



 CC-BY 4.0
CC-BY 4.0
1. Introduction
Structure-privileged phosphonates have attracted considerable attention in the design of biologically active molecules [1]. These compounds have a variety of interesting biological activities. Among their important effects, they are used as antiviral agents; antiretroviral, antitumoral, antibacterial [2], anticancer, anti-HIV [3], and anti-influenza [4] compounds; vitamin D analogues [5]; and as potent antibacterial and antifungal agents [6].
In addition, we have further demonstrated that Morita–Baylis–Hillman (MBH) allylic and cyclic phosphonates exhibit a powerful antioxidant effect [7] and display a remarkable antimutagenesis effect [8]. Conversely, we observed the absence of antifungal effects in these same MBH phosphonates, whereas their MBH acetate precursors exhibit such effects [9].
Beyond their diverse biological applications, phosphonates play pivotal roles in organic synthesis [10], notably in Wittig–Horner reactions [11, 12, 13], and they can be used as complexing agents [14, 15]. In the industrial field, they are recognized as effective inhibitors that protect metals against excessive dissolution due to corrosion [16, 17, 18, 19]. Furthermore, phosphonates serve as phosphorus-based flame retardants, primarily acting in the solid phase of burning polymer materials. By inducing carbonization of the polymer, they inhibit the pyrolysis process crucial for fueling flames [20].
The synthesis of phosphonates is based on several pathways. For example, we synthesized MBH phosphonates by the reaction of phosphite with MBH acetates, using DMAP or imidazole as a catalyst following an SN2′–SN2′ mechanism (Scheme 1, route A) [21]. The same reaction was applied to MBH alcohols to synthesize the same MBH phosphonates (Scheme 1, route B) [22].
Synthesis of phosphonates.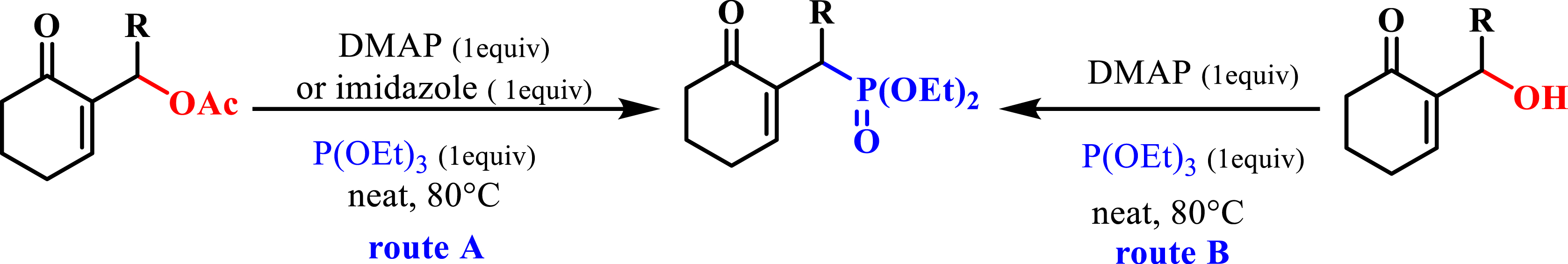
Other researchers have synthesized phosphonates from dialkylmethylphosphonates with n-BuLi as a strong base in the presence of an allylic ester [5].
The Arbuzov reaction is a widely used method for phosphonate synthesis. It involves the reaction of trialkylphosphite with halogenated derivatives or derivatives bearing a leaving group, ultimately yielding the corresponding phosphonate [23].
It is worth noting that the Pudovik reaction, involving the reaction of phosphites or hydrogen phosphonate with aldehydes in the presence of a catalyst, can ultimately lead to hydroxyl phosphonates [23, 24, 25].
While surveying the literature for synthesizing phosphonates from phosphites, we found that quaternary amine salts were used as alkylating agents. Notably, Yaccoubi et al. demonstrated that the synthesis of allylic phosphonates of acyclic MBH adducts was achieved by the reaction of trialkylphosphite with quaternary amine salts [26].
Recently, several authors have synthesized phosphonates using phosphonium salts as starting substrates. Phosphonium salts have been widely used as acceptors of nucleophiles such as amines (Scheme 2) [27, 28], thiols [27, 28], aryls (Scheme 2) [29, 30], and particularly phosphites [31, 32, 33].
Synthesis of phosphonate from phosphonium salts.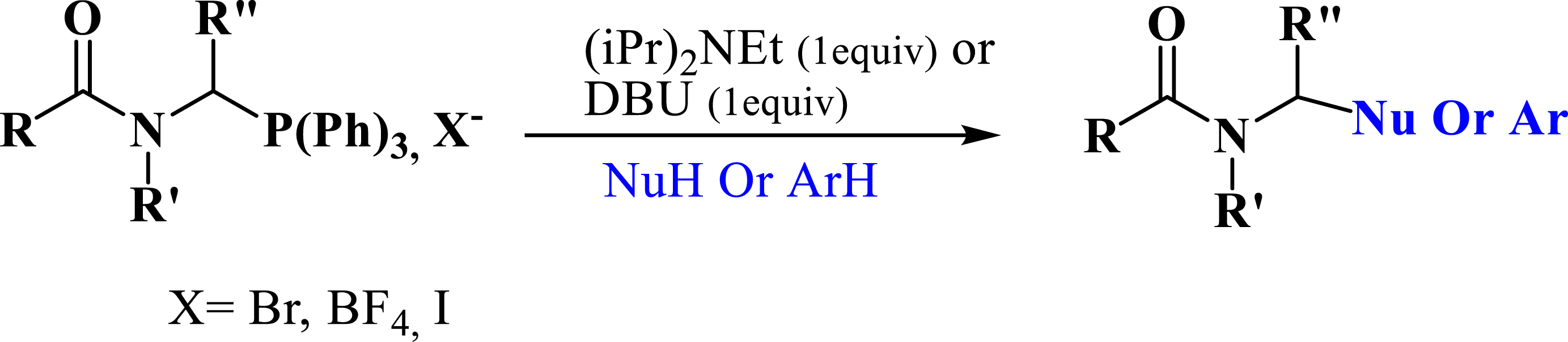
In our study, we synthesized the phosphonium salts of MBH adducts using the Wittig method [34]. It is important to note that the MBH acetates utilized in our previous work necessitate additional reagents for their synthesis, and their purification on silica gel with organic solvents is time-consuming and expensive. Our quest was to find a faster and more cost-effective approach to synthesize phosphine oxides with good yields and without relying on MBH acetates. Recognizing the versatility of our MBH phosphonium salts and seeking to expand their applicability, we report herein a novel method involving the reaction of trialkylphosphites and ethoxydiphenylphosphine as powerful nucleophilic agents with phosphonium salts for synthesizing phosphonates and phosphine oxides from MBH. These compounds hold promise for exhibiting diverse biological properties [7, 8].
2. Result and discussion
2.1. Preparation of phosphonium salts
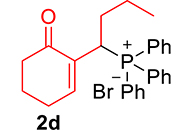
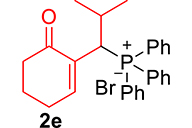
The phosphonium salts were prepared following the method described by our group [34]. Treatment of primary MBH alcohol 1a (R=H) with aqueous HBr (48%) (5 equiv) and triphenylphosphine (1 equiv) in CH2Cl2 at room temperature produces 2a as a white solid, within 20 min, which was isolated and recrystallized with ethyl acetate (Scheme 3). The reaction is general, and 2b–2d were obtained with yields up to 94%.
Synthesis of phosphonium salts 2a–j.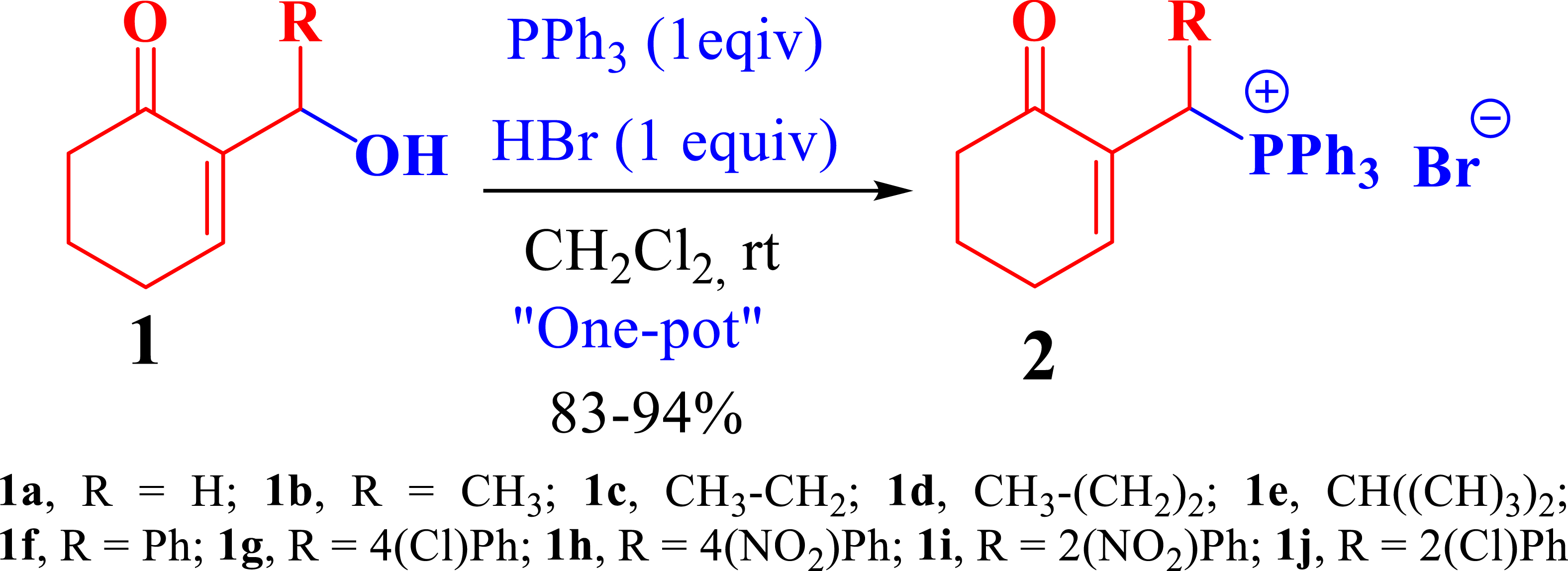
2.2. Alkylation of phosphites with phosphonium salts of MBH
To synthesize the allylic phosphonates of MBH 3 (Scheme 4) from the phosphonium salts of MBH 2, we drew inspiration from prior work [31, 32, 33].
Synthesis of Morita–Baylis–Hillman phosphonates.
First, we used DMAP as an additive to catalyze the reaction as demonstrated in our previous work [21]. Ethoxydiphenylphosphine reacted with the phosphonium 2a salt in the presence of DMAP (1 equiv) in refluxing CHCl3. Unfortunately, despite a 24 h reflux, the reaction did not proceed. Subsequently, we replaced DMAP by imidazole (1 equiv) and repeated the reaction in refluxing chloroform. However, even after 24 h, we recovered the starting materials. Consequently, we concluded that neither DMAP nor imidazole is effective under these reaction conditions.
In our second attempt, we replaced DMAP and imidazole by DBU (1 equiv) and conducted the reaction with the mixture in refluxing CHCl3. Utilizing trialkylphosphite by default allowed us to establish the reaction time, as the phosphonium salts tend to be sticky on thin-layer chromatography (TLC) plates, making it difficult to observe the reaction progress. Under these conditions, the trialkylphosphite reacted with the phosphonium salts of MBH, resulting exclusively, and with very good yield, in the corresponding phosphine oxides and phosphonates.
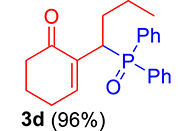
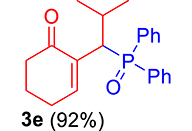
Finally, by determining the reaction time, and to avoid wasting the prepared phosphonium salts, phosphonium 2a–2e salts (1 equiv) were made to react with trialkylphosphite or ethoxydiphenylphosphine (1.1 equiv) in the presence of 1 equiv of DBU in refluxing CHCl3. This reaction is completed after 1 h, yielding MBH phosphonates in very good yields (90–99%) (Scheme 4, Table 1).
Synthesis of phosphine oxides and phosphonates 3a–m from phosphonium salts 2a–m
| Entry | Phosphonium salt 2 | Phosphonate/oxide phosphine 3 (Yield (%)) |
|---|---|---|
| 1 |
 |
 |
| 2 |
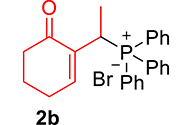 |
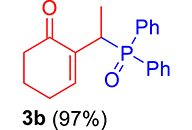 |
| 3 |
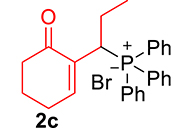 |
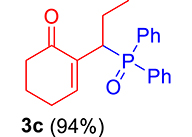 |
| 4 |
 |
 |
| 5 |
 |
 |
| 6 |
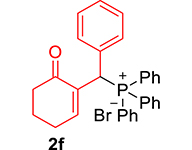 |
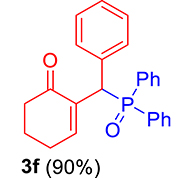 |
| 7 |
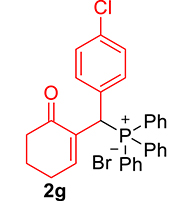 |
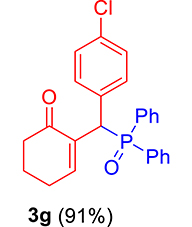 |
| 8 |
 |
 |
| 9 |
 |
 |
| 10 |
 |
 |
| 11 |
 |
 |
| 12 |
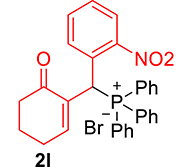 |
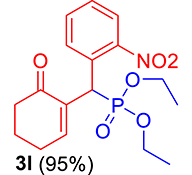 |
| 13 |
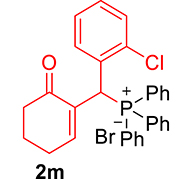 |
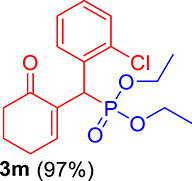 |
The reaction mechanism involves a 1,4-addition of DBU to the cyclohexenone moiety, accompanied by the cleavage of the C–P bond and subsequent elimination of P(Ph)3 via an SN2′ mechanism. Simultaneously, the trialkylphosphite attacks the opposite side of the C=C double bond by following an SN2′ mechanism. This leads to a repositioning of the electrons towards their original configuration, resulting in the elimination of DBU. Ultimately, an Arbuzov-type rearrangement occurs, leading to the formation of phosphine oxide (Scheme 5).
Reaction mechanism and synthesis of Morita–Baylis–Hillman oxide phosphine.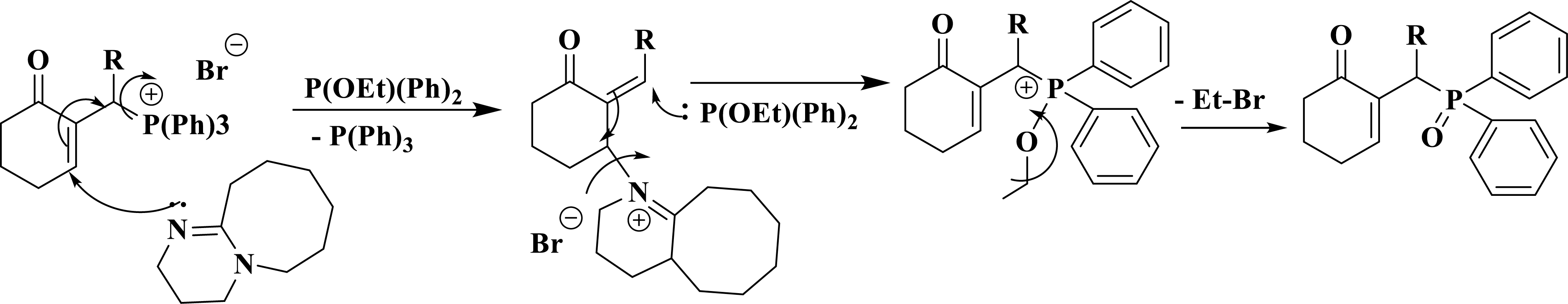
The succession of two SN2′ mechanisms generates an SN2 type mechanism.
This method exemplifies the principles of reducing resource use, such as solvents of the process, to produce MBH acetates efficiently. Conversely, a straightforward purification of the phosphonium salts was achieved through simple washing with a small volume of ethyl acetate or ether. Moreover, their heightened electrophilicity, in comparison to MBH acetates, broadens their applicability in organic chemistry, extending beyond the Wittig reaction to include a wider range of synthetic reactions.
Typically, MBH acetates are synthesized from alcohols 1 (Scheme 6) [21] using acetic anhydride, Et3N, and DMAP. The resulting MBH acetates were then neutralized with HCl and extracted with CH2Cl2. Subsequently, purification involves chromatographic column separation on silica, requiring considerable quantities of the organic solvent (such as petroleum ether or ether) to remove various impurities. In contrast, the phosphonium salts necessitate only a simple washing with ether or ethyl acetate, and in much small amounts for purification. This efficient method proves invaluable for synthesizing phosphine oxides and phosphonates, which can be involved in diverse biological and chemical applications, while ensuring the prevention of secondary reactions. Moreover, it eliminates the need for multiple reagents and catalysts like acetic anhydride, Et3N, and DMAP, thereby simplifying the synthesis process, accelerating it, and reducing costs significantly (Scheme 6).
Synthesis of MBH acetates from MBH alcohols.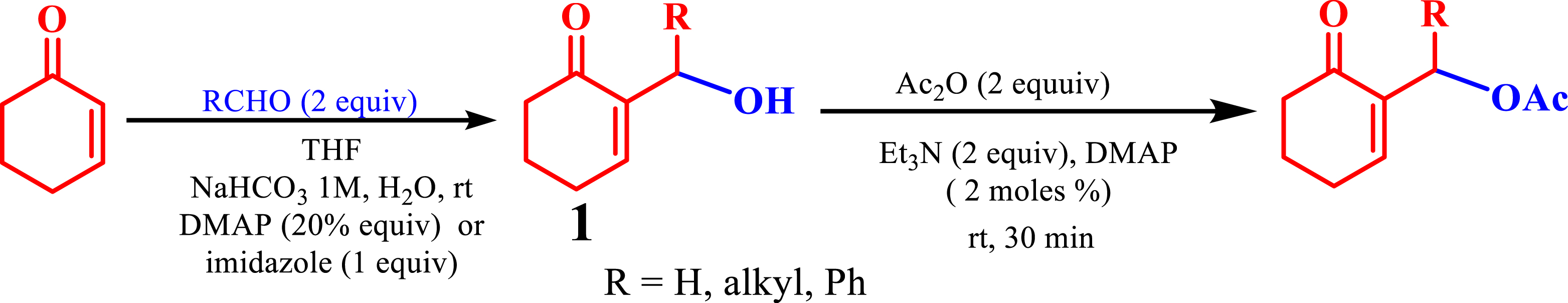
The results and yields are reported in Table 1.
2.3. Conclusion
In this study, we have developed a method that prioritizes atom economy and minimizes the need of a solvent for purification in the synthesis of phosphine oxides and allylic phosphonates of MBH adducts. To this end, we employed phosphonium salts 2 as reagents capable of undergoing a single-step reaction with trialkylphosphites and ethoxydiphenylphosphine. Besides their utility in the Wittig reaction, the resulting products hold promise for their strong complexation abilities. Previous research has emphasized the reliability of similar compounds in facilitating the assembly of complex molecular architectures [14, 15, 35, 36, 37].
3. Experimental section
3.1. General considerations
3.1.1. Materials and methods
1H NMR and 13C NMR spectra were recorded at 300 and 75 MHz, respectively, in CDCl3, using TMS as an internal standard (chemical shifts in δ values, J in Hz). High-resolution mass spectra (HRMS) were recorded as TOF-HRMS on a micromass spectrometer. Analytical TLC was performed using silica gel 60 F254 precoated plates. Visualization was achieved by UV light (254 nm). Flash chromatography was performed using silica gel 60 and a gradient solvent system (petroleum ether/ether) as the eluent.
3.2. Synthesis of phosphonium salts 2
In a round-bottom flask, a combination of 2-hydroxymethylcyclohex-2-en-1-one (1a) (1 g, 7.93 mmol, 1 equiv) and triphenylphosphine (2.49 g, 9.51 mmol, 1.1 equiv) in CH2Cl2 (20 mL) and an aqueous solution containing 48% HBr (5 mL, 39.65 mmol) were introduced. The resulting mixture was stirred at room temperature for 20 min, during which the progress of the reaction was monitored using TLC. Once the reaction was completed, the mixture underwent hydrolysis with H2O, followed by multiple extractions with CH2Cl2. After drying the organic phase over MgSO4, the solvent was evaporated, yielding a white solid. This solid was subjected to recrystallization using EtOAc, filtered through a sintered glass, and rinsed with Et2O, resulting in the isolation of pure 2a–m as a white solid.
3.3. Synthesis of phosphonates 3
In a small round-bottom flask equipped with a stirrer and reflux condenser, phosphonium salt 2 (1.55 mmol, 1 equiv) was dissolved in CHCl3, followed by the addition of trialkylphosphite or ethoxydiphenylphosphine (1.70 mmol, 1.5 equiv) and DBU (0.155 mmol, 0.1 equiv). The mixture is then refluxed for 1 h. The progress of the reaction was monitored using TLC. Upon completion, the reaction mixture was neutralized with a 4 N HCl aqueous solution and extracted with CH2Cl2. The organic phase was dried over MgSO4, and CH2Cl2 was removed under reduced pressure. Subsequently, products 3 were purified by column chromatographic silica gel, using CH2Cl2/Et2O (40:60) as the eluent. The resulting products 3a–m were obtained as solid compounds with excellent purity.
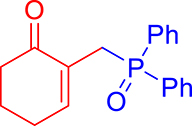
3.3.1. 2-((Diphenylphosphoryl)methyl)cyclohex-2-enone (3a)
mp: 106–107 °C; Yield: 93% (465 mg on 1.62 mmol reaction scale); 1H NMR (300 MHz, CDCl3): δ: 1.79 (m, 2H), 2.22 (t, J = 6.0 Hz, 2H), 2.31 (m, 2H), 3.37 (d, JP–H = 12.0 Hz, 2H), 7.31 (t, J = 6.0 Hz, 1H), 7.44–7.77 (m, 10H); 13C NMR (75 MHz, CDCl3): δ: 22.6, 26.2, 27.8 (d, J = 68.8 Hz), 37.6, 128.2–131.6 (aromatics), 133.1 (d, JC–P = 3 Hz), 150.2 (d, JC–P = 7.5 Hz), 197.2 (d, JC–P = 4.5 Hz); 31P NMR (121 MHz, CDCl3): δ: 30.4; HRMS (ESI-TOF): [M+H]+ calcd for C19H20O2P: 311.1195. Found: 311.1206.
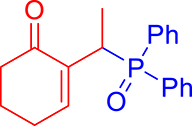
3.3.2. 2-(1-(Diphenylphosphoryl)ethyl)cyclohex-2-en-1-one (3b)
mp: 112–113 °C; Yield: 97% (485 mg on 1.543 mmol reaction scale); 1H NMR (300 MHz, CDCl3): δ: 1.29 (dd, J = 15.9, 7.4 Hz, 3H), 1.66 (m, 1H), 1.86 (m, 1H), 2.26 (m, 2H), 2.35 (m, 2H), 4.15 (dd, J = 7.3 Hz, 1H), 7.28–7.94 (m, 11H); 13C NMR (75 MHz, CDCl3): δ: 14.4 (d, JC–P = 3 Hz), 22.5, 26.3 (d, JC–P = 2.5 Hz), 28.4 (d, JC–P = 69 Hz), 37.7, 128–133.3 (aromatics), 136.9 (d, JC–P = 4.5 Hz), 149.4 (d, JC–P = 6.75 Hz), 197.3 (d, JC–P = 4.5 Hz); 31P NMR (121 MHz, CDCl3): δ: 34.19; HRMS (ESI-TOF): [M+] calcd for C20H21O2P: 324.1279. Found: 324.1264.
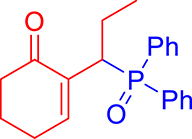
3.3.3. 2-(1-(Diphenylphosphoryl)propyl)cyclohex-2-en-1-one (3c)
mp: 117–118 °C; Yield: 94% (470 mg on 1.48 mmol reaction scale); 1H NMR (300 MHz, CDCl3): δ: 0.81 (t, J = 9 Hz, 3H), 1.63 (m, 1H), 1.81 (m, 3H), 2.02 (m, 1H), 2.35 (m, 3H), 4.00 (td, J = 6.0, JP–H = 9.0 Hz, 1H), 7.28–7.95 (m, 11H); 13C NMR (75 MHz, CDCl3): δ: 12.2 (d, JC–P = 13.5 Hz), 22.6 (d, JC–P = 1.5 Hz), 26.4 (d, JC–P = 2.25 Hz), 30.8, 35.6 (d, JC–P = 69 Hz), 37.7, 127.9–133.6 (aromatics), 135.1 (d, JC–P = 5.25 Hz), 149.1 (d, JC–P = 6.75 Hz), 197.9 (d, JC–P = 5.25 Hz); 31P NMR (121 MHz, CDCl3): δ: 33.30; HRMS (ESI-TOF): [M+] calcd for C21H23O2P: 338.1436. Found (M+H): 339.1502.

3.3.4. 2-(1-(Diphenylphosphoryl)butyl)cyclohex-2-en-1-one (3d)
mp: 124–125 °C; Yield: 96% (480 mg on 1.42 mmol reaction scale); 1H NMR (300 MHz, CDCl3): δ: 0.80 (t, J = 6.0 Hz, 3H), 1.60–2.04 (m, 6H), 2.24–2.38 (m, 4H), 4.09 (ddd, J = 3.0, JP–H = 12.0 Hz, 1H), 7.27–7.95 (m, 11H); 13C NMR (75 MHz, CDCl3): δ: 13.7, 20.7 (d, JC–P = 12.75 Hz), 22.6, 26.4, 31.3 (d, JC–P = 1.5 Hz), 33.6 (d, JC–P = 69 Hz), 37.7, 127.9–133.6 (aromatics), 135.4 (d, JC–P = 4.5 Hz), 149.1 (d, JC–P = 6.75 Hz), 197.97 (d, JC–P = 4.5 Hz); 31P NMR (121 MHz, CDCl3): δ: 33.55; HRMS (ESI-TOF): [M+] calcd for C22H25O2P: 352.1592. Found (M+H): 352.1639.
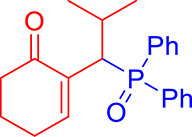
3.3.5. 2-(1-(Diphenylphosphoryl)-2-methylpropyl)cyclohex-2-en-1-one (3e)
mp: 125–126 °C; Yield: 92% (456 mg on 1.42 mmol reaction scale); 1H NMR (300 MHz, CDCl3): δ: 0.81 (d, J = 6.0 Hz, 3H), 0.98 (d, J = 6.0 Hz, 3H), 1.49 (m, 1H), 1.86 (m, 2H), 2.28 (m, 4H), 4.05 (t, J = 6.0, JP–H = 15.0 Hz, 1H), 7.33–7.96 (m, 11H); 13C NMR (75 MHz, CDCl3): δ: 20.3 (d, JC–P = 6.75 Hz), 22.4, 22.9 (d, JC–P = 9 Hz), 26.2, 29.4, 37.6, 39.6 (d, JC–P = 69 Hz), 127.9–134 (aromatics), 134.6 (d, JC–P = 8.7 Hz), 150.1 (d, JC–P = 6.75 Hz), 197.9 (d, JC–P = 6 Hz); 31P NMR (121 MHz, CDCl3): δ: 33.20; HRMS (ESI-TOF): [M+] calcd for C22H25O2P: 352.1592. Found [M+H]+: 352.9818.
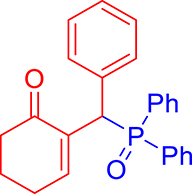
3.3.6. 2-((Diphenylphosphoryl)(phenyl)methyl)cyclohex-2-en-1-one (3f)
mp: 157–158 °C; Yield: 90% (450 mg on 1.295 mmol reaction scale); 1H NMR (300 MHz, CDCl3): δ: 1.64–1.89 (m, 2H), 2.18–2.34 (m, 4H), 5.30 (d, JP–H = 6.0 Hz, 1H), 7.16–7.92 (m, 16H); 13C NMR (75 MHz, CDCl3): δ: 22.4, 26.4 (d, JC–P = 0.75 Hz), 37.8, 41.2 (d, JC–P = 68.25 Hz), 126.9–133.3 (aromatics), 136.3 (d, JC–P = 6 Hz), 150.5 (d, JC–P = 6.75 Hz), 197.0 (d, JC–P = 6.75 Hz); 31P NMR (121 MHz, CDCl3): δ: 32.51; HRMS (ESI-TOF): [M+] calcd for C25H23O2P: 386.1436. Found [M+H]+: 386.1434.
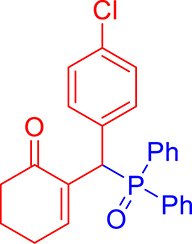
3.3.7. 2-((4-Chlorophenyl)(diphenylphosphoryl)methyl)cyclohex-2-en-1-one (3g)
mp: 181–182 °C; Yield: 91% (455 mg on 1.19 mmol reaction scale); 1H NMR (300 MHz, CDCl3): δ: 1.64–1.87 (m, 2H), 2.10–2.37 (m, 4H), 5.26 (d, JP–H = 9.0 Hz, 1H), 7.13–7.87 (m, 15H); 13C NMR (75 MHz, CDCl3): δ: 22.4, 26.4 (d, JC–P = 0.75 Hz), 37.6, 40.7 (d, JC–P = 148.5 Hz), 128.2–134.9 (aromatics), 136.3 (d, JC–P = 17.25 Hz), 150.7 (d, JC–P = 18.75 Hz), 196.9 (d, JC–P = 12 Hz); 31P NMR (121 MHz, CDCl3): δ: 32.15; HRMS (ESI-TOF): [M+] calcd for C25H22ClO2P: 420.1046. Found [M+H]+: 420.1109.
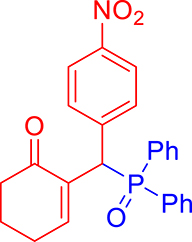
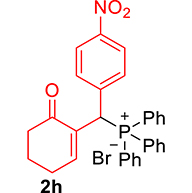
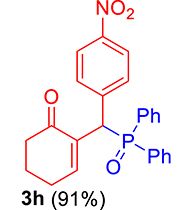
3.3.8. 2-((Diphenylphosphoryl)(4-nitrophenyl)methyl)cyclohex-2-enone (3h)
mp: 214–215 °C; Yield: 97% (485 mg on 1.16 mmol reaction scale); 1H NMR (300 MHz, CDCl3): δ: 1.74 (m, 2H), 2.27 (m, 4H), 5.42 (d, JP–H = 6.0 Hz, 1H), 7.96 (t, J = 6.0 Hz, 1H), 7.30–8.04 (m, 14H); 13C NMR (75 MHz, CDCl3): δ: 22.3, 26.4, 37.7, 41.3 (d, JC–P = 66 Hz), 123.7–135.3 (aromatics), 143.9 (d, JC–P = 4.5 Hz), 151.3 (d, JC–P = 6 Hz), 196.4 (d, JC–P = 6 Hz); 31P NMR (121 MHz, CDCl3): δ: 31.7; HRMS (ESI-TOF): [M+] calcd for C25H23NO4P: 431.1286. Found: 431.1288.
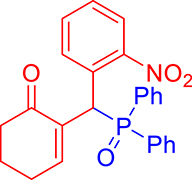
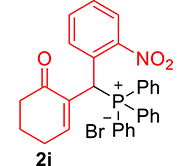
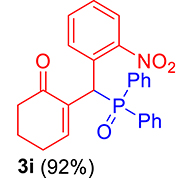
3.3.9. 2-((Diphenylphosphoryl)(2-nitrophenyl)methyl)cyclohex-2-enone (3i)
mp: 213–212 °C; Yield: 92% (460 mg on 1.16 mmol reaction scale); 1H NMR (300 MHz, CDCl3): δ: 1.77 (m, 2H), 2.21 (m, 2H), 2.34 (m, 2H), 6.14 (d, JP–H = 12.0 Hz, 1H), 7.23–8.33 (m, 15H); 13C NMR (75 MHz, CDCl3): δ: 22.3, 26.4 (d, JC–P = 0.75 Hz), 35.4 (d, JC–P = 66.75 Hz), 37.6, 124.4–132.3 (aromatics), 135.7 (d, JC–P = 3 Hz), 151.1 (d, JC–P = 6 Hz), 196.1 (d, JC–P = 6 Hz); 31P NMR (121 MHz, CDCl3): δ: 32.52; HRMS (ESI-TOF): [M+] calcd for C25H22NO4P: 431.1286. Found: 431.8281.
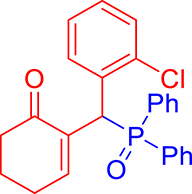
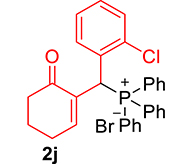
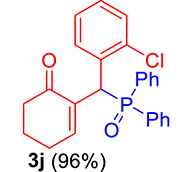
3.3.10. 2-((2-Chlorophenyl)(diphenylphosphoryl)methyl)cyclohex-2-en-1-one (3j)
mp: 106–107 °C; Yield: 96% (1.19 mg on 480 mmol reaction scale); 1H NMR (300 MHz, CDCl3): δ: 1.69–1.91 (m, 2H), 2.22–2.39 (m, 4H), 5.94 (d, JP–H = 9.0 Hz, 1H), 7.06–8.18 (m, 15H); 13C NMR (75 MHz, CDCl3): δ: 22.4, 26.5 (d, JC–P = 1.50 Hz), 37.1 (d, JC–P = 67.50 Hz), 37.7, 126.8–134.7 (aromatics), 135.0 (d, JC–P = 4.50 Hz), 151.3 (d, JC–P = 6 Hz), 195.9 (d, JC–P = 5.25 Hz); 31P NMR (121 MHz, CDCl3): δ: 32.56; HRMS (ESI-TOF): [M+] calcd for C25H22ClO2P: 420.1046. Found: 420.3382.
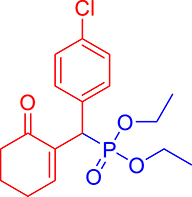
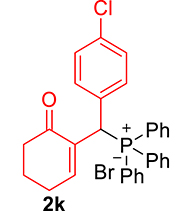
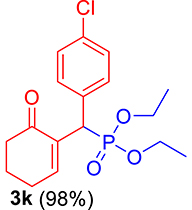
3.3.11. Diethyl ((4-chlorophenyl)(6-oxocyclohex-1-en-1-yl)methyl)phosphonate (3k)
mp: 142–141 °C; Yield: 98% (498.4 mg on 1.40 mmol reaction scale); 1H NMR (300 MHz, CDCl3): δ: 1.09 (t, J = 6.0 Hz, 3H), 1.26 (t, J = 6.0 Hz, 3H), 1.96 (m, 2H), 2.43 (m, 4H), 3.91 (q, J = 6.0 Hz, 2H), 4.04 (q, J = 6.0 Hz, 2H), 4.77 (d, JP–H = 24.0 Hz, 1H), 7.24–7.41 (AB, J = 9.0 Hz, 4H), 7.56 (t, J = 3.0 Hz, 1H). 13C NMR (75 MHz, CDCl3): δ: 16.1, 16.4, 22.5, 26.3, 39.56 (d, JC–P = 141 Hz), 52.2, 62.3, 62.8, 117.5–134.6 (aromatics), 135.65 (d, JC–P = 2.25 Hz), 149.1 (d, JC–P = 6.75 Hz), 196.7 (d, JC–P = 9 Hz). 31P NMR (121 MHz, CDCl3): δ: 25.09. HRMS (ESI-TOF): [M+] calcd for C17H22ClO4P: 356.0944. Found: 356.5342.
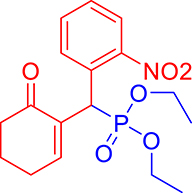
3.3.12. Diethyl ((2-nitrophenyl)(6-oxocyclohex-1-en-1-yl)methyl)phosphonate (3l)
mp: 146–145 °C; Yield: 95% (475 mg on 1.36 mmol reaction scale); 1H NMR (300 MHz, CDCl3): δ: 1.1 (t, J = 6.0 Hz, 3H), 1.28 (t, J = 6.0 Hz, 3H), 1.98 (m, 2H), 2.45 (m, 4H), 3.87 (q, J = 6.0 Hz, 2H), 4.11 (q, J = 6.0 Hz, 2H), 5.39 (d, JP–H = 27.0 Hz, 1H), 7.14–7.81 (m, 5H). 13C NMR (75 MHz, CDCl3): δ: 16.1, 16.2, 21.8, 25.4, 36.7 (d, JC–P = 138.75 Hz), 52.2, 62.7, 62.8, 117.5–134.1 (aromatics), 134.9 (d, JC–P = 8.25 Hz), 147.6 (d, JC–P = 54.75 Hz), 200.2 (d, JC–P = 12.75 Hz). 31P NMR (121 MHz, CDCl3): δ: 26.74. HRMS (ESI-TOF): [M+] calcd for C17H22NO6P: 367.1185. Found: 367.6969.
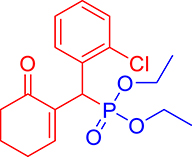
3.3.13. Diethyl ((2-chlorophenyl)(6-oxocyclohex-1-en-1-yl)methyl)phosphonate (3m)
mp: 144–143 °C; Yield: 95% (485 mg on 1.40 mmol reaction scale); 1H NMR (300 MHz, CDCl3): δ: 1.1 (t, J = 6.0 Hz, 3H), 1.28 (t, J = 6.0 Hz, 3H), 1.98 (m, 2H), 2.45 (m, 4H), 3.87 (q, J = 6.0 Hz, 2H), 4.11 (q, J = 6.0 Hz, 2H), 5.38 (d, JP–H = 27.0 Hz, 1H), 7.14–7.81 (m, 5H). 13C NMR (75 MHz, CDCl3): δ: 16.1, 16.4, 22.5, 26.4, 36.6 (d, JC–P = 141 Hz), 53.4, 62.5, 62.8, 126–131 (aromatics), 135 (d, JC–P = 4.5 Hz), 149.8 (d, JC–P = 6.75 Hz), 296.9 (d, JC–P = 5.25 Hz). 31P NMR (121 MHz, CDCl3): δ: 24.38. HRMS (ESI-TOF): [M+] calcd for C17H22ClO4P: 356.0944. Found: 356.8127.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Funding
The authors thank the DGRST and the Ministry of Higher Education, Tunisia, for financial support of this work.
The authors extend their appreciation to the Deanship of Scientific Research at Northern Border University, Arar, KSA for funding this research work through the project number FFR-2024-2939-01.