



 CC-BY 4.0
CC-BY 4.0
1. Introduction
The nuclear fuel cycle includes the front-end steps (mining and milling, conversion, enrichment, and fuel fabrication) that prepare uranium for use in nuclear reactors and the back-end steps that manage spent nuclear fuel (reprocessing and reuse and disposal). Uranium is, in fact, the most widely used raw material as UOX fuel in nuclear fission power plants, sometimes with Plutonium (MOX fuel). Uranium is relatively abundant in the earth’s crust and can be found in various geological deposits (proterosoic unconformity, sandstones, metasomatite, granite-related, carbonates, etc.) [1], rivers and seawater. It can be extracted in a variety of forms and environments, ranging from very low grade to very high grade ores, as well as from mines with a different primary product such as copper or gold. Uranium resources are divided into conventional and unconventional resources. Conventional resources are defined as resources (pitchblende, uranite) from which uranium is recoverable as a primary product, co-product or significant by-product. Unconventional resources are those from which uranium can only be recovered as a minor by-product. This is particularly the case for low-grade deposits such as phosphate rock or phosphorite, which is mined primarily to produce phosphorus pentoxide for fertilizer production, as well as non-ferrous ores, carbonatite, black shale, rare earth elements (REE) deposits, lignite, and even very low levels of uranium, such as in seawater.
Historically, uranium has been produced primarily by open pit and underground mining techniques and then processed by conventional uranium milling, leaching and separation from the leach solutions by solvent extraction or ion exchange. Uranium can thus be recovered from the ore using dynamic or heap leaching processes, in which the preliminary crushed ore is either mixed with oxidizing acid or base solutions in a tank, or placed on an engineered barrier sprayed with acid or base. Other mining methods include in situ leaching (ISL), which involves the extraction of uranium from the deposit using an acid or alkaline solution directly injected into the confined underground zone, followed by chemical treatment to purify it. Sulfuric acid is the most common acid used in the leaching process because uranium sulfate salt is readily soluble in the hexavalent state. An oxidizing agent (sodium chlorate or manganese dioxide) is usually required because most of ores contain tetravalent uranium, which is poorly soluble in the acidic leach solutions. The resulting leach solutions consist of complex acid sulfate solutions containing a wide variety of ions in addition to uranium, including aluminum, calcium, iron, magnesium, molybdenum, vanadium, etc., depending on the composition of the ore being treated. It is therefore necessary to implement a stage for the separation of the uranium present in a sulfuric acid phase. However, uranium can also be found in a phosphoric acid phase if it comes from wet-process phosphoric acid (WPA) or in a nitric acid phase if it comes from the final purification before conversion and enrichment steps.
As a result, uranium can be present in different acidic environments (sulfuric, phosphoric or nitric, etc.), which requires different conditions for its extraction and recovery involving specific extractants.
Despite the efficiency of the current systems, but due to some drawbacks, the search for more efficient and selective systems for uranium extraction and recovery with respect to competing ions is still relevant.
Through this review, we present the research carried out to understand and capture the mechanisms involved in the classical processes, but also the search for new systems.
2. Molecular systems for the extraction of uranium from conventional ores
The marketable uranium product, Yellowcake, can be obtained through various processes, depending on the origin of the ore from which it is produced. Uranium ores are categorized into two types: primary or conventional ores, which are the richest, and secondary ores, from which uranium is sometimes extracted as a by-product. The primary source of uranium production is obtained from conventional ores, achieved through the utilization of sulfuric acid leach liquors. The ore extracted from mine, whose fragments measure some centimeters or more in diameter, is crushed and ground to the consistency of fine sand. Since most of the conventional ores currently processed contain around 0.02% to 0.2% recoverable uranium, it is necessary to process 500 to 5000 kg of ore for each every kilogram of uranium recovered. Most uranium ore processing plants use wet milling, and the resulting sludge is directed to a leaching circuit, where sulfuric acid is added. Acid consumption does not depend on the uranium content of the ore, but on the constituents of the gangue. Thus total acid consumption can range from 10 kg H2SO4 per tonne of ore to over 100 kg/tonne. Leaching times vary from a few hours to more than 24. For some ores, leaching times can be considerably reduced by heating the pulp in tanks at temperatures of between 40 °C to 60 °C. However, these processes require purification of the leach liquor to effectively separate uranium from competing elements such as iron (Fe), molybdenum (Mo), zirconium (Zr), and others. Uranium is then selectively extracted from the leach solutions by solvent extraction or ion exchange. The uranium industry was the first hydrometallurgical industry to make extensive use of both processes. In the solvent extraction process, the active agent is generally an organic molecule diluted in a petroleum fraction such as kerosene, which can selectively extract uranium ions by forming an organic complex that is insoluble in water. The organic phase is separated from the aqueous phase by continuous mixing and settling techniques. The uranium is then re-extracted from the organic complex by contacting it with a solution of inorganic salt, such as sodium chloride or ammonium sulfate. Thus, the marketable uranium product Yellowcake is precipitated from the re-extraction solution by adding ammonia, soda, magnesium hydroxide or hydrogen peroxide, and the resulting solid product is dried and packaged for shipment to a refining facility.
Since current industrial processes predominantly use solvent extraction to reach higher level of purification, this liquid–liquid extraction process consists in contacting the leachate with an immiscible organic solvent, a diluent loaded in lipophilic ligand. Through various interactions (ionic, electrostatic, polar) with the metal cation at the interface, liposoluble metal-ligand complexes are obtained that are then spontaneously transferred from the aqueous leachate to the organic phase. Recovery of the pure metal is then achieved during the back-extraction or stripping step. Depending on the nature of the metal, its release from the solvent can be induced by contact with an aqueous phase that may contain either acid-base or water-soluble ligand before the recovering step by precipitation or electrowinning. The effectiveness of the stripping process is driven by the concentration of the stripping reagent (H+, HO−, etc.) that must be modulated according to the type (neutral or ionizable) of the liposoluble extractant.
Industrial uranium extraction began in 1955 with the development of the DiAlkyl Phosphoric acid EXtraction (DAPEX) process, which utilized di-2-ethylhexylphosphoric acid (HDEHP) as the extractant. This pioneering solvent extraction process was developed and marketed for uranium VI. However, owing to its limited selectivity when it came to competing metals, especially iron, it has been replaced in 1957 by the amine extraction (AMEX) process, which used a mixture of secondary and tertiary amines (in particular Alamine 336, mainly composed of tertiary amines) or more recently pure trialkylamine such as trioctylamine [2].
3. Historical of AMEX and DAPEX processes in sulfuric acid
AMEX and DAPEX processes were actually concomitantly developed at the Oak Ridge National Laboratory (ORNL) during the 50’s. First ideas were about the designs of long alkyl chain polyamines but this concept was quickly dropped for the benefits of alkylamines and phosphorous derivatives [3], Hence, numerous structures such as linear or branched di- or trialkylamines, trialkylphosphines oxide, mono-, di- or triesters of phosphinic, phosphonic or phosphoric acid were considered for uranium extraction. However, only a few of them exhibited the required properties allowing efficient large-scale processing, namely AMEX and DAPEX, discussed below.
3.1. The DAPEX process
The development of the DAPEX process at the ORNL relied on previous results reported by other laboratories where affinity of organophosphorus compounds towards uranium was extensively studied. More specifically, in 1949, it was fortuitously found that the hydrolysis products (mono- and dibutylphosphate) of the tri-n-butyl phosphate (TBP) showed higher affinity than that of the parent compound towards uranium in nitric media. Ensuing works allowed to set a first uranium extraction process based on long-chain monoalkylphosphoric acid at the pilot plant scale in 1954. A year earlier, dibutylphosphate was claimed as a potential ligand for uranium extraction from seawater [4].
Consequently, at the ORNL, emphasis was put on the development of a method involving the use of promising phosphate-based ligands. HDEHP was quickly identified as a good candidate as it was a cheap commercially available compound with very low water solubility [4].
Nevertheless, considering that neutral phosphorus derivatives such as phosphine oxides could provide efficient and applicable extraction properties, investigations were also carried out with a series of symmetrical trialkylphosphine oxide (TAPO) in the same laboratory [3].
Extraction properties of HDEHP towards uranium contained in sulfuric acid leachates were assessed with regards to (i) ions (sulfate, uranium and other metals) and ligand concentrations, (ii) pH of the leachate, (iii) nature of the diluent, (iv) temperature, (v) chemical structure of the phase modifier, (vi) volumetric phase ratio (A:O) of aqueous phase volume (A) to organic phase volume (O), (vii) required stripping conditions and (viii) up-scaling feasibility [4, 5].
HDEHP is an ionizable acidic extractant that allows the transfer of metal by cationic exchange, that is, n protons are released in aqueous phase by the extractant while m metal cations are trapped in the organic phase. Because this exchange verifies electroneutrality, n and m are correlated as follows:
| (1) |
| (2) |
Considering that in the concerned range of concentration, activities could be approximated by concentrations, the equilibrium constant was:
| (3) |
Finally, the distribution coefficient of uranium, which is the ratio at equilibrium of transferred uranium concentration to that of the metal remaining in aqueous phase, could be expressed as:
| (4) |
Although fatty alcohols were found to drastically impair the extraction of uranium, it was shown that the organic phase supplemented with v:v 2% 2-ethylhexan-1-ol as a phase modifier (whose essential role is discussed in later section) could maintain a good level of uranium extraction (DU ∼ 30) without being subjected to the third phase formation (see AMEX process) occurring in pure kerosene during the stripping step. Thus, while operating with leachates partially miming (pH = 1, sulfate concentration range from 0.2 to 0.5 mol⋅L−1, 4 × 10−3 mol⋅L−1 uranium) those recovered with ores processing, first optimal system for uranium extraction could be defined as HDEHP solubilized at 0.1 mol⋅L−1 in 98:2 v:v kerosene:2-ethylhexan-1-ol mixture. The best uranium extraction performance could be achieved with this mixture at 1:1 A:O at 298 K in short contact time (∼2 min).
Efficiency and selectivity of the uranium extraction enabled by numerous other dialkylphosphates were evaluated for this system and this was an unexpected opportunity to preliminary investigate the structure-activity relationship (SAR) of the phosphoric diester ligand (Table 1).
DU obtained for a series of dialkyl phosphoric esters at two concentrations of sulfate in the aqueous phase with [UVI] = 4 × 10−3 mol⋅L−1
| Dialkyl phosphoric ester O ← P (OR)2OH | Formula of R | DU (0.5 mol⋅L−1 ) | DU (1.5 mol⋅L−1 ) |
|---|---|---|---|
| Dioctyl hydrogen phosphate | n-C8H17- | 450 | 57 |
| Bis(3,5,5-trimethylhexyl) hydrogen phosphate |
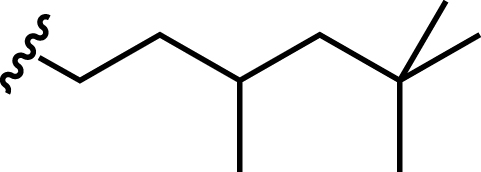 |
260 | 41 |
| Bis(2-ethylhexyl) hydrogen phosphate (HDEHP) |
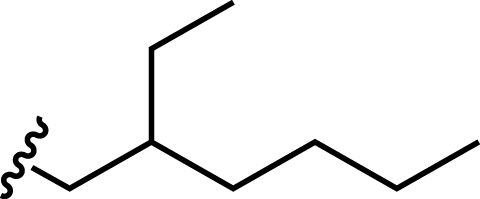 |
135 | 20 |
| Bis(2,6-dimethylheptan-4-yl) hydrogen phosphate |
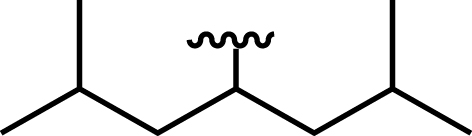 |
10 | 2 |
Organic phase was 0.1 mol⋅L−1 extractant in w:v 98:2 kerosene:ol, contact time was 2 min. with A:O = 1:1. Data extracted from Blake et al. [5].
Actually, ores leach liquors contain, in addition to uranium, many other elements present in higher or even much higher concentrations than those of uranium. Therefore, beside the affinity of the ligand could be lower for those elements than for the uranium, most of them might represent a cumbersome powerful competitor impairing the extraction process.
For instance, with tests performed according to the conditions previously established for U (VI) and with a 10−2 mol⋅L−1 single metal-containing solution, significant extractions were observed for thorium (IV), titanium (IV), iron (III), molybdenum (VI), Vanadium (IV) and cerium (IV) with DM = 26 (Th = Ti), 16, 6, 1.8, 1.1 (V = Ce), respectively. These results also underlined the benefit brought by the fast U extraction equilibrium that should allow to circumvent the huge co-extraction of ferric cations that could take place in real condition at contact time longer than 2 min (DFe ∼ 250 at t = 7.5 h with HDEHP). However, the iron extraction could be drastically lowered in the presence of reducing agents that converted ferric ions in ferrous one.
Stripping could be achieved at high mineral acid concentrations as well as in basic condition with 10% sodium carbonate, which was preferred because of its cheap and fast implementation. Moreover, the uranium carbonate complex is water-soluble whereas those of competitors mostly precipitate, allowing thus to remove co-extracted contaminants from the recovered uranium. Nonetheless, as mentioned above, the main issue was the formation of the third phase that resulted from insolubility of HDEHP sodium salt in both phases and that could be addressed by supplying the kerosene with 2% 2-ethylhexan-1-ol. Once again, SAR could be explored for phase modifier and ligand. A significant impact of both the branching and the length of the alkyl chain was typically demonstrated but without place for some comprehensive explanations (see Table 2).
Structures of various alcohols tested as phase modifiers and their respective amount needed in the diluent to prevent the formation of third phase during the stripping
| Alcohol | Formula | w:v% needed as phase modifier |
|---|---|---|
| n-Octan-1-ol |
 |
0.9 |
| 2-Ethylhexanol |
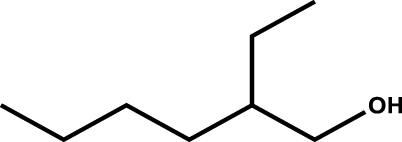 |
1 |
| n-Octan-2-ol |
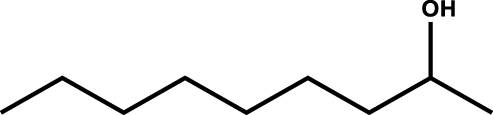 |
1 |
| 4-Ethyloctan-1-ol |
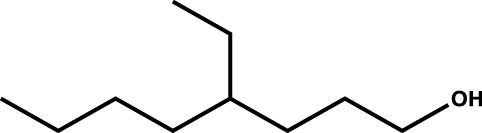 |
0.9 |
| 3-Neopentyl-5,5-dimethyl hexanol |
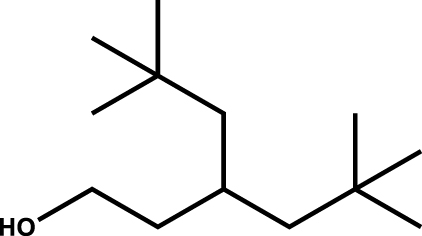 |
1.5 |
| 5-Ethyl-nonan-2-ol |
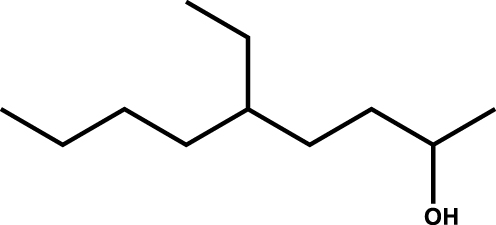 |
2.5 |
| 2,6,8-Trimethylnonan-4-ol |
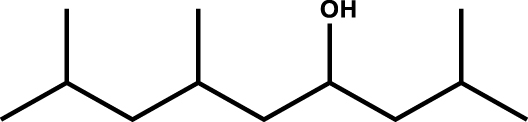 |
7.5 |
| 7-Ethyl-2-methylundecan-4-ol |
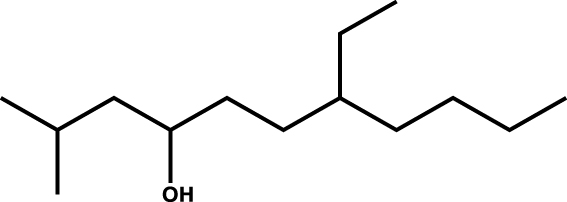 |
7.5 |
| 3,9-Diethyl-tridecan-6-ol |
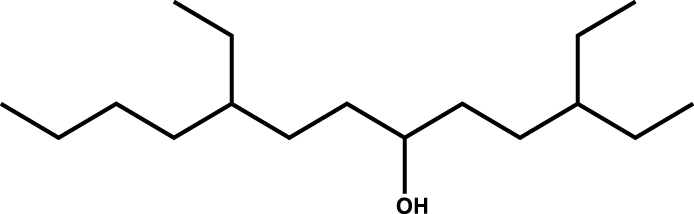 |
>12 |
Organic phase was 0.1 mol⋅L−1 HDEHP in kerosene modified by various w:v percentages of phase modifier. Aqueous phase was 10% Na2CO3 and contact were performed at A:O = 1:1. Data extracted from Blake et al. [4].
In a last stage, synthetic leachate liquors containing U, Fe and Al were processed on a pilot scale to allow the recovery (after acidic hydrolysis of carbonate salts and precipitation with ammonia and finally after calcination) of 98% U under its 78–84% pure U3O8 form.
Subsequent works focused on the use of neutral organophosphorus compounds as phase modifiers. At variance with fatty alcohol, few amount of trialkylphosphates, dialkylphosphonates or TAPO were found to increase the DU by several orders of magnitude when compared to each respective DU (ranging from 0 to 5.9 for various TAPO tested at 0.3 mol⋅L−1 in kerosene, for instance) and to improve the selectivity towards U. TBP, dibutyl butylphosphonate (DBBP) and tributylphosphine oxide (TBPO) provided the strongest synergies (see Table 3).
Values of DU obtained with DEHPH at 0.1 mol⋅L−1 in kerosene modified by few w:v% of neutral organophosphorus compounds
| Phase modifier | Formula | w:v% needed as phase modifier | Synergic DU |
|---|---|---|---|
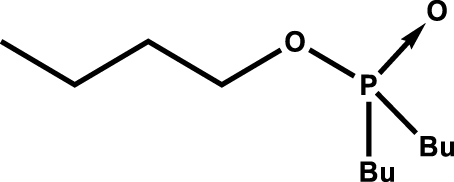
| Tri(n-butyl)phosphite | P(OBu)3 | 7 | 700 |
| Tri(n-butyl)phosphate (TBP) |
 |
2.2 | 800 |
| Dibutyl butylphosphonate |
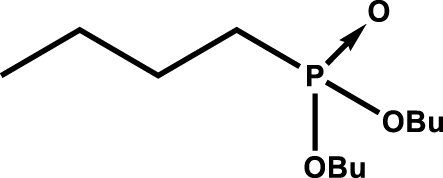 |
1.4 | 3000 |
| Butyl dibutylphosphinate |
 |
0.9 | 2500 |
| Tributylphosphine oxide |
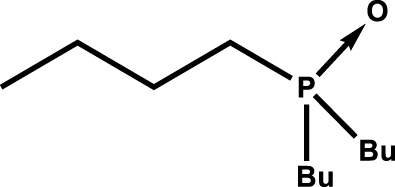 |
1.1 | 7000 |
Aqueous phase was 0.5 mol⋅L−1 SO, 0.004 mol⋅L−1 U (VI), pH = 1. Time contact was 10 min with 1:1 O:A at 298 K. Data extracted from Blake et al. [5].
This synergy was explained by Blake et al. [7] after considering the revisited mechanism of U extraction mediated by HDEHP where the ligand forms dimers in the organic diluent. At low levels (maximum organic uranium concentration four times lower than that of HDEHP) [6] of extraction the associated equilibrium became:
| (5) |
And at a high level of extraction, (minimum organic uranium concentration more than four times higher than that of HDEHP) [6] polymers responding to formula as (UO2)n(R2PO4)2n+2H2 were formed in the organic phase. In the presence of a neutral extractant like TAPO, this did not happen as the neutral extractant played its role of solubilizing agent to lead to (UO2(R2PO4)4H2)(R3PO).
Indeed, neutral extractants do proceed via solvation of ionic species and absolutely not by ionic exchange. In this case, the metal is transferred to the organic phase with its counterions, water and acids. The equilibrium of this transfer may be described as:
| (6) |
Although the processing of sulfuric leach liquors by DAPEX was formerly applied at large scale and remains nowadays topic for studies [8, 9, 10], it has been progressively abandoned for the more selective AMEX process [10].
3.2. The AMEX process
As mentioned above, the development of the AMEX process took place in the early 50’s at the ORNL to be set at large scale at the end of the same decade [2]. Conditions were optimized according to similar approach as that followed for the DAPEX development. Tests were performed on several hundred of various primary, secondary, tertiary amines and tetraalkylammoniums (see some examples in Figure 1). The ligands were selected in respect of their solubility in the organic diluent (before and after contact with acidic leach liquors), their low ability to form a third phase and their extraction properties.
some structures (and related DU) of amines II and amines III that were formerly assessed in the AMEX context. Conditions were 0.1 mol⋅L−1 , pH 1, 4 × 10−3 mol⋅L−1 U(VI), phase ratio 1:1; 0.1 mol⋅L−1 amine in kerosene. TP stands for third phase formation. Amine II: N,N-di(decyl)amine (1), bis(3,5,6,8-tetramethylnonyl)amine (2), bis(2-isobutyl-3,5-dimethylhexyl)amine (3), N-(2-isobutyl-3,5-dimethylhexyl)decan-1-amine (4), N-(cyclohexylmethyl)-4-ethyl-2-(3-ethylpentyl)octan-1-amine (5), 4-ethyl-2-(3-ethylpentyl)-N-((2,4,6-trimethylcyclohexyl)methyl)octan-1-amine (6), N-benzyltetradecan-1-amine (7), N-(3-phenylpropyl)tetradecan-1-amine (8), N-benzyl-4-ethyl-2-isopropyloctan-1-amine (9), N-benzyl-4-ethyl-2-(3-ethylpentyl)octan-1-amine (10), 2-ethyl-N-(1-phenylethyl)hexan-1-amine (11), N-benzyl-2,5,7-trimethylnonan-4-amine (12), 2,5,7-trimethyl-N-(1-phenylethyl)nonan-4-amine (13). Amine III: trioctylamine (14), N-decyl-N-methyldecan-1-amine (15), N,N-dibutyldodecan-1-amine (16), N,N-dihexyldodecan-1-amine (17), tridodecylamine (18), N-dodecyl-N-methyldodecan-1-amine (19), N-benzyl-N-dodecyldodecan-1-amine (20), tris(2-ethylhexyl)amine (21), tris(3,5,6,8-tetramethylnonyl)amine (22), tris(5,5-dimethyl-3-neopentylhexyl)amine (23). Data extracted from Coleman et al. [2].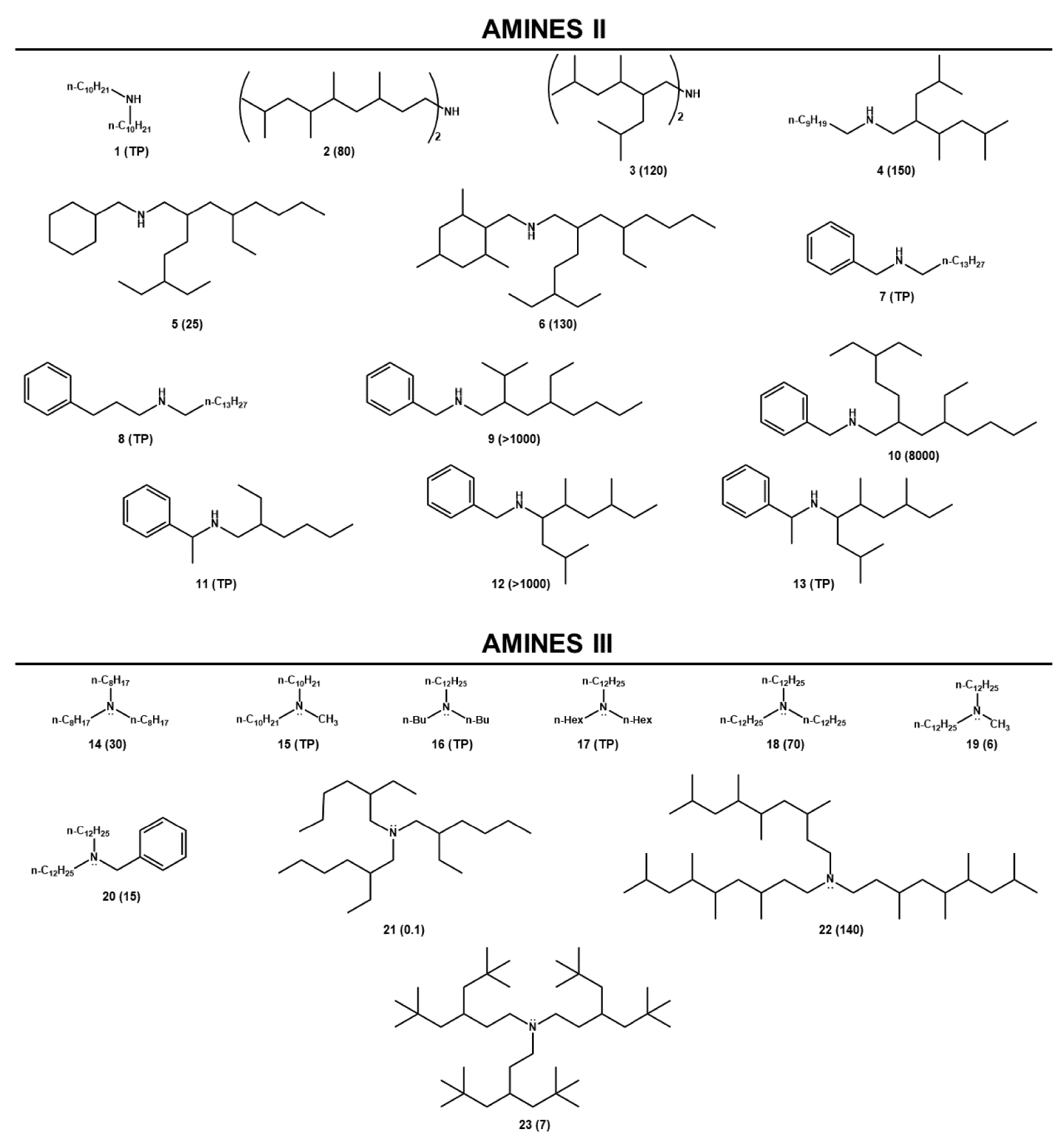
Considering solubility and phase behavior, it was found that, once protonated, only trialkylamines showed acceptable solubility in the diluent. More precisely, branched trialkylammoniums were found to be soluble in kerosene whereas linear one became soluble after supplying the diluent with 3–5% tridecanol as a phase modifier. In sulfuric acid, this protonation could be described as follows:
| (7) |
| (8) |
| (9) |
| (10) |
Efficiency of the extraction was shown to tightly depend on the concentration of aqueous sulfate (too high sulfate concentration led to uranium release), cationic competitors (molybdenum, vanadium, iron, etc.), free amine sulfate, pH of the leachate (too low pH led to ammonium salt hydrolysis) and the temperature (extraction decreased by 20 to 30% for 10 °C temperature rise), while DU was not affected by the uranium concentration. Uranium:amine stoichiometry in the transferred complexes could be determined as 1:6 with linear to moderately branched secondary amines and as 1:4–5 with symmetrical trialkylamines (1:4.7 for tri-n-octylamine, for instance) or highly branched secondary amines. A mass law could be applied [2] showing that DU evolved proportionately to the activity of the uncomplexed amine according to:
| (11) |
Again, dependence of DU on the structure of amines, could not be established with accuracy because the extracting properties were claimed to be driven by solvent effects subsequently to comparative studies carried out in kerosene, benzene and chloroform (partially shown in this paper) that were not fully understood. Indeed, if extraction was certainly mainly ruled by steric hindrance, basicity and lipophilicity, the observed effects seemed to result from a complex combination of these parameters. Moreover, it is interesting to note that while the authors had tried to characterize the colloidal properties of the system, they did not report some theories establishing the link between chemical structure and supramolecular assembly behavior. However, ignoring the supramolecular phenomena and considering the DU values measured in the same solvent, namely kerosene (Figure 1), it was obvious that extraction was enhanced with a longer alkyl chain in the symmetrical linear trialkylamine series. It became more difficult to give proper explanations in the case of their dissymmetric isomers. In the branched trialkylamine series, the presence of an ethyl group on position 2 (tris(2-ethylhexyl)amine 22) generated a drastic decrease of DU while the highest level of extraction was reached with the branched trialkylamine 22 carrying methyl groups on positions 3, 5, 6 and 8. Comparison with the DU obtained for a trialkylamine bearing two bulky neopentyl groups on the same position leads to postulate that steric hindrance affects the process according to the position and the nature of the branching. N-benzyl secondary amine were the best extractants. For instance, the N-benzyl-4-ethyl-2-(3-ethylpentyl)octan-1-amine (10) showed exceptional efficiency in standard conditions (DU = 8000) and was still extractant at very low pH (DU = 1 with [H2SO4] = 3 mol⋅L−1), finally, any attempt of rationalization based on the theoretical basicity of the tested amines should be a failure.
Selectivity of extraction could also be tuned according to the amines structure and the diluent. Some of the more promising and commercially available secondary and tertiary amines were successfully used in the AMEX process developed at large scale. For instance 90% of the uranium contained in the leach liquor could be extracted in countercurrent extraction equipment.
Stripping was easily achieved by displacement of uranyl with either chloride or nitrate ions, or alternatively by simple hydrolysis of the ammonium salt. The precipitated uranium oxide was recovered as predominant component presenting a few percent of contamination.
The main co-extracted competitors are molybdene (VI), vanadium (V) and zirconium. At the time it was already known that vanadium (V) and molybdene (VI) formed polymeric species incorporated in the organic phase. This further but not trivial detail is discussed below.
4. Current issues and progress in the AMEX process
Nowadays, the principle of the AMEX process is still the same but the composition of the system has been lightly revisited as it consists in a mixture of trialkylamines diluted in kerosene with 3% tridecanol. The mixture of trialkylamines, namely Alamine 336, is composed of trioctylamine (TOA), trinonylamine and tridecylamine.
Despite its widespread application and economic efficiency, the AMEX process also has drawbacks that warrant consideration: (i) the uranium separation factor is not optimal when compared to competing elements like Zr, Fe, Mo, and V, which are co-extracted [11, 12], (ii) it requires the use of a volatile phase modifier to prevent the formation of a third phase (organic phase splitting) [2], (iii) the amine extractants undergo degradation due to a radical reaction catalyzed by the co-extracted vanadium in the presence of the phase modifier [13], (iv) industrial facilities require significant quantities of organic diluent, typically kerosene, for solvent extraction carried out in mixer-settlers. Additionally to the resulting dysfunction, these issues have a high economical impact and must be addressed. For instance, the Somaïr plant in Niger maintains an inventory of approximately 900 m3 of solvent, with 75% replacement annually due to issues such as cruds formation, organic entrainment, and solvent evaporation. Moreover, in the same plant, a 50% loss of TOA due to chemical decomposition was noted over a year of activity [14]. Also, the selectivity with respect to molybdenum(VI) is not optimal which results in a decrease of the uranium extraction efficiency and of the loading capacity of the amine [15].
The degradation of tertiary amines by acidic hydrolysis and oxidation by nitrate [16] or radical reaction in the presence of vanadium(V) [13] is one of the main drawback of such systems.
The amines degradation phenomenon has been thoroughly studied by Chagnes and his co-workers [13, 14, 17, 18, 19]. It results in the appearance of growing concentration of dialkylamines and in the decrease of uranium extraction efficiency. First, they tried to provide a concrete solution with an applicability at large scale and showed by computer modeling that the flow-sheet of the AMEX process could be advantageously modified and optimized in order to delay the periodic replacement of the degraded TOA [17]. All their subsequent works allowed to clarify the mechanism of the TOA degradation, which involved transferred vanadium (V) and/or chromium (VI), the phase modifier (tridecanol) and the diluent (be it kerosene, TPH or n-dodecane).
Oxidation of vanadium (IV) to vanadium (V) should be the root of the problem since VIV is not extracted in sulfuric media whereas VV is extracted. Chagnes et al. properly monitored the already known conversion (see Coleman et al. [2]), after incorporation in 95:5 w:v n-dodecane:tridecanol mixture, of monomeric VV to a polymeric species by 51V nuclear magnetic resonance (NMR) and Fourier Transform-Infra Red (FT-IR) spectroscopies [14]. The first step is the formation of the transferable vanadium.sulfate complex taking place in the aqueous phase. The sulfate complex is then transferred by anion exchange into the organic phase where it is not expected to form polynuclear species. However, the formation of decavanadate () was verified and assumed to result from the colloidal properties of the organic phase. Actually, trialkylammoniums form reverse-micelles in oily diluents and can incorporate further components other than the targeted metal such as water, salts and acids. Because the intrinsic properties (dielectric constant, ionic strength, etc.) of the yielded aqueous core might be significantly different from these of the sulfuric acid leach liquor, some redox reactions that were forbidden in usual aqueous media were instead allowed in reverse-assemblies.
Mechanism and kinetic of the degradation were investigated by UV–visible spectroscopy monitoring of the VIV appearance in the aqueous phase (absorption of VIV at 𝜆 = 760 nm) and degradation by-products contained in the organic phase were identified by Gas Chromatography coupled to Mass Spectrometer (CG–MS) [13]. Indeed, when the oxidation occurred, the VIV yielded after reduction of the VV was immediately released in the aqueous phase, signaling the beginning of the phenomenon that was found to evolve slowly. Its rate depended on the concentration of both VV and sulfuric acid in the aqueous phase, and of TOA and solubilized dioxygen concentrations in the organic phase. Against all expectations, the kinetic of oxidation depended only poorly on the concentration of tridecanol whereas this latter was typically involved in the process. Actually, decavanadate and/or VV⋅O2 complex generated radical oxidative species that reacted first with tridecanol to yield tridecanal and then with TOA to produce mainly dioctylamine, octanal and (E)-N,N-dioctyloct-1-en-1-amine (Figure 2A). The role of phase modifier was more specifically studied in the context of another works undertaken by the same team [18]. A series of either commercially available or synthesized alcohols were tested as phase modifier regarding the impact on the kinetic of TOA degradation. Although SAR could not be established with accuracy, it was shown that the kinetic of degradation became slower even extremely slower in the presence of a long branched alkyl chain alcohol (Figure 2B).
(A) Schematic representation of the two mechanisms of extractant degradation reported to occur in the AMEX process by Chagnes et al. [13, 14]. Main degradation by-products are tetradecanal (24), dioctylammonium (25), (E)-N,N-dioctyloct-1-en-1-aminium (26) and octanal (27). (B) Structures of phase modifier assessed by Chagnes et al. [18] with related values of the degradation kinetic constant k observed before/after degassing of the organic phase by nitrogen bubbling. Values are factors of 10−7 and k is expressed in s−1. Tested alcohols were tridecan-1-ol, nonan-2-ol (28), 9-methylnonadecan-9-ol (29), 9-hexylnonadecan-9-ol (30), 10-octylicosan-10-ol (31), 9-octylheptadecan-9-ol (32), 3-methyl-1-phenylpentan-3-ol (33), 4-nonylphenol (34), 1,1,2,2,3,3,4,4,5,5,6,6,7,7,7-pentadecafluoroheptan-1-ol (35), 3,3,4,4,5,5,6,6,7,7,8,8-dodecafluoro-2-methyloctan-2-ol (36).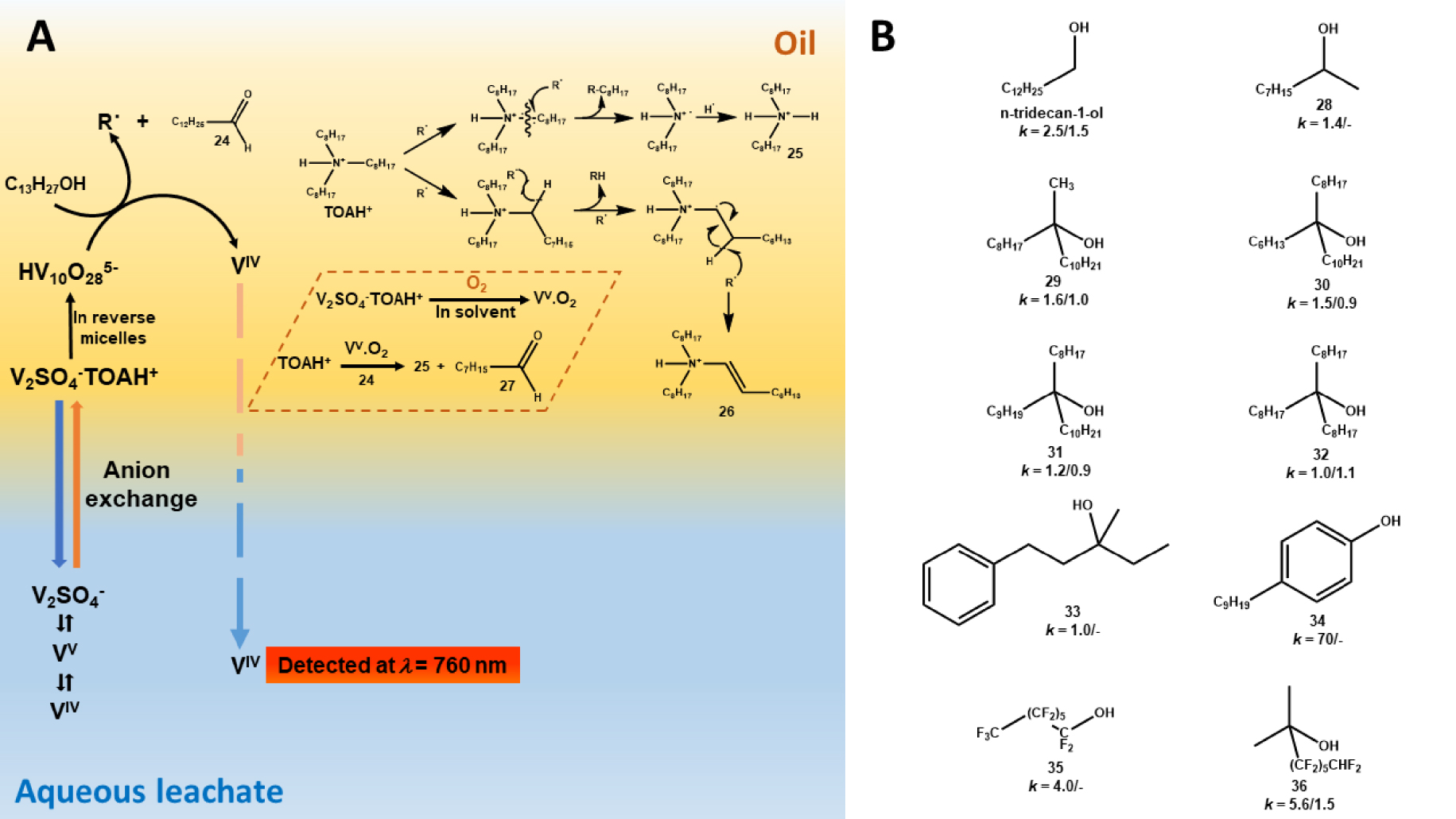
Therefore, to optimize the AMEX process and enhance its efficiency, it is essential to gain a deeper insight into the mechanisms of this solvent extraction process and to focus on optimizing the composition of extractants employed. This process uses extractant mixtures consisting of a range of secondary and tertiary amines. While numerous studies have investigated the extraction capabilities of these secondary and tertiary amines, there has been a conspicuous lack of research dedicated to comprehending their inherent properties. Notably, most of these studies has addressed their aggregation characteristics, which are a consequence of their amphiphilic nature.
In the context of uranyl extraction in sulfuric acid solutions using tertiary amine extractants, an in-depth examination of the microstructure of uranyl complexes was conducted through a combination of Extended X-ray Absorption Fine Structure (EXAFS) analysis and Molecular Dynamics (MD) simulations [20]. The TOA extractant was used as a reference for tertiary amines. The MD simulations and classical fitting methods yielded consistent results, revealing an unprecedented structure in the organic phase: the uranyl complex consists of three sulfate anions coordinated to the uranyl cation through U–O bonds in its first coordination shell (Figure 3a).
(a) Snapshots issued from MD simulations performed in n-dodecane presenting the complex UO2(SO4)3(TOA)4 with 3 in the bidendate coordination. The uranium atom is colored in green. (b) k 3–weighted Fourier transformed EXAFS spectra of U-SO4-TOA complex. Insert: corresponding k 3–weighted EXAFS spectra [20]. Reproduced from Ref. [20] with permission from the Royal Society of Chemistry.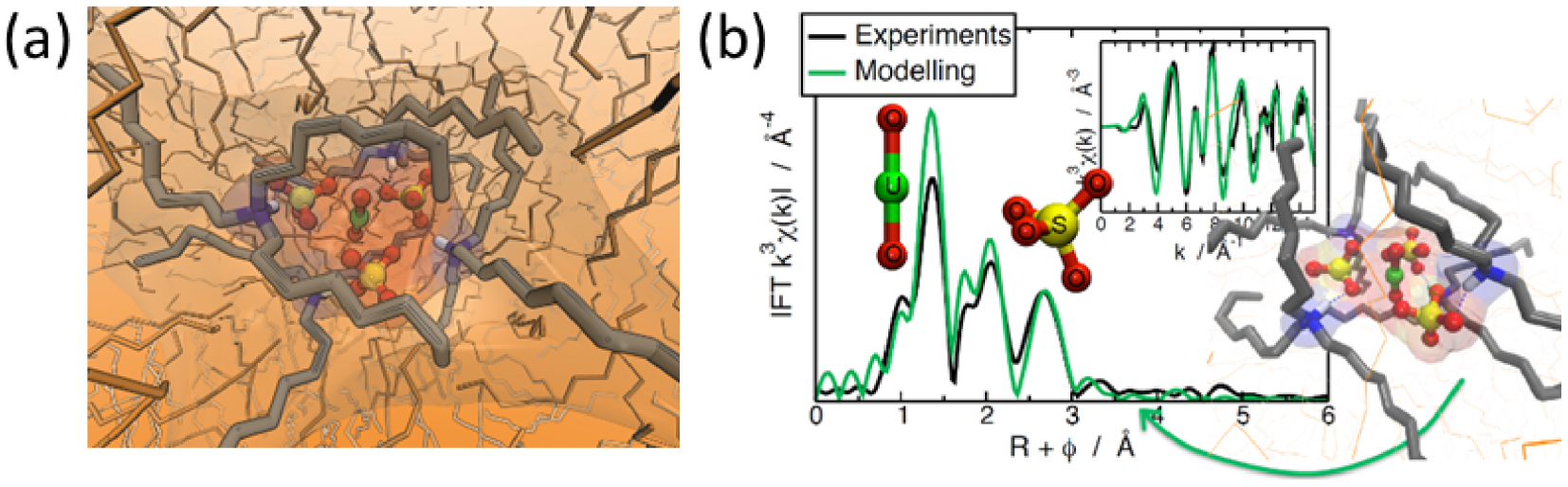
Further analysis of the EXAFS spectra demonstrated that uranyl trisulfate complexes exhibit dynamic behavior, transitioning between a configuration in which all three sulfate ions are bidentate and another configuration where two sulfate ions are bidentate, and one is monodentate. The MD simulations corroborated these findings, providing evidence that the configuration with three bidentate sulfate anions is energetically more favorable than the alternative arrangement with two bidentate and one monodentate sulfate ions (Figure 3b). Behind this sulfate coordination aspect, the distribution of exchangeable hydrogen bonds observed through MD simulations also revealed a significant interplay between the inner- and outer-sphere environments of the uranyl complex.
The effect of alkyl chain configuration on the phase stability and the aggregation properties of tertiary amines has further been investigated [21]. The results showed that tertiary amines with longer or slightly branched alkyl chains exhibit enhanced phase stability and maintain their effectiveness in extracting uranium (Figure 4a).
(a) Illustration of effect of tertiary amines alkyl chain length and branching on organic phase organization and stability [21]. (b) Main thermodynamic contributions to the uranium transfer energy plotted as a function of alkyl chains length and branching [22].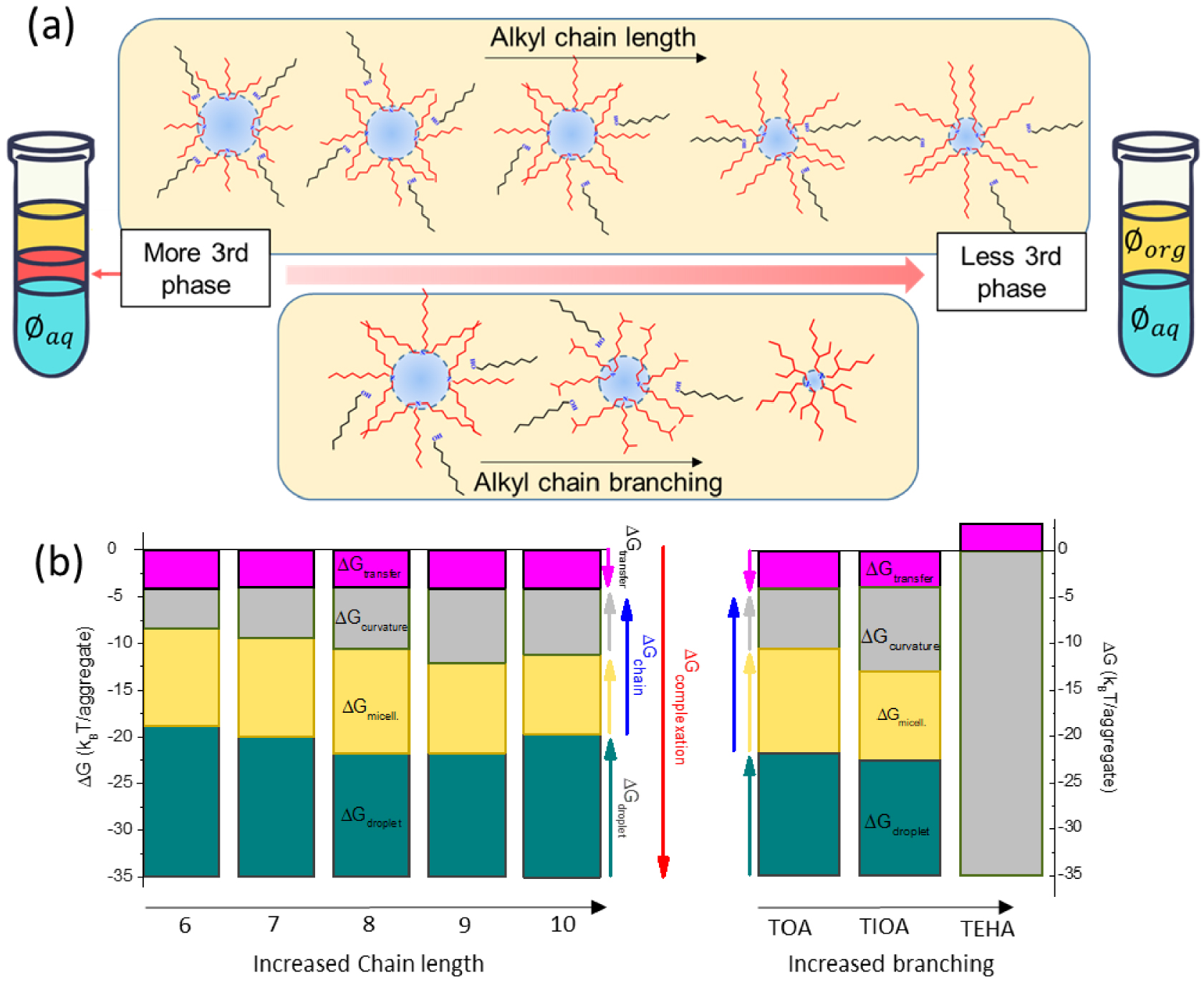
Through a combination of techniques such as small-angle scattering to determine nanostructures in the organic phase and measurements of oil–water surface tension, it has been revealed that these effects on extraction and phase stability are linked to the presence of reverse aggregates characterized by smaller polar core volumes and reduced co-extraction of water. When the alkyl chains exhibit more pronounced branching, they prevent the formation of a swollen reverse aggregates, thereby hindering efficient extraction. This suggests that the arrangement and packing of the alkyl chains directly influence the efficiency of the solvent extraction system.
Taking such a structural approach into account, this study has not only demonstrated that smaller aggregates effectively prevent the formation of a third phase but also elucidated how reduced attractive interactions among these aggregates play a pivotal role in achieving this outcome.
An in-depth thermodynamic analysis, based on the “ienaic” approach, also provided a comprehensive rationale for this phenomenon [22]. It highlighted the significant impact of the alkyl chains on the curvature free energy of the aggregates (Figure 4b). Notably, this curvature free energy emerges as a primary inhibitory factor affecting the transfer free energy of uranium, primarily due to the significant entropic contributions arising from the branching and packing of the extractants.
It is worth noting that this study was conducted using commercially available tertiary amines. However, for a more nuanced understanding, we synthesized a family of branched TOA isomers with different degrees of branching in terms of length, number, and location. Interesting complementary conclusions could be established. This work will be reported elsewhere [23].
An eco-friendly solvent extraction process has been innovatively developed for uranium extraction using diluent-free tri- and tetraalkylammonium-based extractants. A series of ammonium mixtures were systematically tested in absence of diluent to evaluate the influence of ammonium cations and anions on extraction efficiency, viscosity and phase separation [24].
Tetraalkylammoniums were readily accessible as they were commercially available as chloride (Cl−) or bis(trifluoromethanesulfonyl)imide () salts of N-methyltrioctylammonium and Aliquat 336 mixture (Figure 5a). Trialkylammoinium-based ionic liquids (ILs) required synthesis. They were easily obtained by neutralizing the TOA diluted in ethanol with stoichiometric (n = 0.5 in Equation (12)) or half amount (n = 1 in Equation (12)) of sulfuric acid.
| (12) |
| (13) |
Numerous binary mixtures of each IL were yielded and their properties were tuned according to the molar ratio of each partner, the nature and the molar ratio of each counterion (, Cl−, , ) (Figure 5a).
(a) Structure of the tested cations and anions. (b) Distribution ratios of Uranium obtained for mixte ammoniums plotted as a function of the molar ratio of the sulfate anion. The initial aqueous solution (pH = 1) contains 0.1 mol⋅L−1 sulfuric acid, 1 mol⋅L−1 ammonium sulfate, 250 mg⋅L−1 U(VI). It was contacted to the organic phase with an aqueous to organic volume ratio A:O = 2:1 [24].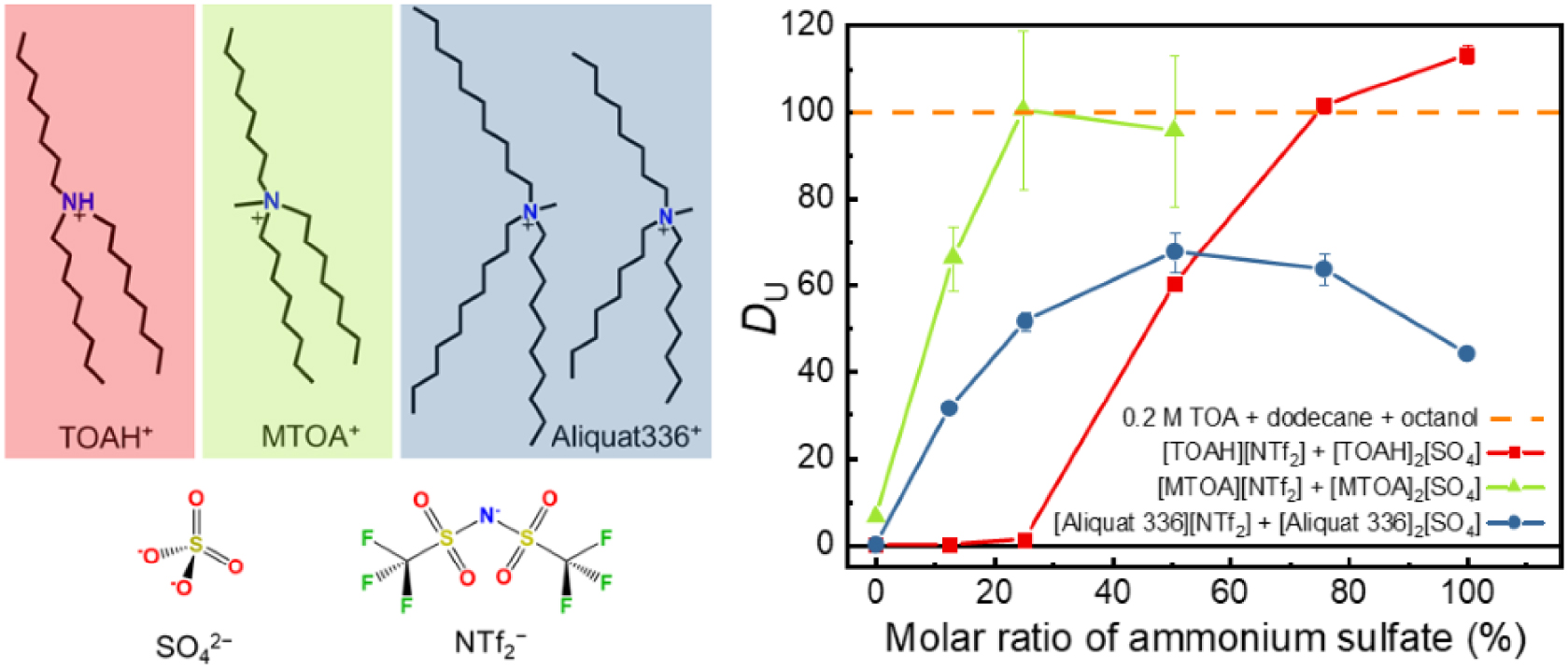
These systems have proven to be exceptionally effective and straightforward to implement, as they employ the same chemical components as those employed in the AMEX process. As presented in Figure 5b, distribution ratios of uranium obtained with some of these ammoniums salts and mixtures allow either comparable or much higher extraction than the classical TOA extractant (taken as reference for the AMEX process). Uranium extraction increases with sulfates concentrations, which is consistent with the recognized extraction mechanisms for such systems [20]. An interesting synergistic behavior with the molar ratio of one ammonium to another was further investigated as it could be advantageously considered to adjust physicochemical properties of the final solvent (as viscosity or density) [25]. Furthermore, it has been established that adopting such alternatives not only serves to avoid the formation of a third phase and subsequent extractant degradation but also leads to a remarkable reduction in solvent evaporation losses. To provide a comprehensive evaluation of their sustainability, a comparative life cycle assessment (LCA) was carried out between this novel system and the conventional solvent [24]. The results indicate that the new system is 18 times less environmentally impactful.
Beyond focusing on the improvement of some historical and well established processes that still need optimization, developing new methods and/or new tools is certainly the more innovative and attractive alternative intended by organic chemists. This point is discussed in the following section.
5. Development of new ligands working in sulfuric media
The development of new, more efficient extractants than those currently used in the AMEX and DAPEX processes is an important issue for the uranium mining industry. Knowing that these processes are based on trialkylated tertiary amines for AMEX and organophosphorus compounds (HDEHP and tri-n-butylphosphate (TBP)) for DAPEX, various studies have investigated the possibility of combining amine or amido to organophosphorus functional group to offer bifunctional extractants. These molecules are designed to have at least two distinct functional groups that enable them to selectively bind and extract uranium from a complex mixture in order to improve the efficiency and selectivity.
Organophosphorus compounds are very widely used in the context of separation chemistry in hydrometallurgy. The classification of organophosphorus extractants can vary based on different criteria, such as their structural features, chemical properties, or extraction mechanisms [26].
The coordination strength of the organophosphorous extractant can be enhanced with an increase in their basicity. This can be achieved by varying the oxidation state of phosphorus. Phosphines [R3P], for instance, are generally more basic than phosphine oxides [R3P(O)] due to the presence of an electron pair on the phosphorus atom in phosphines. The number of O atoms linked to the P atom plays also a role in the basicity of the phosphine oxides (R3P(O)). Indeed, these compounds with one substituted oxygen atom are more basic than phosphinates [(R′O)P(O)R2], phosphonates [(R′O)2P(O)R], and finally phosphates [(R′O)3P(O)]. The nature of the substituents (R) attached to the P atom can significantly influence the basicity of the compound. By introducing electron-donating groups or electron-withdrawing groups on the alkyl groups attached to phosphorus, the overall electronic environment can be altered and consequently, the basicity of the compound. Bulkier substituents can decrease the basicity due to steric hindrance effects. In addition, they are usually categorized into neutral (R′ = organic moiety) and acidic compounds (R′ = H or R′ = H and organic moiety).
Taking into account the versatility of organophospohorous compound, their combination with amine allowed proposing various α-amino-functionalized organophosphorus compounds with a strong potential for the extraction of lanthanides and actinides including uranium [27, 28].
One of the first systems based on the use of α-amino-functionalized organophosphorus extractant described in the literature was proposed by Jagodić and Grdenić with the use of N-substituted mono-esters of α-aminophosphonic acid (ethyl and octyl mono-esters of α-anilinobenzylphosphonic acid) (see Table 4 entry #1) as complexing agents for the extraction of various metal including uranium from sulfuric acid solutions [29]. Interestingly, it turned out that this category of extractant exhibits a different behaviour depending on the degree of oxidation of uranium. Uranium (IV) is extracted at lower sulfuric acid concentration while uranium (VI) is extracted over a wide concentration range (0.25 to 4.5 mol⋅L−1 H2SO4).
An advantage of α-aminophosphonate-, phosphinate-, and phosphine oxide-based extractants is that they can be easily synthesized through Kabachnik–Fields, Mannich and Pudovik pathway [30, 31, 32].
The Kabachnik–Fields reaction is mainly used since it allows obtaining α-amino-functionalized organophosphorus derivatives in a single step by a three-component condensation reaction involving a carbonyl (aldehyde or ketone), an amine (primary or secondary amine) and a phosphoryl compound (phosphite, phosphinate, or phosphine oxide) [33].
Extractant and related extraction condition for the extraction of uranium from sulfuric acid solution
| Entry | Extractant struture | Extraction condition | Reference |
|---|---|---|---|
| #1 |
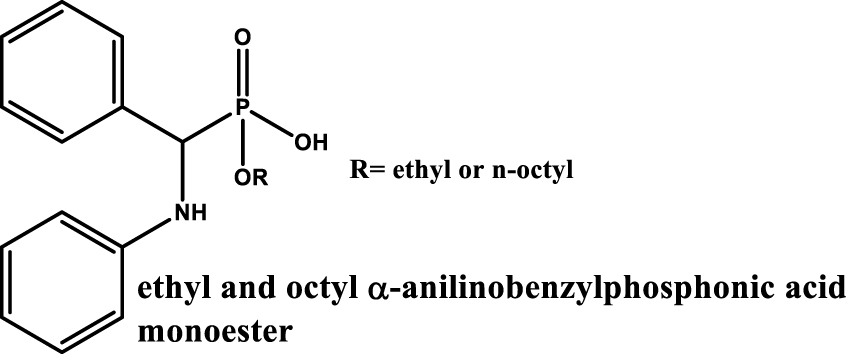 |
Sn (II), UO2 (II), VO (II), Sb(III), Bi(III), Ce(III), Ti(III), Fe(III), Ce(IV), Zr(IV), Sn(IV), Th(IV), Ti(IV), U(IV) Neutral and H2SO4 ⩾ 1 mol⋅L−1 | [29] |
| #2 |
 |
U(VI), Fe(III), Mo(VI), Ce(III), La(III), Gd(III), Yb(III), Nd(III), Ti(IV), Zr(IV) H2SO4 1 mol⋅L−1 + Li2SO4 1 mol⋅L−1 | [34] |
| #3 |
 |
Th(IV), U(VI), RE(III), Fe(III) and Al(III) Leach solution of ion-adsorption rare earth (RE) ores by aluminum sulfate H2SO4 ⩽ 1 mol⋅L−1 | [35] |
| #4 |
 |
Separation by precipitation of thorium, uranium, and scandium from RE | [36] |
| #5 |
 |
U(VI) Mo(VI), Zr(IV), Ti(IV), La(III), Ce(III) and Fe(III) 0.1 mol⋅L−1 H2SO4 | [37] |
| #6 |
 |
U(VI) Mo(VI), Zr(IV), Ti(IV), La(III), Ce(III) and Fe(III) 0.1 mol⋅L−1 H2SO4 | [38] |
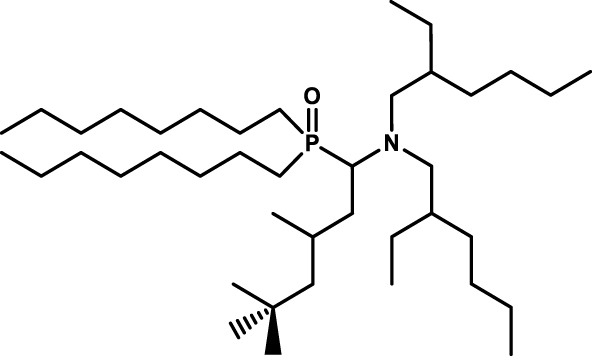
According to this approach, Leydier et al. proposed different amino phosphine oxide type extractants for the extraction and recovery of uranium from sulfuric acid [34], The condensation of an aldehyde, a dialkylamine, a dialkylphosphite or a dialkylphosphine oxide in the presence of para-toluene sulfonic acid in catalytic quantity in toluene leads to aminophosphine oxide (Figure 6). Among all the extractants synthesized, an interest was demonstrated for the (1-(bis(2-ethylhexyl)amino)-4,6,6-trimethylheptan-2-yl)dioctylphosphine oxide (R1 = R2 = Octyl; R3 = 2,4,4,-triMethylPentyl; R4 = 2-EthylHexyl) (see Table 4 entry #2). Indeed under the conditions of the study this extractant shows extraction performance for uranium much higher with much greater selectivity than HDEHP or TOA used as a reference system.
Kabachnik–Fields pathway for the synthesis of aminophosphine oxide extractant studied for the extraction and recovery of uranium from sulfuric acid [34].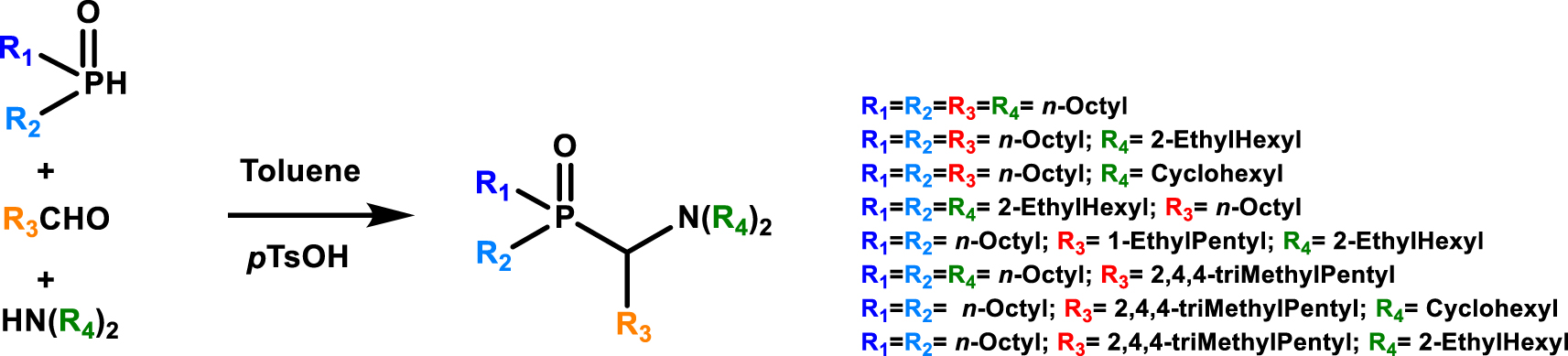
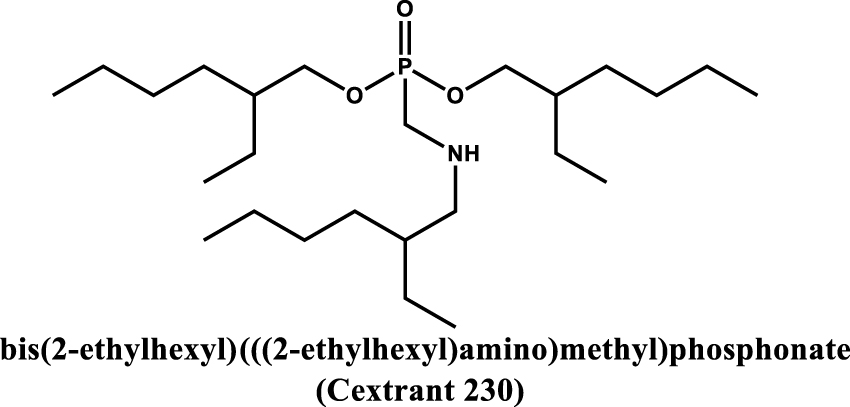
More recently, Liao and coworkers showed that neutral aminophosphonate extractants exhibit high extraction ability towards high-valence metal ions such as U(IV). Indeed, bis(2-ethylhexyl) (((2-ethylhexyl)amino)methyl)phosphonate also called Cextrant 230 (see Table 4 entry #3) can form a UO2SO4⋅2[Cextrant 230] complex in which phosphoryl oxygen atom are involved in the coordination of and the extraction of U(VI) is an exothermic process [35]. It has been shown that the extraction capacity can be influenced with steric hindrance and that the Cextrant 230, which has a relatively low steric hindrance, shows a strong affinity for high valence uranium.
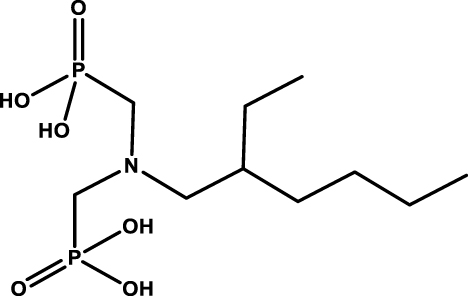
Usually the amino phosphorylated-based extractants are functionalized by long alkyl chains that can increase their lipophilicity in order to facilitate the separation between the aqueous feed phase and the organic phase. Recently, Moilanen and coworkers proposed water-soluble aminobis(phosphonate) ligands (see Table 4 entry #4) as precipitating agents for REEs, Th, and U allowing a selective separation [36].
Beyond organophosphorous extractant compounds, extractants with amino or amido moieties have been also investigated for the extraction and recovery of uranium from sulfuric leachates. The carbonyl oxygen atoms of the amide groups and the nitrogen atom of the amino groups allow to bind lanthanide and actinide ions.
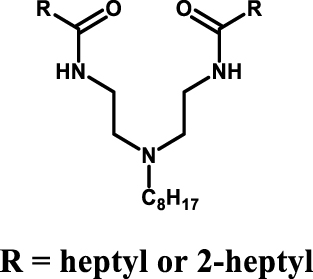
Iminodiamides which combine amides and amine functionalities have been proposed for this purpose (see Table 4 entry #5) [37]. The chelating behaviour of the iminodiamides was evaluated through complementary analysis (FT-IR, NMR, Mass spectroscopy) and molecular modelling (DFT calculation) and the extraction performances showed the potential of this system for the extraction of uranium at low concentration in sulfuric acid.
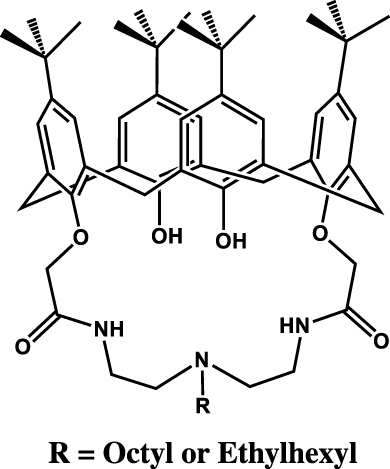
In the same way, it could be demonstrated that iminodiamides could be attached on calixarene platform leading calix[4]azacrowns (see Table 4 entry #6) [38]. Such macrocycle is able to selectively extract uranium at low sulfuric acid concentration but in a different way in comparison to the “free” iminodiamide. Indeed, the stoichiometry of complexation is different with the demonstration of complex 1:1 for the iminodiamide-functionalized calixarene while it was found to be 2:1 for the iminodiamide. Also, a lower efficiency can be noticed when the iminodiamide is tethered on calixarene.
6. Molecular systems for the extraction of uranium from unconventional ores
Wet process phosphoric acid is a potential source of uranium as significant concentrations of uranium ranging from 20 to 200 mg⋅L−1 can be found in phosphate rock. It represents an alternative resource for uranium production as large quantities of phosphate rock are processed annually to produce phosphoric acid [39, 40, 41, 42, 43, 44].
Several hydrometallurgical processes such as precipitation [45, 46], membrane separation [47, 48, 49], and liquid–liquid extraction [50, 51] have been investigated to remove or recover uranium from the WPA.
Among these processes, liquid–liquid extraction has emerged as the benchmark technology for the industrial recovery of uranium from phosphoric acid [44].
Three processes have reached commercial status between the late 1970s and 2000 with varying degrees of success:
- The URPHOS process (uranium from industrial phosphoric acid), developed at the ORNL, using a synergistic combination of HDEHP and trioctyl phosphine oxide (TOPO) as extractants for the recovery of uranium(VI) from 4–6 mol⋅L−1 phosphoric acid solution [52].
- The OctylPhenyl Acid Phosphate (OPAP) process, also from ORNL but using a mixture of mono- and dioctylphenylphosphoric acid as the extractant for the recovery of uranium(IV) from WPA [53].
- The OctylPyroPhosphoric Acid (OPPA) process, developed by Dow for the recovery of uranium(IV) by using octylpyrophosphoric acid as the extractant [53].
In each of these processes, organophosphorus extractants are used, as shown in Figure 7.
Chemical structure of extractant involved in URPHOS [52], OPAP [53] and OPPA [54] processes.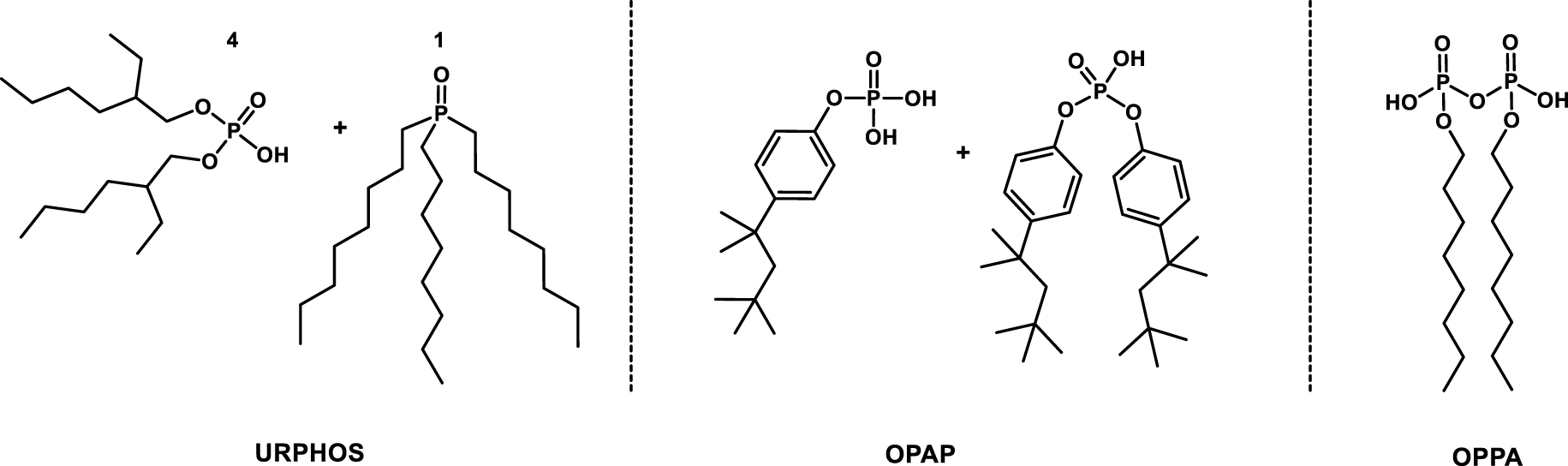
In the case of the URPHOS process, a mixture of a cationic exchanger HDEHP with a solvating agent TOPO is used in the following proportions 0.5 mol⋅L−1 HDEHP + 0.125 mol⋅L−1 TOPO in aliphatic diluents. Indeed, Hurst et al. [52] have shown that a synergistic effect is observed for a HDEHP/TOPO ratio of 4/1 with a significant increase in the uranium extraction compared to extractants used independently.
A number of studies have been carried out in order to gain a better understanding of the mechanisms involved in this synergistic extraction. Among the various mechanisms proposed to predict and gain insights into synergism in solvent extraction, studies with a molecular approach were usually proposed, explaining the extraction of species by the mechanism of addition, substitution, and solvation [55, 56, 57, 58, 59]. The possibility of pre-organizing a mixture of extractant molecules has further been explored on the system HDEHP/TOPO [60]. This concept encompasses scenarios such as synergistic aggregation of the extractants, promoting molecular interactions between extractant molecules and to enhancing the solubilization into the reverse micelles. To examine this hypothesis, the aggregates formed by the extractant molecules were characterized with neutron and X-ray scattering, but also thermodynamically in order to determine the critical aggregation concentation (CAC) for each ratio of the mixture (Figure 8). This unprecedent study revealed that lower CAC, which can be related to favored micellar energy, is concomittant with the synergistic extraction of uranium.
(a) SAXS data showing the aggregation of the extractant molecule. The top inset shows the deduced aggregate diameter. (b) CAC as a function of TOPO ratio determined from tensiometry, SAXS and SANS [60].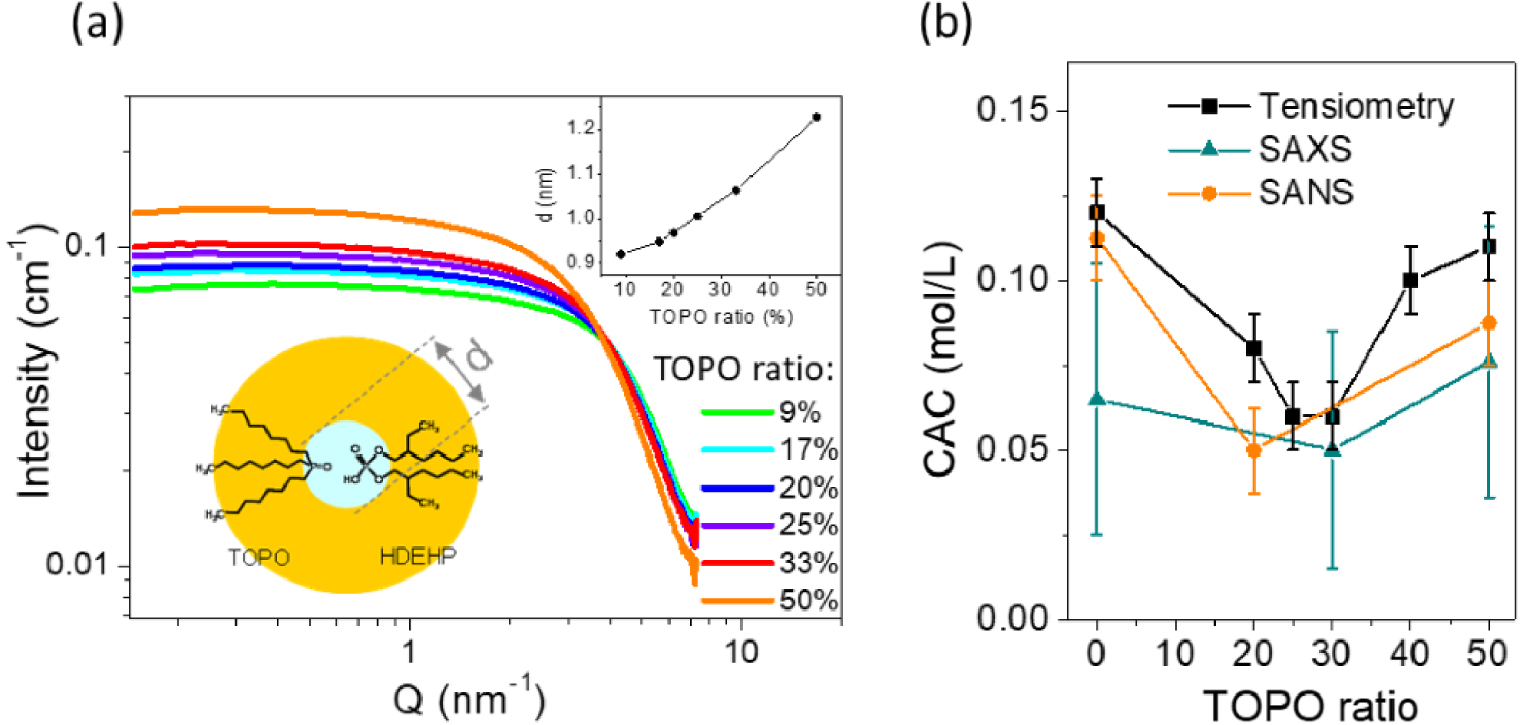
Conducted in the absence of any cations, this innovative study further demonstrated that there is no discernible preorganization of the extractants, suggesting that the synergistic aggregation observed in the presence of cations is a result of the chelation mechanism [61]. Finally, the synergistic extraction of uranium by HDEHP/TOPO was shown to be controlled by an entropic effect (offering more configurations to extract), by a favored extractant association (in presence of acid and cations), and by the curvature energy of the aggregates, this latter depending on both the extractant alkyl chains geometry (packing parameter) and the diluent penetration in the apolar shell of the aggregates [62]. These results were further confirmed on another extractant mixture thanks to a complete thermodynamic balance perfomed with the so-called ineaic approach [63, 64].
7. Research on new extractants for uranium extraction from phosphoric acid medium
In spite of the accomplishments in uranium recovery from WPA (4–6 mol⋅L−1 phosphoric acid solution), the three processes commercialized exhibit significant potential for enhancement, prompting further research. The primary shortcomings of these developed methods are classically an inadequate selectivity and limited capacity.
Efforts have been dedicated to enhancements, particularly through the exploration of novel extractant systems surpassing the efficiency of URPHOS. The initial approach involved combining extractants in different ways: (i) mixing cation exchangers with TOPO [59, 65, 66, 67], (ii) integrating solvating agents with HDEHP [68, 69, 70], or (iii) new synergy system [71, 72, 73, 74, 75, 76].
However, the use of mixtures is limited by the need to control the composition during the extraction phase, particularly when recycling the extraction phase. To overcome this challenge, the search for multifunctional molecules has become a priority with a particular focus on autosynergistic molecules.
Such autosynergistic molecules refer to bifunctional extractants that incorporate both a cationic exchanger and a solvating agent within their structure.
For this purpose, the combination of phosphine oxide and phosphoric acid groups has been proposed Figure 9.
Bifunctional molecule bearing a phosphine oxide and phosphoric acid groups [75, 77].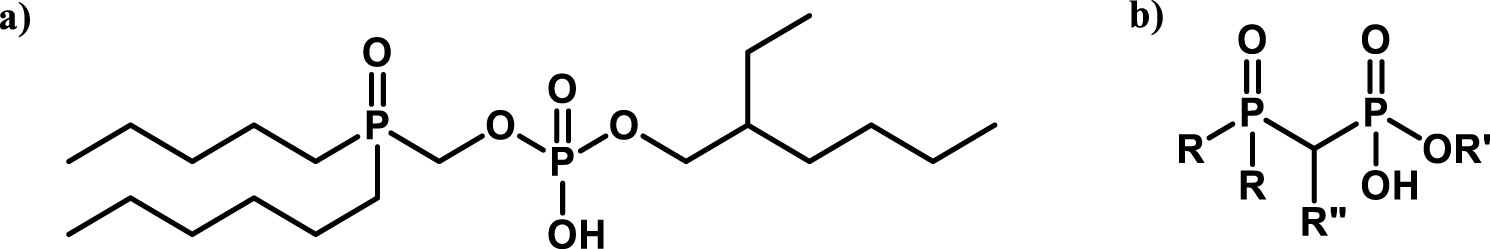
One of the first examples studied was proposed by Warshawsky et al. using a O-methyldihexylphosphine oxide O′-hexyl-2-ethyl phosphoric acid however a third-phase formation occurs during the extraction (Figure 9a) [75]. In a comparable approach, molecules that contain a phosphoric acid group, similar to HDEHP, and a phosphine oxide group, similar to TOPO, have been described (Figure 9b) [77].
A number of different extractants were synthesized on the basis of a reaction scheme (Figure 10) involving di-n-octylphosphine oxide or di(2-ethylhexyl)phosphine oxide obtained from di-n-butylphosphite using the appropriate (alkyl)magnesium bromide. The introduction of an alcohol group, which is then substituted in a chloride derivative, allows an Arbuzov reaction to be carried out with a suitable trialkyl phosphite (P(OR′)3). The resulting (dialkylphosphoryl)-methyl-(dialkylphosphonate) can then be treated to become monosaponified or alkylated.
General synthesis of bifunctional molecules bearing a combination of phosphine oxide and phosphoric acid groups [77].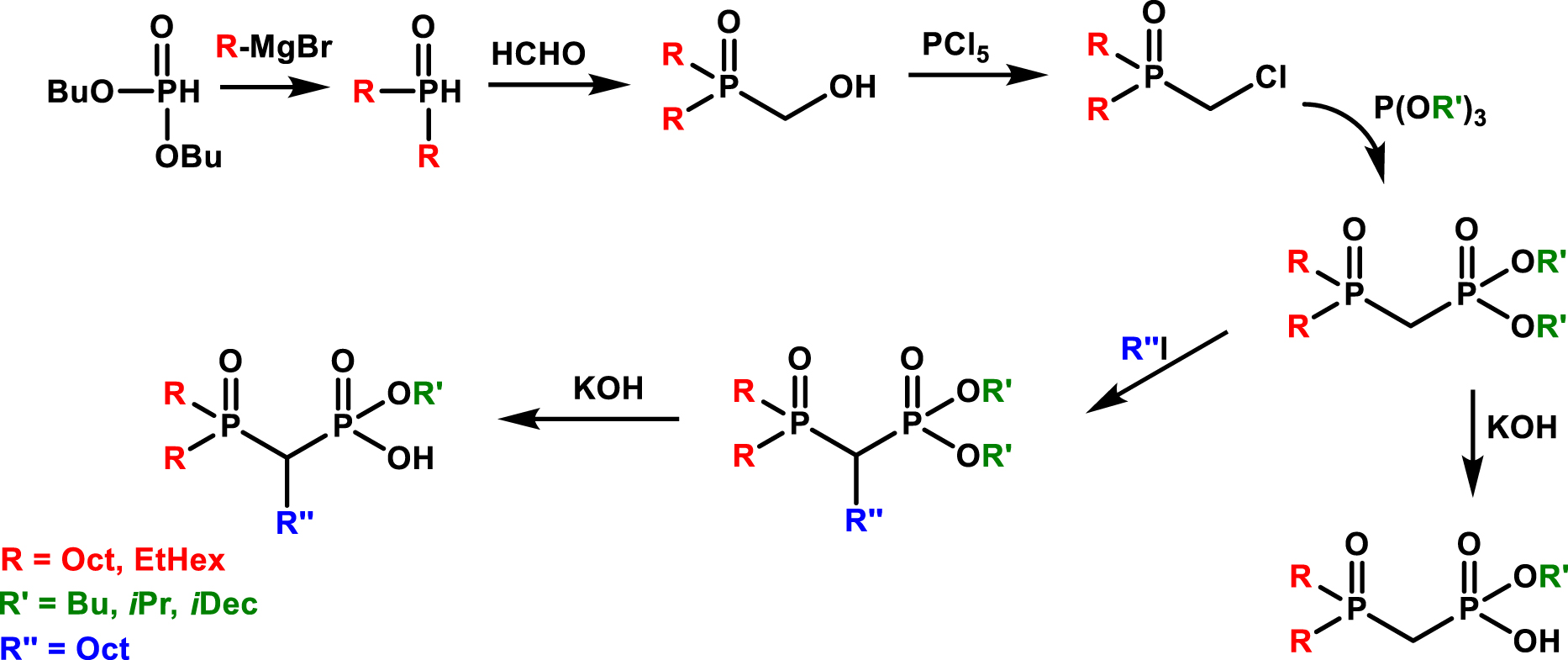
These “autosynergistic” molecules show an increased extraction efficiency for U(VI) compared to Fe(III) from concentrated phosphoric acid solutions (5 mol⋅L−1). This efficiency exceeds that obtained by using two molecules in a synergistic ratio. Furthermore, the selective extraction of uranium from phosphoric acid proves to be superior to the HDEHP/TOPO system, even in the presence of numerous impurities. With such an extractant it is possible to achieve a distribution ratio for U(VI) that is more than 10 times higher than the one achieved with the HDEHP/TOPO system. This superiority has been confirmed by investigations on an authentic industrial phosphoric acid solution.
The exploration of bifunctional molecules, involving the innovative combination of a neutral donor and an acidic extractant, that achieve the selective extraction of uranium from phosphoric acid has been extended to other molecule families. A novel family of bifunctional molecules has been successfully synthesized and extensively examined for their capability to extract U(VI) from phosphoric solutions. This approach is built upon amidophosphonic acid [78] and amino-phosphonate functional groups [79].
The reaction scheme depicted in Figure 11 illustrates the straightforward synthesis of these bifunctional molecules, starting with the amidation of N,N-dialkylamine with halogeno-acylchloride followed by an Arbusov reaction with triethylphosphite to lead to the N,N-dialkylcarbamoylalkylphosphonate. The complete or partial hydrolysis of phosphonate respectively performed with bromotrimethylsilane or sodium hydroxide results in phosphonic acids or monophosphonate molecules. The possibility of introducing an alkyl group on the methylenic bridge was also considered before the hydrolysis step. Many structures have been synthesized according to this approach and then subjected for the extraction and recovery of uranium from phosphoric acid.
General synthesis of bifunctional molecules bearing a combination of amido and phosphoric acid groups and structure of butyl-1-[N,N-bis(2-ethylhexyl)carbamoyl]nonyl phosphonic acid (DEHCNPB) [78, 79].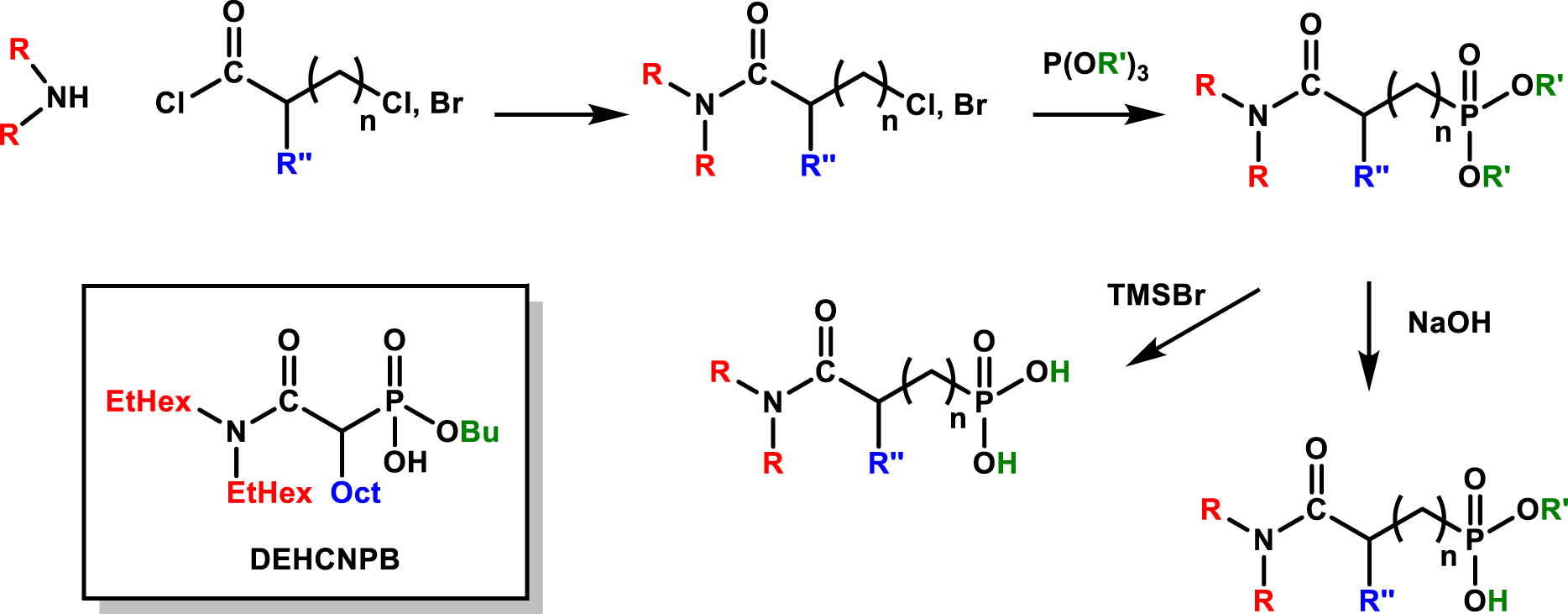
Taking a structure-activity perspective, a specific ligand named butyl-1-[N,N-bis(2-ethylhexyl)carbamoyl]nonyl phosphonic acid (DEHCNPB) has been subject to in-depth study due to its remarkable performance in selectively extracting and quantitatively recovering uranium [79, 80]. These results stand in contrast to those of the URPHOS reference system. Indeed, the extraction efficiency reaches levels up to 90 times greater, and the selectivity is amplified by 150 times compared to the synergistic HDEHP/TOPO reference system. Importantly, this enhanced performance is achieved without any occurrence of third-phase formation during both the extraction and back-extraction processes which is totally reached by using ammonium carbonate.
Currently, this extractant appears to be most effective for the selective extraction and recovery of U(VI) from industrial phosphoric acid solution.
Further advancement includes the design of a comprehensive flow sheet and the ongoing development of a solvent extraction process using DEHCNPB as the central molecule [81].
As with the URPHOS reference system, mechanistic studies at the molecular and supramolecular scale were investigated for the DEHCNPB [79, 80, 82].
Slope methods and electronspray ionization mass spectroscopy (ESI-MS) analysis revealed the presence of complexes with four DEHCNPB [82]. Density functional theory (DFT) coupled to IR studies showed moreover that DEHCNPB molecules solely interact through their phosphonate groups by forming hydrogen bond, while the amide group does not participate to the association. The carbonyl group seems to participate primarily by forming hydrogen bond in the outer coordination sphere. Even if such an outer-sphere bond is not as strong as an uranium-oxygen bond, it was supposed to stabilize the uranyl complex and to favor the extraction of uranyl species. These effects combined with the favorable aggregation properties of the system was associated to the very high extraction efficiency of DEHCNPB toward U(VI).
Classical MD coupled with experimental data from Small-Angle Neutron Scattering (SANS) and Small-Angle X-ray Scattering (SAXS) techniques were further employed to elucidate the aggregation behavior of the bifunctional extractant DEHCNPB [83].
It showed that in the absence of uranium, the organic phase primarily consists of dimers and trimers held together by hydrogen bonds formed through the phosphonate groups, without any water molecules. However, in the presence of uranium, it is observed that two to three extractant molecules directly coordinate with the uranyl cation through their phosphonate groups. Notably, uranyl remains partially hydrated in this organic solution, with the amide groups of the extractants forming hydrogen bonds with the water molecules bound to uranyl.
These intricate hydrogen bond networks surrounding the metallic cation serve to stabilize the complexes and facilitate the extraction process. These findings underline the significance of considering weak interactions to understand extraction processes and highlight how molecular simulations play a pivotal role in providing crucial insights into the intricate chemistry of organic phases characterized by a multitude of species.
8. Molecular systems for the purification of uranium in nitric acid
Uranium refining and conversion, which transforms yellow cake into different types of enriched uranium (uranium dioxide (UO2), natural metallic uranium (U), etc.), is one of the most important steps in the nuclear fuel cycle, as it enables the purity levels required for use in nuclear reactors to be achieved. Uranium purification process consists of three sub-processes: dissolution, solvent extraction, solvent regeneration. The yellow cake is first dissolved in nitric acid to produce a feed solution of metal nitrates. Uranium is then selectively extracted from this acidic feed with TBP diluted in kerosene or another suitable hydrocarbon mixture. Finally, uranium is stripped from the TBP extract in low acidified water to produce a highly purified uranyl nitrate, UO2(NO3)2 [84, 85].
Today, TBP extraction technology and processes are used worldwide for the purification of uranium from nitric acid feed solution. The success of TBP solvent extraction over other uranium purification processes is due to the fact that TBP (i) is highly selective for uranium and provides excellent decontamination from most impurities, (ii) is relatively stable to degradation under conditions normally used for uranium purification, (iii) has physicochemical properties tailored to the process in terms of density, viscosity, corrosion, etc [86].
In addition, TBP is one of the most versatile extractants, used at various stages of the nuclear fuel cycle, most notably in the Plutonium Uranium Reduction Extraction (PUREX) process for reprocessing spent nuclear fuel. In the context of this back-end of the nuclear fuel cycle, despite the performance of TBP, some drawbacks such as: (i) the co-extraction of neptunium and technecium, (ii) the difficulty of performing the back-extraction stage, with a necessary addition of U(IV) and hydrazine to assure the change in the oxidation state of the Pu(IV) to Pu(III), and (iii) the significant degradation of the TBP through radiolysis and the water-solubility of the corresponding degradation products, led researchers to develop alternative extraction agents. Then, several research groups have carried out in-depth studies on uranium extraction with TBP [87, 88, 89, 90].
If alternatives to TBP have been proposed in the context of the PUREX process, some are also proposed for the upstream part of the cycle [51]. Naturally, several organophosphorus derivatives have been proposed, such as HDEHP [91, 92], TOPO [93], OPAP [94], mono-2-ethylhexyl (2-ethylhexyl)phosphonate (PC88A) [95], cyanex compounds [96, 97, 98], but it is the nitrogen-based extractants that have shown the most significant results. Among them, amides and diamides [99, 100] and in particular monoamides have a high potential [101, 102, 103, 104]. Symmetrical and non-symmetrical monoamides with linear or branched chains have been studied. Some of them are grouped in Table 5 [105, 106, 107, 108, 109]. The results of these studies show that depending on the alkyl group on the monoamides, the performance and selectivity of uranium extraction with respect to other cations can be modulated.
General formula of N,N-dialkylamides used for uranium extraction from nitric acid medium
| Name |
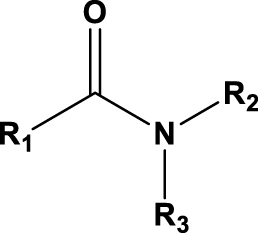 |
Reference | |
|---|---|---|---|
| N,N-di-2-ethylhexylacetamide | R1 = CH3 | R2 = R3 = (C4H9)(C2H5)CHCH2 | [105] |
| N,N-di-2-ethylhexylpropionamide | R1 = CH3CH2 | R2 = R3 = (C4H9)(C2H5)CHCH2 | [105] |
| N,N-di-2-ethylhexylbutyramide (DEHBA) | R1 = CH3CH2CH2 | R2 = R3 = (C4H9)(C2H5)CHCH2 | [105] |
| N,N-di-2-ethylhexylisobutyramide (DEHiBA) | R1 = (CH3)2CH | R2 = R3 = (C4H9)(C2H5)CHCH2 | [105, 106, 107, 108] |
| N,N-di-2-ethylhexylpivalamide | R1 = (CH3)3C | R2 = R3 = (C4H9)(C2H5)CHCH2 | [105] |
| N,N-dihexyloctanamide | R1 = C8H17 | R2 = R3 = C6H13 | [108] |
| N-methyl-N-octyl-2-ethylhexanamide | R1 = (C4H9)(C2H5)CHCH2 | R2 = C8H17 R3 = CH3 | [109] |
Another approach is the design of multifunctional molecules; various multifunctional compounds have shown promising properties (Table 6). For example, the combination of sulfur and nitrogen functions with amidosulfoxide structures [110] as well as the combination of phosphorus and nitrogen functions with iminomethylenediphosphonic acids [111] or aminophosphonic acid and aminophosphonate extractant [112] were suggested for the extraction and purification of uranium from dilute or concentrated nitric acid solutions. Recognizing the potential of this approach, the multifunctional ligands proposed for the extraction of uranium from the phosphoric medium such as amidophosphonates showed interest for the purification of uranium from the nitric medium. The importance of the presence of a biphosphonate moiety rather than a monophosphonate moiety was demonstrated for high extraction efficiency and selectivity. In addition, the bifunctional extractant was found to improve the uranium loading capacity by a factor of about 1.3 compared to TBP and to have a slightly lower solubility in the aqueous phase. As with TBP, uranium extraction is favoured in high nitric acid concentrations, suggesting that back-extraction at low acidity is also possible [113].
General formula of multifunctional molecules used for uranium extraction from nitric acid medium
| Entry | Extractant struture | Extraction condition | Reference |
|---|---|---|---|
| #1 |
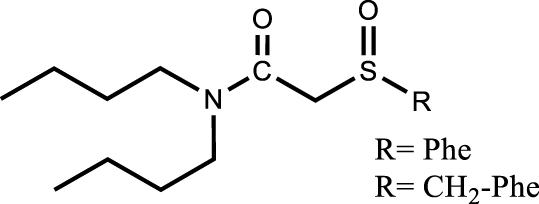 |
[extractant] = 0.5 mol⋅L−1 [U] = 10−10 mol⋅L−1; [HNO3] = 0.5 −−9.5 mol⋅L−1 | [110] |
| #2 |
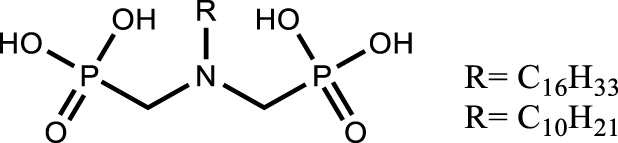 |
[extractant] = 0.03 −−0.3 mol⋅L−1 [U] = 0.015 mol⋅L−1; pH = 2 | [111] |
| #3 |
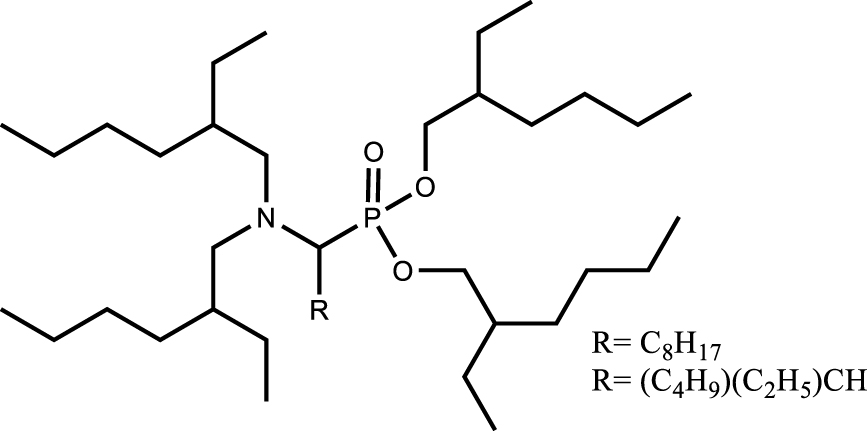 |
[extractant] = 0.1 mol⋅L−1 [UVI = FeIII = MoVI = CeIII = LaIII = GdIII = YbIII = NdIII = TiIV = ZrIV = ThIV] = 250 mg⋅L−1; [HNO3] = 2 mol⋅L−1 | [112] |
| #4 |
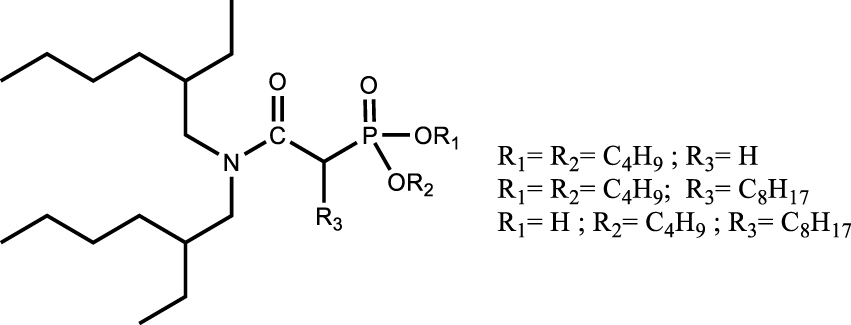 |
[extractant] = 0.2 mol⋅L−1 [UVI = FeIII = MoVI = VIII = ZrIV = ThIV] = 1×10−3 mol⋅L−1; [HNO3] = 4 mol⋅L−1 | [113] |
The extraction mechanisms of two bifunctional extractants, differing only by the grafting of an alkyl chain between their two functions, were studied at molecular and supramolecular scales to investigate the origin of their very different separation factors for uranium and zirconium (Figure 12a) [114]. This investigation aimed to uncover the reasons behind their significantly different separation factors when it comes to uranium and zirconium. By scrutinizing the complex structure through spectroscopic analyses, including FTIR, ESI-MS, and EXAFS, it was established that the alkylation process does not alter the chelation mechanism for uranium and zirconium. The stoichiometries of the formed complexes remained identical for both extractants, resulting in the formation of UO2L2(NO3)2 complexes and zirconium polynuclear complexes upon extraction into the organic phase. Consequently, the investigation shifted its focus to discerning the origins of selectivity by examining the supramolecular self-assembly behavior of these two bifunctional molecules.
(a) U/Zr distribution coefficients and corresponding separation factors for the alkylated and non-alkylated extractants (Alk and NAlk) diluted at 0.5 mol⋅L−1 in dodecane after contact with an aqueous phase: [HNO3] = 4 mol⋅L−1 [U] = [Zr] = 10 mmol⋅L−1. (b) Schematic illustration of the aggregates formed with Alk and NAlk extractants according to their polar and apolar part distribution [114].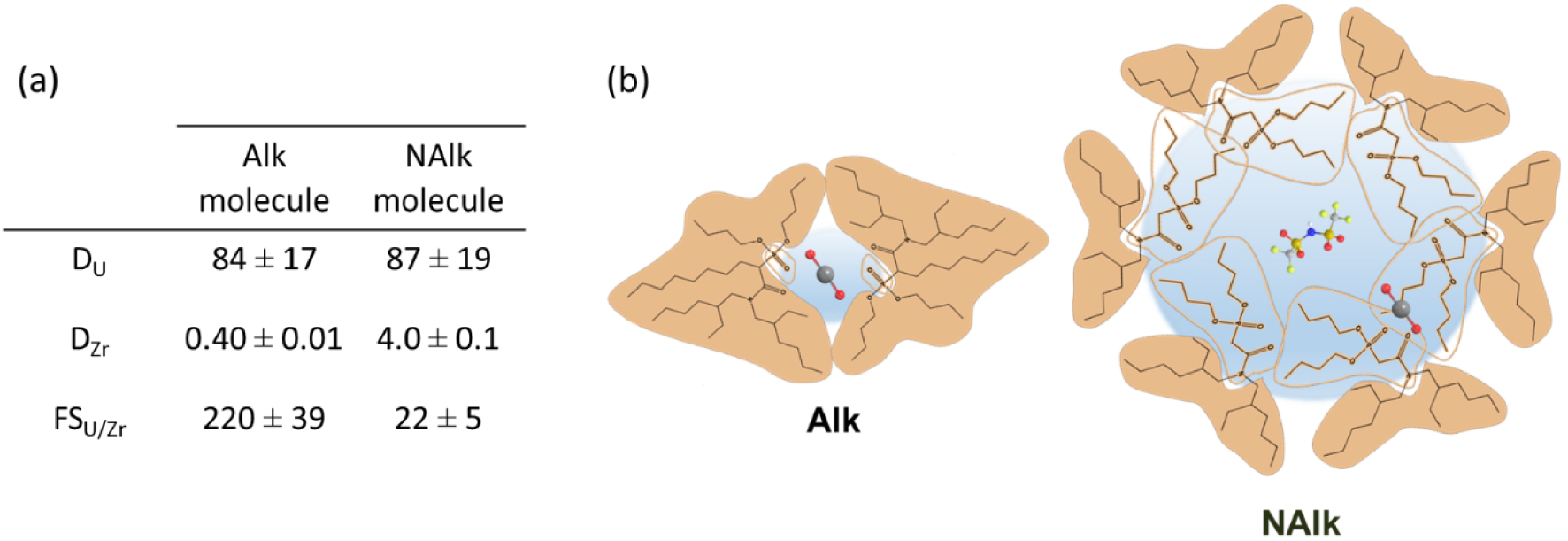
SAXS and SANS showed that the most significant separation factors between uranium (U) and zirconium (Zr) are achieved when smaller aggregates are present (Figure 12b). Consequently, this study strongly suggests that the selectivity is governed by the supramolecular self-assembly of the two extractant molecules. In the case of the non-alkylated molecule, its larger packing parameter leads to the formation of larger aggregates, akin to reverse micelles. These larger aggregates have the ability to extract additional polar species into their polar core through a solubilization effect, which, in turn, reduces the separation factor between uranium and zirconium.
Here it has been demonstrated de novo that the efficiency and the selectivity of a liquid–liquid extraction process may rely on a complex combination of various parameters, among which the colloidal properties of the extraction system are certainly one of the main contributors. However, ability to self-assemble is itself driven by a peculiar combination of the lipophilic-hydrophilic balance of the ligand, the composition of the aqueous phase, the solvophobic effect and the geometry of the ligand [63, 115, 116, 117]. As the geometry of the ligand is tightly linked to its stereochemistry, one interesting open insight was to investigate the SAR between stereochemistry and complexation properties at supramolecular scale. This aspect is discussed below.
9. The influence of the ligands stereochemistry on the liquid–liquid extraction process
9.1. A foreword about significance of the stereoisomerism in the context of hydrometallurgy
By observing carefully all the ligands structures presented in previous sections (see entries 3 and 4 Table 4, for instance) or in the cited literature, the questioning about how extraction may be affected by the stereochemistry of the ligand is perfectly understandable. Indeed, branched alkyl chains and unsymetrically substituted methylene bridges bring chirality and the possibility for isomery of position. Therefore, some studies have been carried out to demonstrate the effect of the diasteroisomerism and/or the regioisomerism on the solvent exchange process. Nevertheless, as ambitious and challenging as they were, these studies have only been sparsely reported during the last four decades.
Pionnering works that concerned the use of two diastereosiomers (cis-syn-cis- and cis-anti-cis-isomers) of dicyclohexano-18-crown-6 ether (Figure 13A) in the context of uranium extraction [118] and reprocessing [119], were achieved during the 80’s and the 90’s by different teams. It was found than the cis-syn-cis-diastereoisomer had typical higher affinity for uranium than that of its cis-anti-cis-diastereoisomer. This was explained by the different orientation of the lone pairs of the oxygen atoms in both isomers as reported by Wang et al. [118], and a decade later, Dietz et al. [120] considered rather the difference of free energy reorganization needed for each complexed isomer. A year later, Tsukube et al. reported that the selectivity towards Ag (I) of a series of tridendate pyridine podans (Figure 13B) could be easily adjusted by tuning the chirality of these derivatives [121]. Short explanations were only given at the molecular scale. Finally, a major step forward was done with the works of Wilden and Weßling who showed that diastereoisomerism of some chiral diglycolamide (DGA) derivatives (Figure 13C) could significantly affect the organization of the ligand and its interaction with other partners than the metal cation (lanthanide or actinide), on the inner- and the outer-sphere of coordination [122, 123].
(A) Structure of the cis-syn-cis- (37) and cis-anti-cis- (38) isomers of dicyclohexano-18-crown-6 ether and respective uranium extraction equilibrium constant Kex [118]. (B) Structure of two diastereosiomers of a chiral podand and respective silver extraction equilibrium constant Kex [121]. (C) Structure of some achiral and chiral DGAs. N,N,N′,N′-tetraoctyldiglycolamide (TODGA) 41 and N,N,N′,N′-tetradecyldiglycolamide (TDDGA) 42 are achiral whereas their respective bis-C-methylated derivatives TODGA-Me2 and mTDDGA can be distinguished in cis-(or meso) (43 and 45) and (44 and 46) trans-isomers. Americium coefficient distribution DAm were obtained with extraction of 10−5 mol⋅L−1 of Am in 4.3 mol⋅L−1 HNO3 performed at 298 K, with 0.1 mol⋅L−1 ligand in TPH [122]. Stability constants for the complexation of Cm (III) was measured in 2-propanol with 5 vol% H2O and 10−2 mol⋅L−1 HClO4 [123]. (D) Structure of various DGAs and associated samarium to lanthanum factor separation (SFSm/Ln = DSm∕DLn) determined by extraction of 5 × 10−4 mol⋅L−1 of lanthanides in 3 mol⋅L−1 HCl with 0.1 mol⋅L−1 ligand in Isopar L with 30 vol% Exxal 13 at 298 K [124].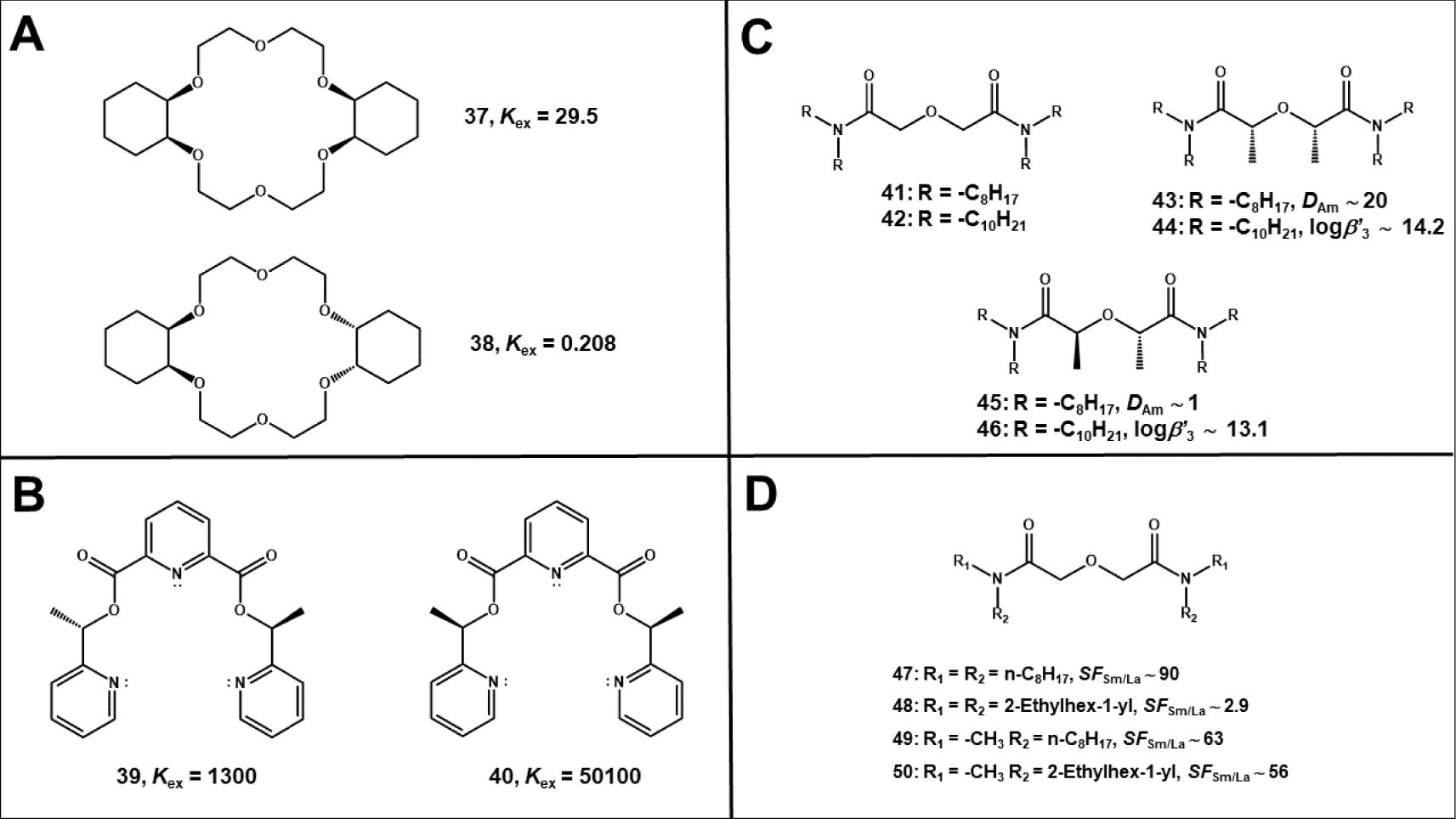
Beside the influence of the DGAs chirality, the effect induced on the liquid–liquid extraction (LLE) process by their regioisomerism was also highlighted by Stamberga et al. who assessed the intra-lanthanide selectivity of several DGAs derivatives (Figure 13D) whose nitrogen substituents were either linear or branched alkyl chains [124]. Although it was not the main goal of these researches that focused more generally on the effect of chains lenght and the position of the branching, conclusions about regioisomerism could be established by comparing the results yielded by some of those DGAs derivatives that carried regiomers of a same alkyl chains (n-octyl, 2-ethylhexyl for instance). Explanations were given at the scale of the aggregate. Sterical hindrance and the interfacial properties varying according to the considered compound were claimed as the main parameters affecting differently the process. In the same way, the studies undertook with linear and branched trialkylamines by our team in the AMEX context (see Section 4) comes to sustain these conclusions.
To summarize, some ensuing questions would be: if both diastereoisomerism and regioisomerism affect the process, which one is predominant? How? Why? What are the challenges afforded by chirality in the context of the fuel cycle?
The answer to the last question seems obvious if one considers that some current and extensive work aims to replace advantageously the TBP at the down-stream of the cycle by other ligands with more complex structures (see Section 8), thus potentially chiral as it is the case of the N,N-di(2-ethylhex-1-yl)butyramide (DEHBA) (Table 5) claimed as promising candidate.
Thus, in order to address the aforementioned resulting concerns, a partnership was established some years ago between the CEA Marcoule Atalante and the ICSM. Our first results and conclusions are briefly reported in the following section.
9.2. Impact of the stereochemistry of the DEHBA on the plutonium (IV) extraction [125]
The aims of this work was to: (i) clarify if the diastereoisomers and the regioisomers of the DEHBA presented marked differences of extraction properties towards uranium and plutonium, (ii) understand and explain the mechanism involved.
DEHBA is a ternary monoamide carrying two chiral centers and presenting three stereoisomers, which are the (R,R)-DEHBA, its enantiomer (S,S)-DEHBA and the achiral meso (R,S)- or (S,R)-DEHBA (Figure 14).
Structure of the stereoisomers of DEHBA (a) and EHOBA (b) [125, 126].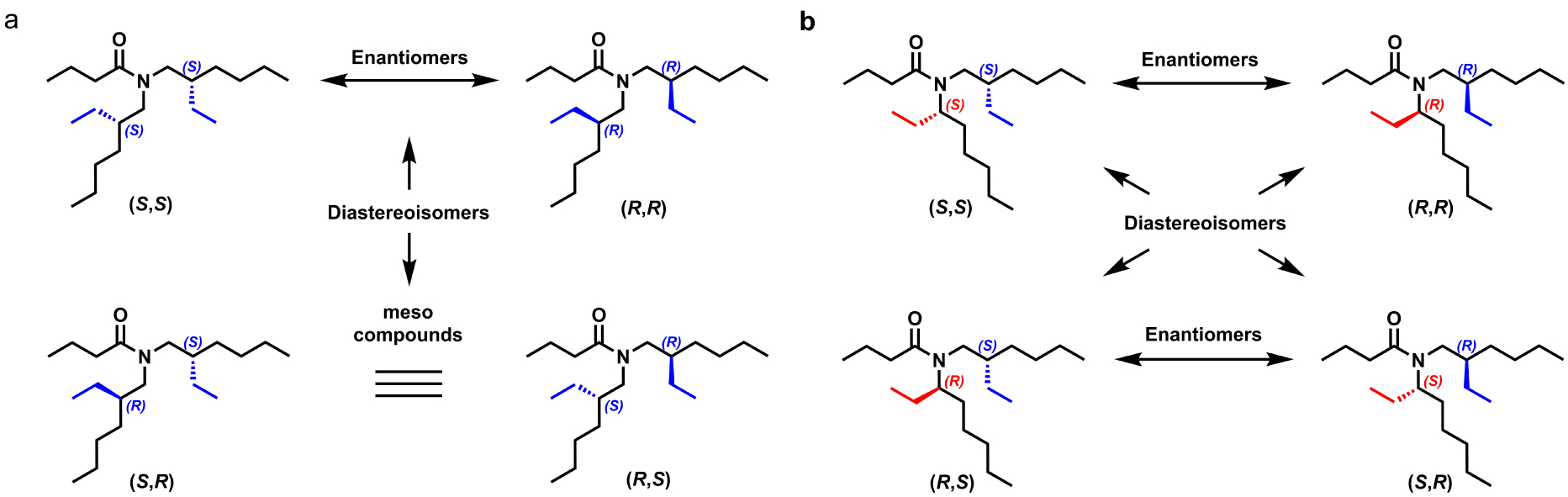
This molecule is commonly obtained as an achiral mixture via a Schotten–Baumann reaction starting from butyryl chloride and commercially available diastereomeric mixture of bis(2-ethylhexylamine). The first part of the project consisted of the development of diastereopure (S,S)- and (R,S)-forms of both the DEHBA and one of its regioisomer, namely the N-(2-ethylhexyl)-N-(octan-3-yl)butyramide, (EHOBA) [125, 127]. The interest in providing EHOBA was to access to a DEHBA isomer for which one ethyl side chain was closer to the complexing amide function. Therefore, if there was one, the impact of chirality was expected to be more pronounced for the EHOBA. The synthesis of the diastereopure forms of the monoamides consisted to react activated butyric acid with the diastereopure forms of the N,N-dialkylamine. These latter were obtained after a multi-steps synthesis. The diastereopure bis(2-ethylhexylamine) was yielded after reaction of the enantiopure 2-ethylhexylamine with enantiopure 3-(bromomethyl)heptane. Both synthons were derived from one of the two enantiomers of 2-ethylhexanoic acid resolved by diastereoselective recrystallization. The diastereopure N-(2-ethylhexyl)octan-3-amine was obtained after reaction of the enantiopure 2-ethylhexylamine with the racemic 3-bromooctane, followed by resolution of both yielded diastereoisomers on a chromatography column (Figure 15).
Retrosynthetic scheme of the synthesis of unresolved and diastereopure DEHBA (A) and EHOBA (B). Butanoyl-X may be a butanoyl halide or a reactive ester of butanoic acid. Unresolveld form of EHOBA is obtained from the diastereoisomers mixture of N-(2-ethylhexyl)octan-3-amine, itself yielded by condensation of racemic 2-ethylhexylamine with racemic 3-bromooctane [125, 126, 127, 128].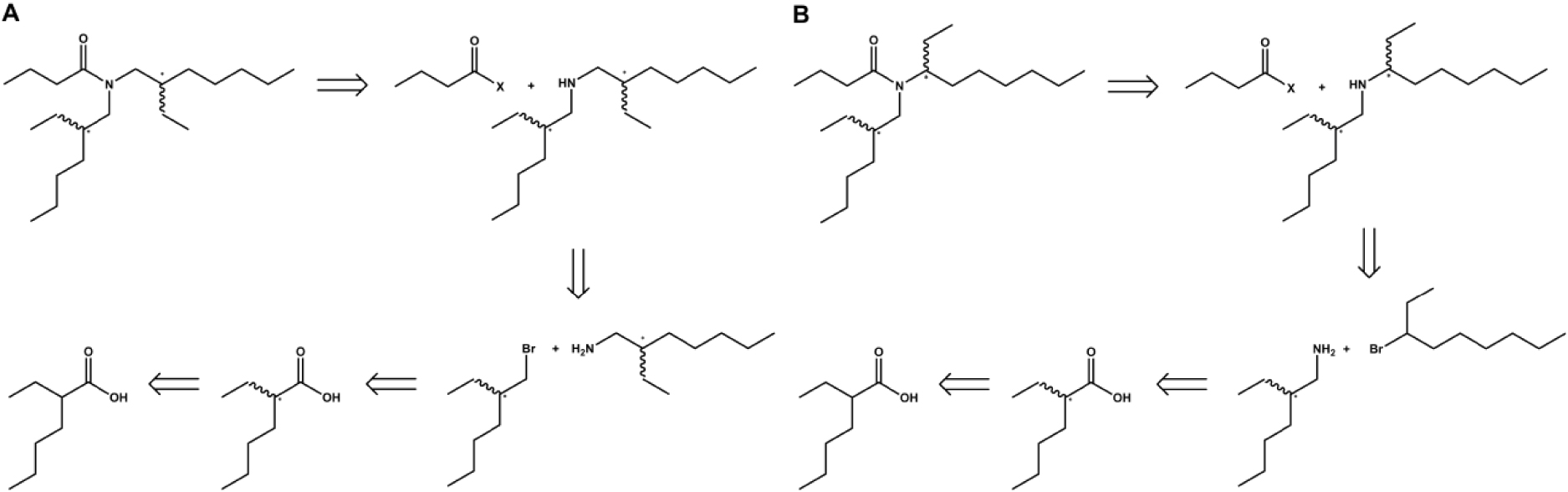
Each diastereoisomers mixture and the diastereopure form of DEHBA and EHOBA were evaluated in the context of U and Pu extraction in nitric media. It was found that the stereochemistry of DEHBA did not affect U extraction (not shown), whereas it impacted the extraction of Pu (Figure 16A).
(A) DPu obtained after extraction of a Pu solution ([PuIV]aq,ini = 2.23 × 10−2 mol⋅L−1, [HNO3]aq,ini = 4 mol⋅L−1) by the various achiral mixtures (mix) or diastereopure monoamides ([Ligand] = 1.2 mol⋅L−1) in hydrogenated tetrapropylene (TPH) at 298 K with 1:1 A:O. The proportion of each type of complex (inner- or outer-sphere) involved in the extraction mechanism was determined UV–vis spectroscopy (not shown). (B) Density functional theory (DFT) model of Pu.monoamide inner- (left) and outer-sphere complex. For simplification, the simulation was achieved with the N,N-diethylpropanamide. Pu:ligand:nitrate stoichiometry was also given for each type of complex [125] (source: Figures 4b and 3 from Ref. [125] under licence CC BY-NC-ND).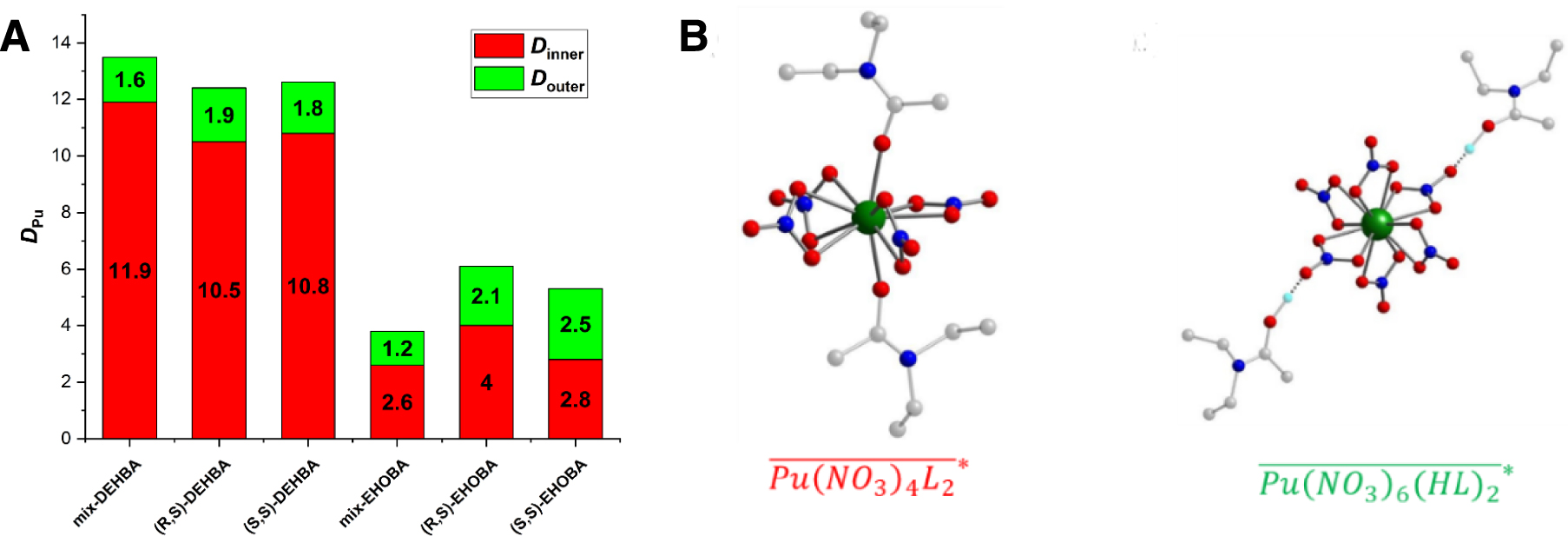
Effect on Pu extraction was shown to be more significant for the EHOBA extractant as the branching is closer (α position) to the complexing site. In both cases, this stereochemical effect could be related to the formation of different inner or outer-sphere complexes (Figure 16B), which are demoted or promoted by steric hindrance.
To summarize, ligands stereochemistry which intervenes at different level in the solvent exchange process should be one of the major concerns to be clarified and addressed for a better understanding of the process. Moreover, once mastered, this key-parameter should allow to finely adjust the process. This last statement paved the way for further fundamental studies aiming to verify the impact of the stereochemistry of branched ligands, be it commonly used at large scale in hydrometallurgic process (HDEHP for instance) or at lab scale (monoamides, amino- and amidophosphonates). These works are currently under development at the ICSM.
10. Conclusion
As the uranium industry has expanded, research efforts have focused on optimizing techniques, especially leaching and liquid/liquid extraction in order to improve both the yield and selectivity toward the targeted uranium as well as the environmental footprint. Future challenges include recovering uranium from more complex resources, which may often be of lower grade and at greater depth than the deposits currently being processed. Thus, research has focused on the improvement of the recovering of uranium from conventional and unconventional resources, including sulfuric, phosphoric and nitric acid, as well as low-grade ores and seawater. The uranium industry aims to develop this wide variety of mining and processing techniques because uranium ores are extremely diverse. For that, processing operations must be designed to take into account the specific composition and mineralogical characteristics of the ore to be treated. Extraction mechanisms of any given extraction operation should be investigated to comprehensively understand and quantify such variability. This understanding is essential for the selection of a combination of unit operations that can economically and environmentally accommodate this variability.
With the common goal of improving and understanding uranium extraction processes, this review aimed to put in regards the various extractant synthesis strategies and the mechanistic studies of uranium extraction in sulfuric, phosphoric and nitric media. As evidenced by the literature, these investigations are fundamental and should encompass a comprehensive characterization that includes both molecular and supramolecular descriptions of the organic phase structure. The alkyl chains of the synthesized and examined extractants, along with those of the diluent, were for instance modified to investigate their impact on the organic phase structure, uranium extraction efficiency, and on the third-phase formation. A study of a system in the absence of diluent was also described, considering the analogy of such a system to ionic liquid media.
Overall, this review shows that synthesizing and studying new extractants for uranium extraction remains essential for improving the efficiency, selectivity, and sustainability of nuclear fuel production while minimizing its environmental impact. These efforts are critical to the continued growth and advancement of nuclear energy technologies.
Abbreviations
| AMEX: |
amine extraction |
| DAPEX: |
dialkylphosphoric acid extraction |
| DBBP: |
dibutyl butylphosphonate |
| DEHBA: |
N,N-di(2-ethylhex-1-yl)butyramide |
| DEHCNPB: |
butyl-1-[N,N-bis(2-ethylhexyl)carbamoyl]nonyl phosphonic acid |
| DGA: |
diglycolamide |
| DU: |
uranium distribution coefficient |
| EHOBA: |
N-(2-ethylhexyl)-N-(octan-3-yl)butyramide |
| ESI-MS : |
electrospray ionization mass spectroscopy |
| EXAFS: |
extended X-ray absorption fine structure analysis |
| FT-IR: |
Fourier-transform infrared spectroscopy |
| HDEHP: |
di-2-ethylhexylphosphoric acid |
| Ils: |
ionic liquids |
| ISL: |
in situ leaching |
| Kex: |
exchange equilibrium constant |
| LCA: |
life cycle assessment |
| LLE: |
liquid–liquid extraction |
| MD: |
molecular dynamics |
| MS: |
mass spectroscopy |
| MOX: |
mixture of oxides |
| NMR: |
nuclear magnetic resonance spectroscopy |
| ORNL: |
Oak-Ridge National Laboratory |
| OPAP: |
octyl phenyl acid phosphate |
| OPPA: |
octyl pyro phosphoric acid |
| PC88A: |
mono-2-ethylhexyl (2-ethylhexyl)phosphonate |
| PUREX: |
Plutonium Uranium Reduction EXtraction |
| REE: |
rare earth element |
| SAR: |
structure-activity relationship |
| SANS: |
Small-Angle Neutron Scattering |
| SAXS: |
Small-Angle X-ray Scattering |
| TAPO: |
trialkylphosphine oxide |
| TBP: |
tributylphosphate |
| TBPO: |
tributylphosphine oxide |
| mTDDGA: |
2,2′-oxybis(N,N-didecylpropanamide) |
| TDDGA: |
N,N,N′,N′-tetradecyldiglycolamide |
| TOA: |
trioctylamine |
| TODGA: |
N,N,N′,N′-tetraoctyldiglycolamide |
| TODGA-Me2: |
2,2′-oxybis(N,N-dioctylpropanamide) |
| TOPO: |
trioctylphosphine oxide |
| TPH: |
hydrogenated tetrapropylene |
| UOX: |
uranium oxide |
| URPHOS: |
uranium from industrial phosphric acid |
| WPA: |
wet-process phosphoric acid |
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Acknowledgements
The authors acknowledge the GDR PROMETHEE, the NEEDS program as well as the CEA for their support. They also thank Tamir Sukhbaatar, Alexandre Artese, Zijun Lu, Nicolas Felines, Julien Rey, Moheddine Wehbie, Olivia Pecheur, Raphael Turgis, Antoine Leydier, Beatrice Baus-Lagarde, Thomas Dumas, Cécile Marie, Manuel Miguirditchian, Thomas Zemb, Jean-Francois Dufrêche, Magali Duvail for their fruitful discussions and contributions on the subject of uranium extraction. The authors are also grateful to the Editors Elsevier, American Chemical Society, Taylord & Francis, Royal Society of Chemistry and Wiley for their applied copyright policy allowing free reproduction and/or modification of all original figures (sometimes only for authors of the original articles, please check those publishers’ policies).