13C12 121.72(19), C13C14C9 118.82(18), C15C16C17 120.7(2), C18C17C16 122.22(18), C17C18C19 121.59(18), C18C19C20 122.25(19), C21C20C19 121.0(2), F3C21F2 107.90(17), F3C21F1 107.89(18), F2C21F1 106.59(16), F3C21S 111.89(14), F2C21S 110.90(14), F1C21S 111.45(14), O3SO2 115.64(10), O3SO1 114.73(10), O2SO1 115.14(9), O3SC21 103.02(9), O2SC21 103.64(9), O1SC21 102.00(9). Symmetry transformations used to generate equivalent atoms (i): –x + 2,–y + 2,–z; b: the crystal packing of 4 in the unit cell (DIAMOND representation). Hydrogen bonding interactions [] are shown by dashed lines. Colour code: sulphur: yellow; oxygen: red; nitrogen: blue; fluoride: green; carbon: grey; hydrogen: white (online).
Furthermore, the π-π stacking occurring between the acridinium and anthracene rings brings the CF3 groups of trifluoromethanesulfonate anions in close proximity, in a face-to-face orientation. The resulting distance (2.892(2) Å), shorter than that observed in the crystal packing of 3, can be described as a type I conformation according to the Desiraju's classification ( and 120.95°, angle dihedral 74°) [21,22]. Two fluorine atoms of each trifluoromethanesulfonate anions are involved in such an interaction. A MERCURY representation focusing on this contact is depicted in Fig. 4. Synthetically, the sandwich structure of 4 can be also obtained mixing two equivalents of 3 with one equivalent of anthracene in dichloromethane solution (Scheme 2, pathway (iii)).
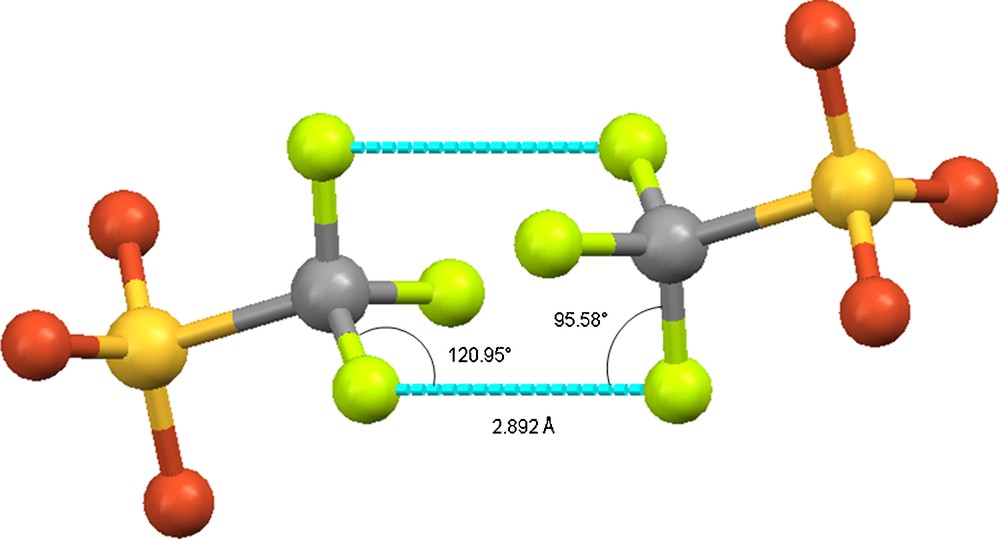
interactions (dashed lines) involving the CF3 groups of the trifluoromethanesulfonate anions of 4 (MERCURY representation). Colour code: sulphur: yellow; oxygen: red; fluoride: green; carbon: grey.
Thus, the three types of supramolecular stacks described in this study and built from acridinium trifluoromethanesulfonates result from the simultaneous involvement of different types of intermolecular interactions: π-π aromatic interactions, hydrogen bonds that can be qualified as strong and weak [23], and also contacts. All modes of interaction which are implicated in the crystal packing of 2, 3 and 4 are listed in Tables 2 and 3. In the three architectures, acridinium cations are directly involved in π-π aromatic stacking and in strong hydrogen bond interactions (), with water molecules (2) or F3CSO3− anions (3 and 4). According to us, these interactions can be considered as the main driving forces leading to the reported self-assembled edifices. In addition, the trifluoromethanesulfonates anions through their fluorine atoms and their oxygen atoms non implicated in strong intermolecular interactions strengthen the cohesion of the crystal packing building bridges between aromatic stacks via the formation of secondary (weak) and hydrogen bonding interactions and short contacts. However, these weaker interactions can be also considered, owing to their orientation, as one of the factors responsible for the slippage, more or less marked (2.79 Å for 2, 1.52 Å for 4), observed between the acridinium rings in π-π interaction.
π-π stacking parameters for 2, 3 and 4.
| Compound direction | Interplanar distance (Å) | Centroïd to centroïd distance (Å) | Slip angle (°) | Slippage distance (Å) | Direction |
| 2 | 3.35 | 4.35 | 39.75 | 2.79 | b |
| 3 | 3.22 | 4.58 | 45.35 | 3.26 | a |
| 3.35 | 3.70 | 25.03 | 1.56 | a | |
| 4 | 3.42 | 3.74 | 23.90 | 1.52 | c |
Geometry of hydrogen bonds and short contacts for 2, 3 and 4.
| Compound | Interaction | Da-H (Å) | b (Å) | (Å) | (°) | Direction |
| 2 | 0.86 | 1.82 | 2.68 | 179 | c | |
| 0.87 | 1.88 | 2.75 | 177 | b | ||
| 0.93 | 2.44 | 3.28 | 149 | c | ||
| 0.93 | 2.49 | 3.30 | 146 | c | ||
| 0.93 | 2.52 | 3.20 | 130 | a | ||
| 3c | 0.86 | 1.89 | 2.74 | 170 | c | |
| 0.93 | 2.50 | 3.34 | 149 | c | ||
| 0.93 | 2.50 | 3.41 | 165 | c | ||
| 0.93 | 2.63 | 3.20 | 120 | b | ||
| 0.93 | 2.60 | 3.47 | 155 | c | ||
| 0.93 | 2.64 | 3.48 | 150 | c | ||
| 4c | 0.86 | 2.02 | 2.77 | 146 | a | |
| 0.86 | 2.60 | 3.08 | 117 | c | ||
| 0.93 | 2.63 | 3.50 | 157 | a | ||
| 0.93 | 2.48 | 3.26 | 141 | a | ||
| 0.93 | 2.69 | 3.61 | 169 | a | ||
| 0.93 | 2.59 | 3.45 | 153 | b | ||
| 0.93 | 2.52 | 3.19 | 130 | a | ||
| 0.93 | 2.52 | 3.40 | 159 | b |
a Hydrogen bond donor.
b Hydrogen bond acceptor.
c In addition to these intermolecular interactions, the crystal packing of 3 and 4 exhibits also short contacts occurring between CF3SO3− anions.
4 Conclusion
So far, the main crystal engineering strategy employed for the synthesis of supramolecular co-crystalline materials based on acridine and phenazine synthons has required the use of suitable hydrogen bond donors as co-reactant. Thus, several examples have been previously reported in the literature describing the co-crystallisation of acridine or phenazine with 2,2′-dihydroxybiphenyl [24], 5,10-dihydrophenazine [25], dicarboxylic acids [20], 2,5-dibromo-3,6-dihydroxy-1,4-benzoquinone [26], hydroxyquinone and 1,5-dihydroxynaphthalene [27], N,N′-bis(2-pyridyl)aryldiamines [28], phloroglucinol [29], vanillin [30]. The resulting solid-state architectures have underlined the predisposition of such N-heterocyclic molecules to generate π-π stacking and hydrogen bonding interactions. In this paper, we have shown that the supramolecular synthon [31], in our case the acridinium trifluoromethanesulfonate, can be generated in situ in the presence of an organotin complex. To our knowledge, this approach involving an organometallic complex is innovative in the domain of crystal engineering. Further studies are in progress in our laboratory to extend the reported reaction for the construction of new π-π self-assembled molecular stacks.
Acknowledgements
Financial support from the Centre National de la Recherche Scientifique (CNRS, France) is gratefully acknowledged. The authors thank also Prof. Enrique Espinosa for his clarification on interactions, Dr. Philippe Richard for his consideration of the structural part and Dr. Richard Decréau for the correction of the manuscript.