



 CC-BY 4.0
CC-BY 4.0
1. Introduction
The operation of the reactors of the French nuclear power plants is based on a fuel composed of natural uranium oxide, enriched in isotope 235, and for some reactors, of a mixture of uranium and plutonium oxides, known as MOx (Mixed Oxide) fuel. The production of MOx fuel enables recycling both uranium and plutonium present in waste nuclear fuel, and thus a sustainable management of natural nuclear resources [1].
The treatment of spent nuclear fuel is currently carried out in France following the PUREX (Plutonium, Uranium, Reduction, Extraction) process [2]. This consists in dissolving the spent nuclear fuel in nitric acid HNO3, followed by the separation of uranium(VI) and plutonium(IV) from the aqueous medium through liquid–liquid extraction using tributylphosphate (TBP) as an extracting molecule [2, 3]. Afterwards, two back-extraction (stripping) steps toward aqueous phases are performed to separate U from Pu, with the stripping of Pu relying on the reduction of Pu(IV) to Pu(III). Pu(III) is then precipitated from the aqueous phase using oxalic acid (C2O4H2). U(VI) is precipitated into uranium(VI) oxalate using the same technique. The solids obtained are then calcined so as to form the corresponding oxides, mixed (in controlled proportions) to form MOx and shaped into pellets to be used as fuel for the nuclear reactors. Inspired by the PUREX process, the COEX™ process was developed by the CEA (Commissariat à l’Energie Atomique et aux Energies Alternatives) with the aim of decreasing the number of steps during uranium and plutonium recycling, in addition to preventing the generation of pure-plutonium effluents, which may be used in non-civil applications [4, 5]. Similar to the PUREX process, the COEX™ process involves in the first two steps the dissolution of waste nuclear fuel in nitric acid followed by extraction of both uranium and plutonium through liquid–liquid extraction using also TBP. The back-extraction step towards the aqueous phase has been optimized in the COEX™ process, so as to generate, in addition to an aqueous flux containing only uranium, another one containing both uranium and plutonium (U:Pu < 20%) [1]. Once uranium and plutonium from the mixed aqueous flux have been purified from minor impurities by liquid–liquid extraction, this flux can be directly used for the production of MOx fuel. This is accomplished through the oxalic coprecipitation of uranium(IV) and plutonium(III) from the aqueous phase [6], followed by a calcination step, which results in the formation of a mixed oxide (U,Pu)O2 powder. Whether in the PUREX or COEX™ processes, oxalic acid (co)precipitation is carried out in aqueous medium, after a back-extraction step of Pu involving Pu(III). Thus, even though these two recycling methods are efficient in the production of MOx, they still involve numerous steps, especially due to the need to proceed to Pu(IV) reduction into Pu(III). The latter reduction is usually performed using U(IV) generated using hydrazine as a reducing agent, a carcinogenic substance.
Back-extraction of metallic elements from an organic phase through precipitation, also known as precipitation stripping, has been reported in the literature [7, 8, 9, 10, 11]. This process combines both the concentration of metal ions in the aqueous phase (stripping) and precipitation stages when bringing the metal-containing organic solution into contact with the aqueous solution containing the precipitating agent. Most of these examples rely on oxalic acid use, and have been applied to the separation and purification of heavy metals and lanthanides. The process results in the formation of metal oxalate precipitates in the aqueous (C2O4H2) phase, from which they can be collected by filtration. The extraction of plutonium (IV) from a plutonium-containing calcium fluoride slag issued from the production of plutonium metal was realized through direct oxalic precipitation from the organic phase [12]. Plutonium was first extracted in an organic solution containing TBP at 20% (v/v) in kerosene. The plutonium-charged organic phase was then put in contact with an aqueous solution of 0.25 mol/L C2O4H2 in HNO3 (1.5 to 3.0 mol/L). After 30 min of stirring, plutonium was precipitated in the aqueous phase in the form of a wet cake of plutonium oxalate [12]. Later on a process for recovering/separating plutonium(IV) from a mixture of plutonium(IV) and uranium(VI) or americium(III) was reported in the literature, through oxalic precipitation from an organic phase of TBP at 15–30% (v/v) in n-dodecane [13]. This resulted in the precipitation of plutonium in the form of plutonium oxalate in the aqueous phase, whereas uranium(VI) and americium(III) remained mainly concentrated in the organic solution.
To our knowledge, the oxalic coprecipitation of uranium and plutonium from the organic phase has not yet been reported in the literature. Therefore, with the aim of reprocessing uranium and plutonium issued from spent nuclear fuel through the production of MOx, we have studied the possibility of coprecipitating both actinides directly from the organic phase using oxalic acid as a precipitating agent through precipitation stripping. This approach aims to reduce the number of steps required for recycling U and Pu compared to the PUREX and COEX™ processes, by avoiding the back-extraction into the aqueous phase through reduction of Pu(IV) into Pu(III). The key points presented in this study are (1) the efficiency of the precipitation of both actinide(IV) and actinide(VI), and (2) the possibility of controlling the ratio of both metals in the precipitate obtained. The latter objective was motivated by the need to develop non-proliferating spent fuel reprocessing sequences, which avoid isolation of pure plutonium, as detailed in one of our previous studies [14]. This proof-of-concept study was carried out using thorium(IV) as a substitute for plutonium(IV). The solids obtained were characterized to assess the mechanism of precipitation and their suitability for use in the further reprocessing sequence. The presence of an unwanted by-product was evidenced in the case of uranium(VI) precipitation stripping, and the experimental conditions were optimized to inhibit its occurrence.
2. Experimental section
All the reagents were used as received and without any further modification. Uranyl nitrate hexahydrate UO2(NO3)2⋅6H2O and Th(NO3)4⋅5H2O were supplied by the CEA. Tri-n-butylphosphate (TBP, 97% pure), n-dodecane (<99% pure) and oxalic acid dihydrate were purchased from Sigma aldrich/Merck. Powder XRD (PXRD) patterns were obtained on a Bruker D8 Advance diffractometer, using CuKα radiation (𝜆 = 1.5418 Å). Scanning electron microscopy (SEM) images were acquired using a FEI Quanta 200 environmental scanning electron microscope, the morphology of the samples was recorded with a secondary electron (SE) detector, whereas a backscattered electron detector (BSED) was used to detect the presence of the different elements constituting the precipitates through energy dispersive X-ray (EDX) analyses. Fourier transform infrared (FT-IR) spectroscopy analyses were performed on a IR-FT100 spectrometer from Perkin Elmer, equipped with an MIR source, a DTGS detector and an attenuated total reflectance (ATR) measurement mode. Analyses were carried out by placing a drop of the liquid or a pinch of the solid on the surface of the diamond analyzer. Thermogravimetric and thermodifferential analyses coupled with mass spectrometry (TGA-TDA-MS) were carried out on a SETSYS EVO 16 machine supplied by Setaram and coupled with a Hiden QGA300 mass spectrometer for gas analysis.
2.1. Liquid–liquid extraction: obtaining an organic phase highly charged in actinides
The general liquid–liquid extraction protocol involved an aqueous solution of UO2(NO3)2⋅6H2O (0.326 M) or a mixture of UO2(NO3)2⋅6H2O (0.326 M) and Th(NO3)4⋅5H2O (0.18 M) in HNO3 6 M. An organic solution of TBP (1 M) in n-dodecane was prepared and pre-equilibrated with 6 M HNO3 by mixing the two solutions in an ampoule, shaken manually for 10 min. Afterward, both phases were separated (A:O volume ratio equal to 5:1), and the aqueous phase was discarded. The organic solution of TBP and the aqueous solution containing U(VI) and Th(IV) were introduced into an Eppendorf tube and shaken in an Eppendorf Thermomixer at 20 °C and 100 rpm during 10 min. At the end of the extraction step, the organic solution, loaded in actinides, was separated from the aqueous phase. The former was stored for use in organic-phase oxalic precipitation.
2.2. Precipitation stripping of uranium and thorium
An aqueous C2O4H2 solution in HNO3 (see details in the Results and discussion section for the concentrations of C2O4H2 and HNO3, volume 500 μL) was introduced into a 5 mL glass vial equipped with a magnetic stirrer. This was followed by the addition of the organic solution recovered from liquid–liquid extraction in a dropwise manner (500 μL) while stirring at 500 rpm at 20 ± 2 °C. Stirring was maintained for 1–2 h (see the Results and discussion section for the precise stirring time for the different experiments), after which the mixture was centrifuged, and the aqueous and organic phases, in addition to the solid precipitate, were collected. The liquid phases were stored in Eppendorf tubes for further ICP analyses (see below). The solid precipitate was washed with n-dodecane, then with n-heptane, and then dried at 40 °C for a few hours. The precipitation yield was determined from the mass balance after analysis of both remaining liquid aqueous and organic phases.
2.3. Quantification of uranium and thorium in solutions
Quantification of metals in solution was performed using inductively coupled plasma atomic-emission spectrometry (ICP-AES) with a Spectro Acros apparatus from Ametek. Aqueous phases were directly analyzed, using an aliquot directly diluted in a matrix solution consisting of 1% aqueous HNO3 and 1% aqueous HCl, with a minimum HNO3:HCl volume ratio of 9:1. In order to determine the concentration of the actinides remaining in the organic phase after precipitation, these were back-extracted by a solution of HNO3 0.01 M (A:O volume ratio equal to 100:1). The organic and aqueous solutions were introduced into an Eppendorf tube and shaken at 20 °C and 1000 rpm for 10 min. The aqueous phase was then recovered and diluted in a matrix solution consisting of 1% aqueous HNO3 and 1% aqueous HCl with a HNO3:HCl volume ratio of 9:1.
3. Results and discussion
The experimental studies were conducted on an organic phase designed in order to mimic the extraction phase obtained during the first stage of the PUREX process [1]. In the PUREX process, after spent fuel dissolution in nitric acid, uranium(VI) and plutonium(IV) are extracted during a multistage counter-current extraction process, leading to a ca. 100 g/L load in U(VI). In the present study, we employed as organic phase a 1 M TBP solution in n-dodecane, which was loaded with uranium(VI) and thorium(IV) (as a substitute for plutonium(IV)) after a one-stage liquid–liquid extraction of a synthetic 6 M HNO3 aqueous phase, prepared from U(VI) and Th(IV) nitrate salts. The load in U(VI) in the synthetic aqueous phase was close to saturation (0.326 M UO2(NO3)2⋅6H2O), in order to approach the targeted 100 g/L U(VI) concentration in the organic phase. The Th(IV) concentration in the synthetic aqueous phase (0.18 M Th(NO3)4⋅5H2O) was chosen in order to reach a U:Th ratio of about 9:1 in the organic phase after liquid–liquid extraction, a value close to what is expected to occur (for Pu(IV)) when waste MOx fuel is recycled). Concentrations of U(VI) and Th(IV) in the organic phase were therefore 0.28 mol/L and 33 mmol/L respectively, i.e., 66 and 7.6 g respectively.
From a general point of view, actinides(IV) and actinides(VI) are well extracted into TBP-based organic solvents, contrary to actinides(III) (and actinides(V)) [1]. As a consequence, during the PUREX process, efficient stripping of plutonium is performed after reduction of Pu(IV) into Pu(III), which is considered inextractible, with distribution coefficients of about 10−2. This step is required to get complete decontamination of organic phase from plutonium. Use of a low concentration HNO3 aqueous solution leads to complete stripping only when a high aqueous–to–organic phase volume ratio is employed. This leads to complete stripping of U(VI) also, as exploited for the quantification of both U and Th in organic phases employing an aqueous–to–organic phase volume ratio equal to 100. Using a phase volume ratio equal to 1, with a 0.1 M HNO3 aqueous solution, stripping of Th(IV) is incomplete (ca. 90%) and 35% U(VI) is stripped as well. Complete actinide(IV) stripping was therefore approached using oxalic acid. As oxalic acid is not soluble in the organic phase, the stripping of U and Th from the organic phase via precipitation was performed by putting in contact the organic phase with an aqueous C2O4H2 solution. The latter was prepared by dissolving oxalic acid dihydrate in a dilute HNO3 solution or in water.
The coprecipitation of U and Th was carried out through dropwise addition of the actinide solution into the aqueous C2O4H2 solution, using a volume ratio of the precursor solutions equal to one. All experiments were conducted at room temperature in a controlled environment (20 ± 2 °C), and temperature effect was not studied. In a first set of experiments, the concentration of C2O4H2 in the initial aqueous solution was varied between 0 and 0.214 M, while fixing that of HNO3 at 0.1 M (in anticipation of work with Pu(IV) which readily forms hydroxides unless in very acidic media). In these experiments, the quantity of C2O4H2 was maintained lower than the quantity required for the total precipitation of both actinides present in the organic solvent (i.e., the sum of the molar amount of U(VI) and twice the molar amount of Th(IV)), in order to observe selectivity between U and Th precipitation. It was rapidly established that Th precipitates preferentially, even in the presence of U. Based on analysis of both aqueous and organic phases, at [C2O4H2] < 0.114 M, thorium was totally precipitated (Table 1), whereas no traces of uranium were detected in the precipitate. When the amount of C2O4H2 required for complete Th(IV) precipitation (0.07 M expressed in concentration, twice the amount of Th(IV)) was reached, U(VI) precipitation was not observed, and U(VI) was simply stripped into the aqueous phase. At 0.085 M C2O4H2 concentration, about 37% of U(VI) were stripped from the organic phase, with no U(VI) detected in the precipitate. Only at [C2O4H2] ⩾ 0.114 M did uranium start precipitating, with the precipitation yieldincreasing as expected with the increase in C2O4H2 concentration until it reached 46% at 0.214 M. Th was always found in the precipitate (as long as sufficient C2O4H2 was added), but even at high C2O4H2 concentration, U(VI) was distributed between the 3 phases, with non-negligible amount in the aqueous phase. The change in order of addition (i.e., addition of aqueous phase into organic solution) did not change the outcome of the reaction. The U(VI) precipitation was also found to have no kinetic limitation since rapid precipitation was observed, with no evolution after 30 min. The latter observation also revealed that it is difficult to decipher the detailed mechanism of precipitation stripping, i.e., to make the difference between direct precipitation in the organic phase after extraction of C2O4H2 into the organic phase, or stripping of metal cations into the aqueous phase followed by precipitation with oxalic acid in the aqueous phase. As oxalic acid is poorly soluble in organic media, it can be suspected however that the precipitation occurs in the aqueous phase, or at least at the interface between both phases.
Variation of the mass percentage of uranium and thorium in the solid precipitate and organic phase as a function of the concentration of C2O4H2 after organic-phase precipitation
| [C2O4H2] | 0 M | 0.057 M | 0.085 M | 0.114 M | 0.142 M | 0.214 M | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| U | Th | U | Th | U | Th | U | Th | U | Th | U | Th | |
| Organic | 65% | 9% | 68% | N.D. | 63% | N.D. | 56% | N.D. | 51% | N.D. | 37% | N.D. |
| Solid | 0% | 84% | 0% | 99% | 13% | 99% | 23% | 99% | 46% | 99% | ||
| (U:Th)s | 0 | 0 | 1.1 | 1.9 | 3.9 | |||||||
Notes: [HNO3] is fixed at 0.1 M, and the complement to 100% represents the fraction of the considered element in the aqueous phase.
PXRD analysis of the solids obtained (Figure 1) revealed the precipitation of thorium in the form of thorium oxalate hexahydrate (Th(C2O4)2⋅6H2O) in all cases. At the concentration at which uranium starts precipitating (i.e., when [C2O4H2] ⩾ 0.114 M), in addition to the peaks corresponding to Th(C2O4)2⋅6H2O, peaks between 8 and 9° (2𝜃) started to appear, which surprisingly do not correspond to uranyl oxalate trihydrate (UO2(C2O4)⋅3H2O). It is also important to note that the intensities of these peaks increased with increasing concentrations of C2O4H2. Furthermore, these peaks could not be identified based on the ICDD (International Center for Crystal Data) and, thus correspond to an unknown phase. The expected peaks for uranyl oxalate were not detected in the diffractogram (Figure 1).
PXRD patterns of the precipitates obtained from organic-phase precipitation of uranium and thorium at variable C2O4H2 concentrations and a fixed HNO3 concentration of 0.1 M. Reference diffractograms of UO2(C2O4)⋅3H2O and Th(C2O4)2⋅6H2O correspond to JCPDS No. 01-073-7325 and No. 00-022-1485, respectively.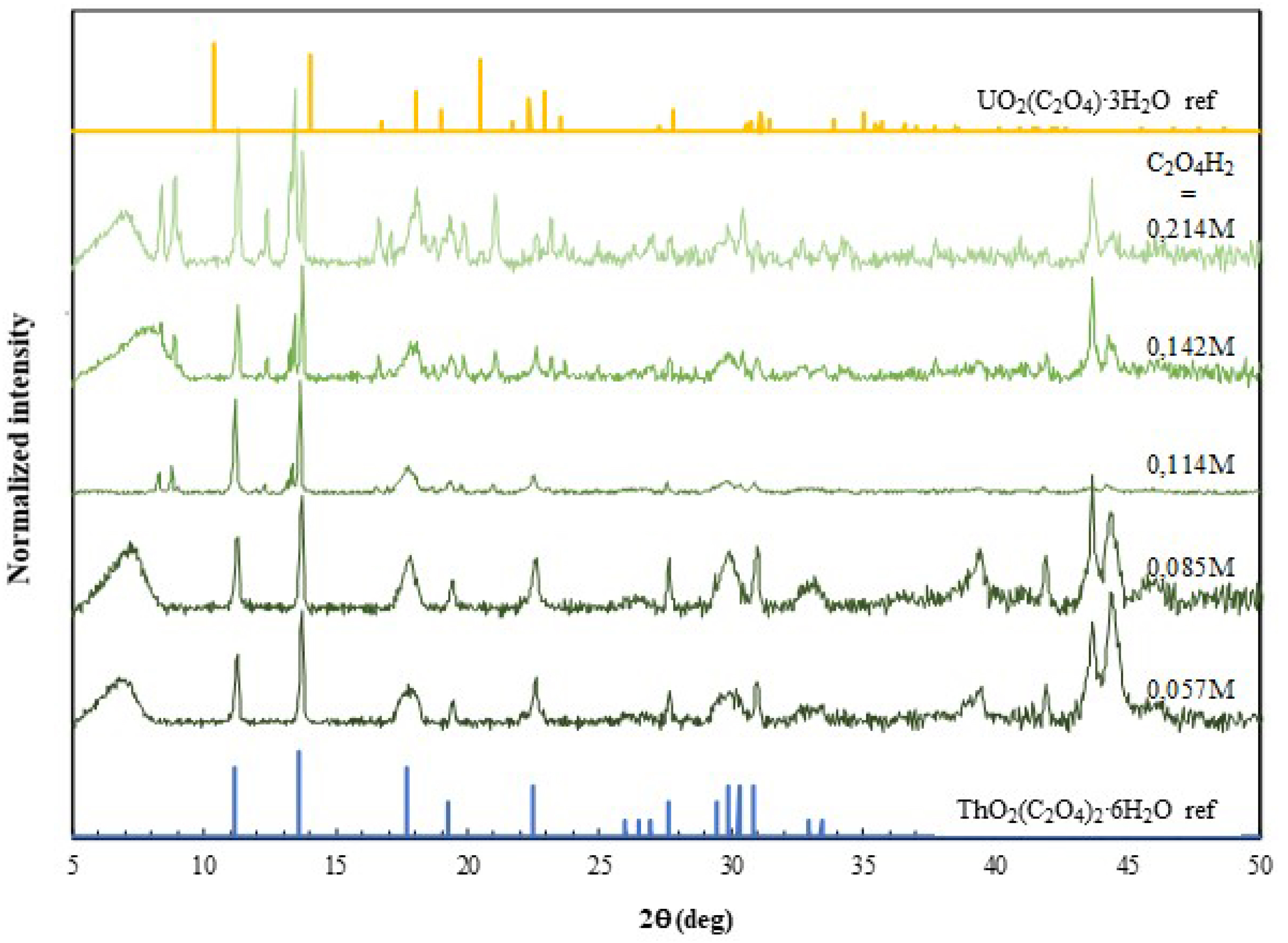
With the aim of determining the nature of the elements constituting the precipitates resulting from the organic-phase precipitation at different C2O4H2 concentrations, EDX analyses were carried out. In addition to Th, U, C, and O, the spectra obtained revealed the presence of phosphorus in the powder obtained for [C2O4H2] ⩾ 0.114 M, whereas no phosphoros was detected at lower concentrations for which only Th was precipitated. These results may indicate that uranium, phosphorus and oxalate precipitate simultaneously. Furthermore, as TBP is the only source of phosphorus in the precipitation medium, it can be assumed that the uranium solid obtained probably contains TBP. Solids composed of uranium(VI) and analogs of TBP, including trimethylphosphate (TMP) and triisobutylphosphate, have already been reported in the literature [15, 16]. Furthermore, Rahman et al. reported the formation of a TBP-containing solid during the precipitation stripping of U(VI) from TBP/kerosene by NH4OH [10]. The presence of TBP in the solid was confirmed through complementary experiments that involved carrying out the precipitation of only uranium under the same precipitation stripping conditions (in terms of C2O4H2 and HNO3 concentrations). The results obtained are presented in the supporting information along with further characterization of the uranium precipitate. These analyses proved the presence of oxalate, nitrate or nitric acid, and uranium in the solid, but could not allow determination of its precise composition. Thus, the mixed-composite uranium solid will be referred to as U–TBP–Ox in the following.
When a phosphorus-containing uranium compound is calcined, a uranium phosphate is expected. Such a product is incompatible with the objectives of this work, which aims at obtaining precursors of actinide dioxides for MOx fuel. Uranium phosphates are minerals known for their relatively high chemical stability [17], once formed, and their transformation into uranium oxide is not possible. It is therefore essential to obtain only actinide oxalates at the end of the precipitation from the organic phase, so as not to introduce any phosphate impurities during the step of conversion into oxide by calcination, which could be difficult to remove [18].
Neither the mode of addition (organic into aqueous or vice versa) nor the stirring speed affected the nature of the precipitate obtained. Therefore, attempts towards precipitating uranyl oxalate from the organic phase without the precipitation of U–TBP–Ox were mainly focused on varying the concentration of C2O4H2 and HNO3 in the precipitation medium. To this end, a new set of experiments was carried out by increasing the HNO3 concentration in the precipitation medium to 2 M (compared to 0.1 M in the previous set of experiments), whereas the oxalic acid concentration was varied between 0.08 and 0.24 M. The results obtained showed that thorium still totally precipitates at all C2O4H2 concentrations, whereas uranium partially precipitates for [C2O4H2] ⩾ 0.2 M (Table 2). It is important to note that, compared to the previous set of experiments (realized at a lower HNO3 concentration), the precipitation yield of uranium decreased, probably as U(VI) is more stabilized in the organic phase. This can be related to the fact that increasing aqueous HNO3 concentration leads to an increase in the distribution ratio of U(VI) between an aqueous nitrate phase and a 30% TBP in n-dodecane organic phase. Consequently, the maximum U(VI):Th(IV) mass ratio reached 1.8, compared to 3.3 at lower HNO3 concentrations. However, compared to the previous set of experiments realized at [HNO3] = 0.1 M, in addition to the formation of Th(C2O4)2⋅6H2O, XRD analyses revealed the formation of UO2(C2O4)⋅3H2O at [HNO3] = 2 M, with the intensity of the peaks corresponding to U–TBP–Ox being relatively low and decreasing with the increase in C2O4H2 concentration (Figure 2). This indicates that increasing the concentrations of both C2O4H2 and HNO3 in the precipitation medium favors the precipitation of uranyl oxalate compared to U–TBP–Ox.
PXRD patterns of the precipitates obtained from organic-phase precipitation of uranium and thorium at variable C2O4H2 concentrations and a fixed HNO3 concentration of 2 M.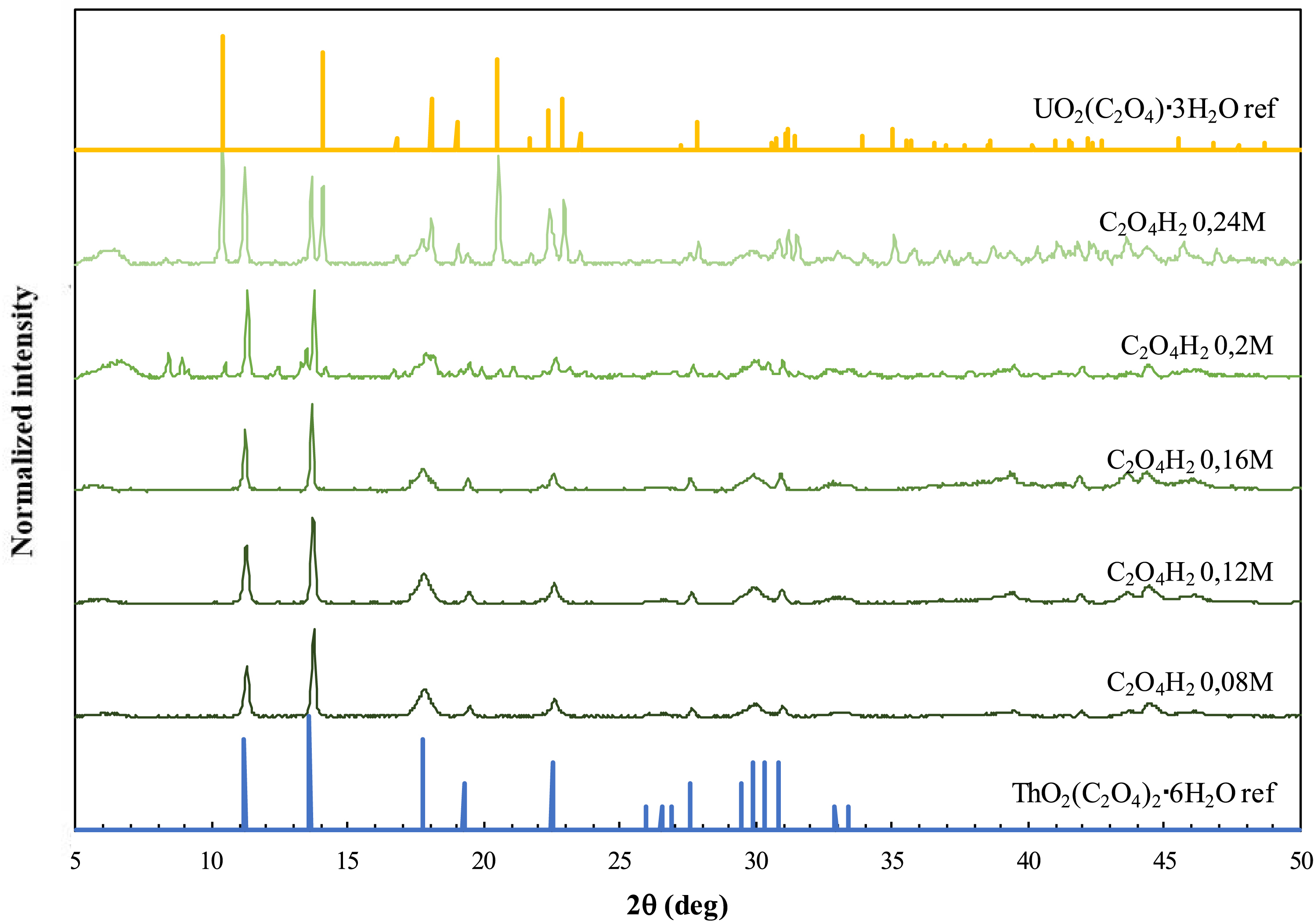
Variation of the mass percentage of uranium and thorium in the solid precipitate and organic phase as a function of the concentration of C2O4H2 after organic-phase precipitation
| [C2O4H2] | 0.08 M | 0.12 M | 0.16 M | 0.2 M | 0.24 M | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| U | Th | U | Th | U | Th | U | Th | U | Th | |
| Organic | 81% | N.D. | 78% | N.D. | 74% | N.D. | 61% | N.D. | 51% | N.D. |
| Solid | 0% | 99% | 0% | 99% | 5% | 99% | 24% | 99% | 33% | 99% |
| (U:Th)s | 0 | 0 | 0.4 | 1.8 | 2.5 | |||||
Notes: [HNO3] is fixed at 2 M, and the complement to 100% represents the fraction of the considered element in the aqueous phase.
Therefore, with the aim of precipitating uranyl oxalate while limiting the precipitation of the U–TBP–Ox impurity, additional experiments were carried out while further increasing both C2O4H2 and HNO3 concentrations to 0.3 and 4 M, respectively. The XRD pattern of the precipitate formed is shown in Figure 3 (top). The results obtained show the precipitation of both uranium and thorium in the form of oxalates, with the complete absence of the U–TBP–Ox impurity, which was also confirmed by EDX analysis, which revealed the complete absence of phosphorus. Moreover, increasing the concentration of C2O4H2 and HNO3 increased the precipitation yield of uranium to 95%, with Th still being totally precipitated and the U–TBP–Ox impurity not detected.
PXRD patterns of the precipitate obtained from organic-phase precipitation of uranium and thorium under optimized conditions ([C2O4H2] = 0.3 M and [HNO3] = 4 M) after washing with dodecane (top) and washing with water (bottom).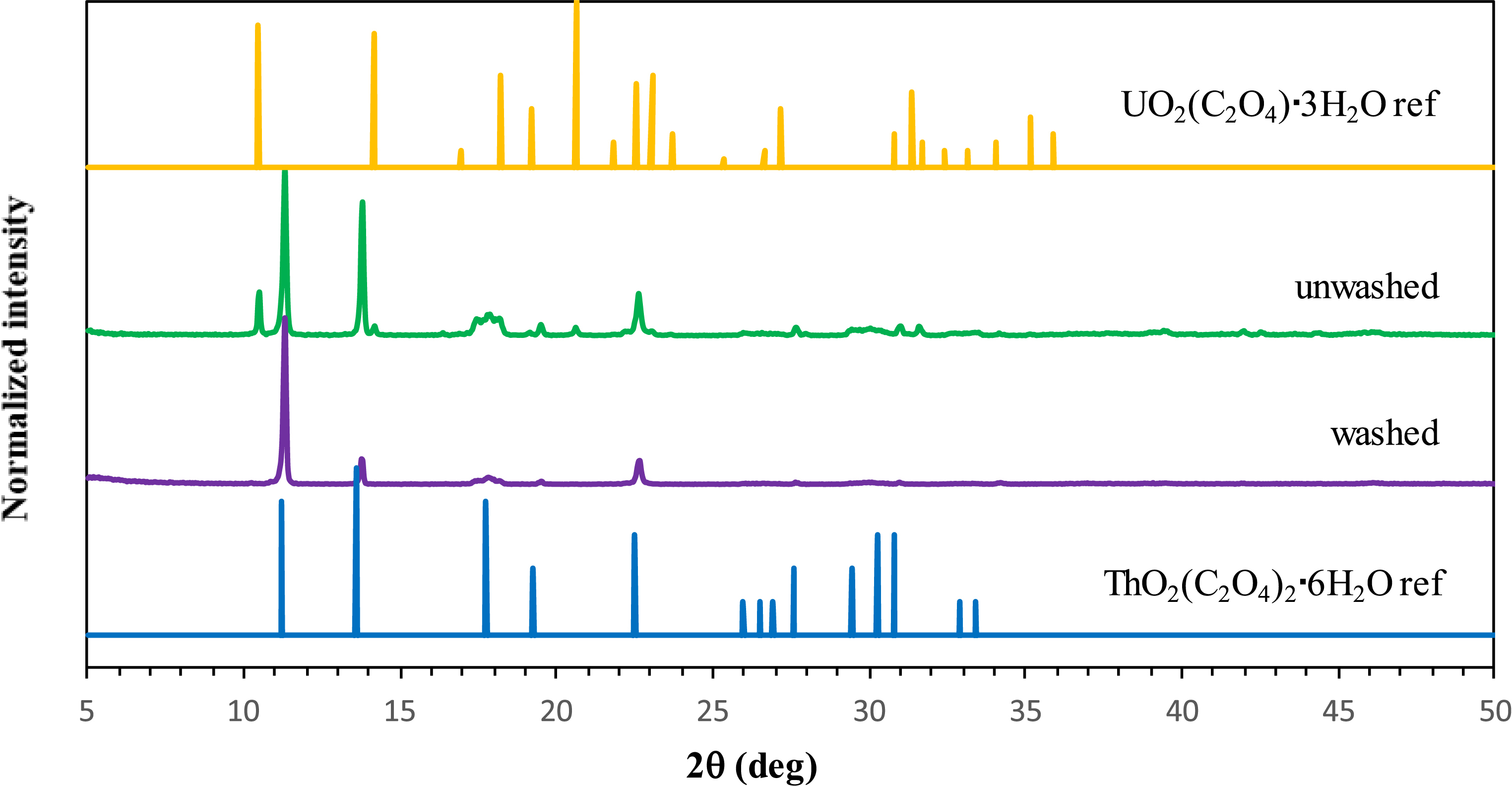
The control of the U:Th mass ratio during the precipitation experiment seems to be the result of a higher solubility of uranyl oxalate (LogKs = −8.5 [19]) compared to thorium oxalate (LogKs = −24 [20]). As the latter compound is more stable, it is reasonable to suppose that its precipitation occurs more quickly. Furthermore, uranyl oxalate is expected to be slightly soluble in water at 20 °C, with a solubility of about 23 mg/L (0.05 mmol/L, corresponding to 13 mg/L uranium). On the contrary, thorium oxalate has a very low solubility, in the nanomolar range. Thus, a post-synthetic treatment of the precipitate was proposed in order to remove uranyl oxalate from thorium oxalate through washing: the precipitate was washed with distilled water. The elimination of uranyl oxalate could be observed by the color of the precipitate which changed from pale yellow to white after the second wash, as uranyl oxalate is characterized by its pale yellow color. Moreover, ICP analysis performedon the supernatant revealed the presence of uranium (no traces of thorium) after the first and second wash. XRD analysis (Figure 4) performed on the precipitate revealed the presence of only Th(C2O4)2⋅6H2O with no traces of UO2(C2O4)⋅3H2O.
SEM analysis of the precipitate obtained from organic-phase precipitation of uranium and thorium under optimized conditions ([C2O4H2] = 0.3 M and [HNO3] = 4 M): SE mode (left) and EDX-based elemental mapping of Th and U (right).
Therefore, following the oxalate precipitation from the organic phase, the uranium- and thorium-based precipitates can be separated through washing with water, and the U:Th ratio in the solid material can be further adjusted with Th always remaining in the solid. This allows a perfect control of the oxalate mixture composition before calcination. Uranium can be recovered from the remaining phases, and recovered through precipitation after addition of further C2O4H2. The proposed approach thus enables rapid recovery of Th(IV) from the U:Th mixture in the organic phase, either as such, or in the presence of a controlled amount of U(VI). Such studies pave the way towards the reprocessing of U(VI)/Pu(IV) phases, in order to enable multirecycling of Pu(IV) into MOx fuel.
These results suggest that uranium and thorium precipitate separately and do not coprecipitate to form a single material. Based on SEM analyses, the precipitate obtained exhibits a platelet shape (Figure 4). Moreover, EDX analysis reveals that U and Th are not systematically colocalized: some microdomains are Th-rich, whereas some others are U-rich (Figure 4). Altogether, whether the precipitation occurs in organic or aqueous phase, or at the interface, Th(IV) precipitates first as thorium oxalate, followed by U(VI) as uranyl oxalate as soon as acid content of the aqueous phase is sufficient. The U:Th ratio is monitored through either adjustment of the C2O4H2 concentration, or after precipitation via selective leaching of uranyl oxalate.
4. Conclusion
During this study, we established the possibility of recovering Th(IV) by precipitation stripping using oxalic acid C2O4H2, in the presence of a controlled amount of U(VI), from an organic phase mimicking that which arises from the classical PUREX process. Th(IV) is quantitatively stripped as long as sufficient C2O4H2 is introduced, whereas U(VI) only partially precipitates unless excess C2O4H2 is employed. Furthermore, the results obtained show that clean precipitation of U and Th from the organic phase in the form of pure actinide oxalates was attained by controlling the concentrations of C2O4H2 and HNO3 in the precipitation medium. The U:Th ratio in the final solid is controlled by the amount of C2O4H2 employed, and can be further adjusted through washing with water as UO2(C2O4)⋅3H2O is more soluble in water than Th(C2O4)2⋅6H2O. Compared to the PUREX process currently employed at industrial scale to recover U and Pu from waste nuclear fuel for the further production of MOx, precipitation stripping seems to be highly promising, as it allows the stripping of actinide(IV) without the aid of a reducing agent. Furthermore, this work opens the door for the application of precipitation stripping using other organic ligands as precipitating agents, which may lead to various morphologies of the uranium- and thorium-based precipitates and, eventually, to better control over the calcination and pelletization steps.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.