



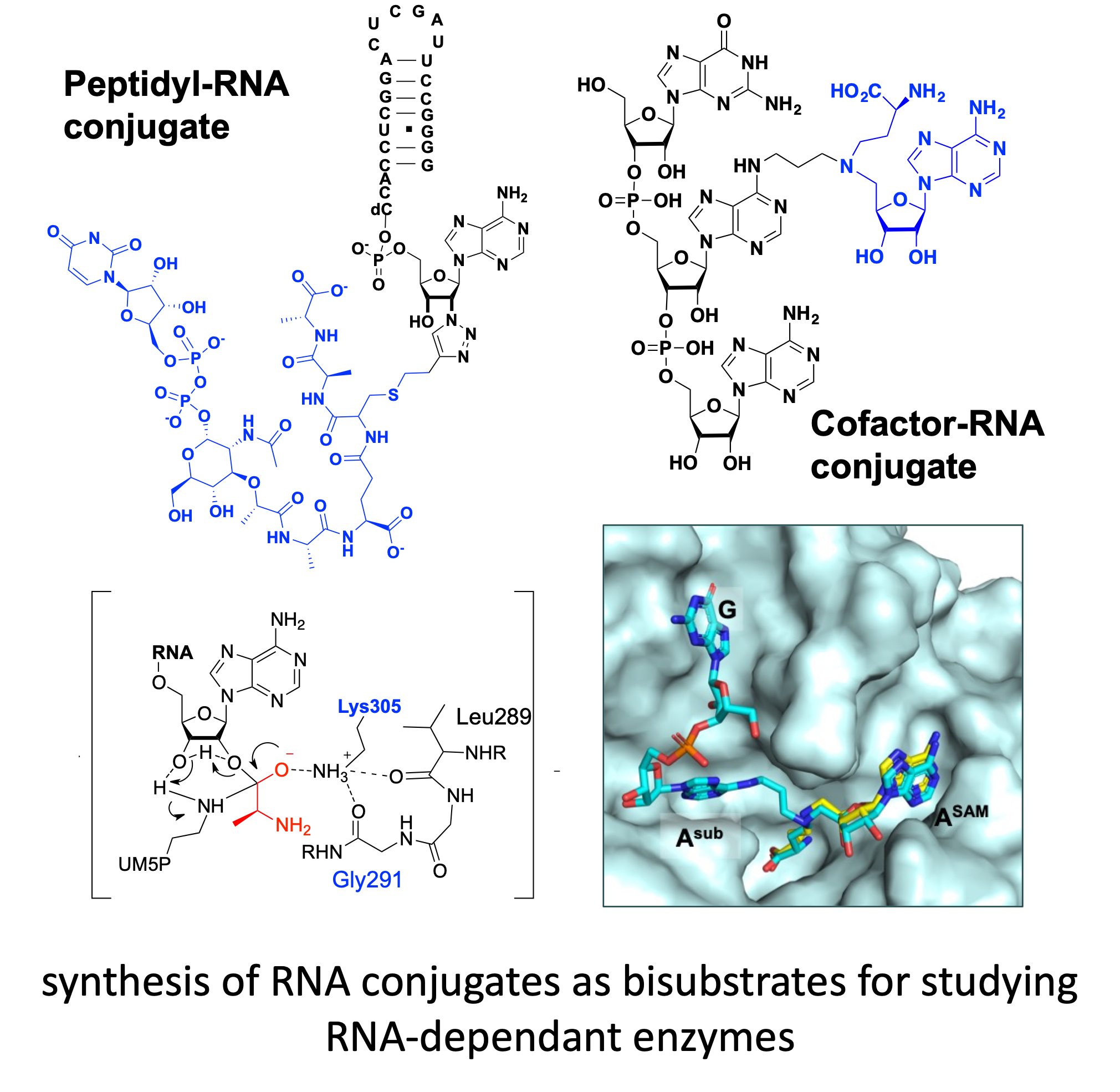
 CC-BY 4.0
CC-BY 4.0
1. Introduction
The bisubstrate strategy is recognized as a powerful approach for developing inhibitors and chemical tools to study enzyme function. This concept leverages the natural interaction of enzymes with multiple substrates to design molecules that can simultaneously engage more than one substrate-binding site. This approach often yields highly selective and potent inhibitors, with selectivity arising from targeting two distinct binding sites and potency enhanced by the combined binding energies of both moieties and an entropy gain from a single molecule interaction. By designing molecules that mimic the interactions of both substrates, researchers can also create highly specific tools that provide insights into the enzyme mechanism of action. This dual engagement makes bisubstrate inhibitors valuable compounds for both therapeutic development and fundamental research.
Based on this concept, the bisubstrate molecules contain two units mimicking the two substrates attached together by a covalent linker (Figure 1). The linker, which is crucial for the correct binding and selectivity of the bisubstrate, must be optimized based on the distance between the two binding sites and the key residues involved in the catalytic pocket.
Bisubstrate-binding mode.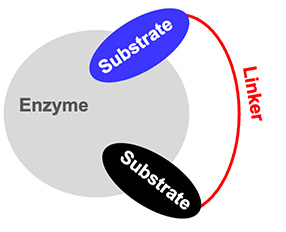
RNA-dependent enzymes, particularly those involved in RNA modifications or in non-ribosomal peptide synthesis, are especially amenable to bisubstrate strategies [1]. In this account, we focus on the use of bisubstrate strategies to explore two families of RNA-dependent enzymes: the Fem transferases and the m6A methyltransferases. We first explain the design of the bisubstrates, then describe their synthesis, and finally their use to explore the mechanism of the reaction catalyzed by the enzymes or as inhibitors of the transferases.
2. Peptidyl–RNA conjugates for the study of Fem transferases
2.1. The Fem transferases
Fem transferases are aminoacyl transferases that participate in peptidoglycan synthesis in Gram-positive bacteria. They catalyze the transfer of amino acids from aminoacyl-tRNA (aa-tRNA) to the amino group of L-Lys of peptidoglycan precursors (Scheme 1) [2, 3]. Enzymes of the Fem family are attractive targets for the development of antibiotics active against resistant bacteria since the residues incorporated by the enzymes are essential for the last cross-linking step of peptidoglycan polymerization in several important β-lactam-resistant pathogens such as staphylococci, pneumococci, and streptococci [4]. Inhibiting Fem activity would therefore result in the production of incomplete precursors acting as chain terminators that block the formation of the essential peptidoglycan layer of the bacterial cell wall [5, 6, 7].
Mechanism of the transfer of Ala from Ala-tRNAAla to the amino group of Lys at the C-3 position of peptidoglycan precursors catalyzed by the aminoacyl transferase FemXWv from Weissella viridescens.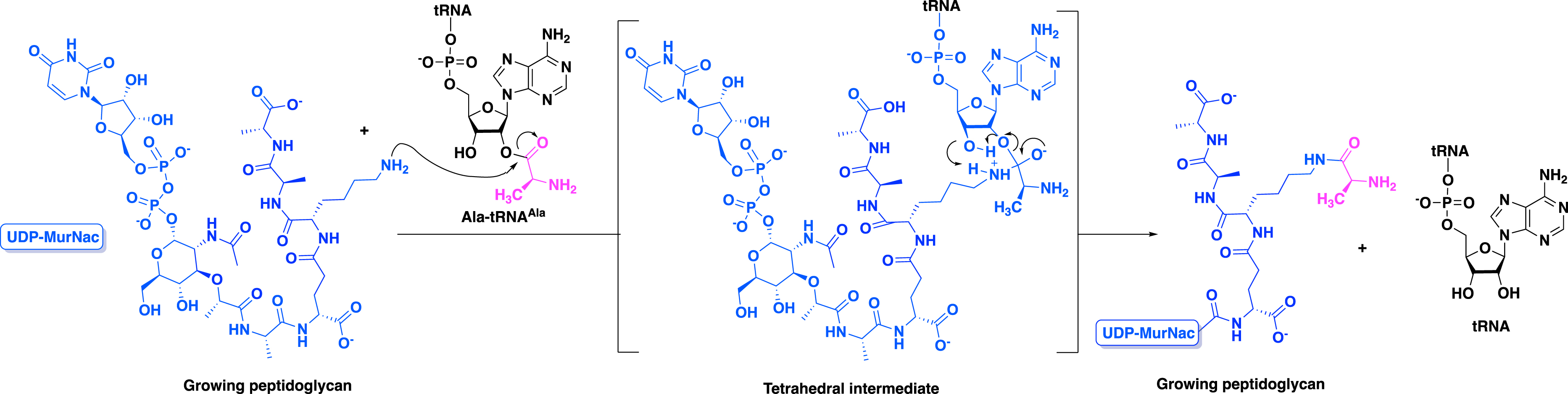
In order to access chemical tools to achieve the resolution of the 3D structure of complexes comprising the enzyme and the aa-tRNA or to obtain inhibitors, the design and the synthesis of bisubstrates have been described for two Fem enzymes: FemXWv [8, 9] and FmhB [10] from Weissella viridescens and Staphylococcus aureus, respectively.
In Weissella viridescens, the transferase FemXWv uses an ala-tRNAala to transfer an alanine to a peptidoglycan fragment consisting of a pentapeptide tethered to a uridine diphosphate N-acetyl-muramic acid moiety [11] (UDP–MurNAc) as described in Scheme 1.
To mimic the tetrahedral intermediate formed during the reaction catalyzed by FemXWv, peptidyl–RNA conjugates were designed as bisubstrates. These compounds contain double-stranded RNA of different lengths simulating the natural substrate tRNAala and a pentapeptide (L-Ala-D-iGlu-L-Lys-D-Ala-D-Ala) tethered to a UDP–MurNAc to mimic the growing peptidoglycan (Figure 2). Linkers based on 1,2,3-triazole and squaramide were chosen to link the two units of the conjugates.
General structure of peptidyl–RNA conjugates to mimic the intermediate formed during the reaction catalyzed by FemXWv.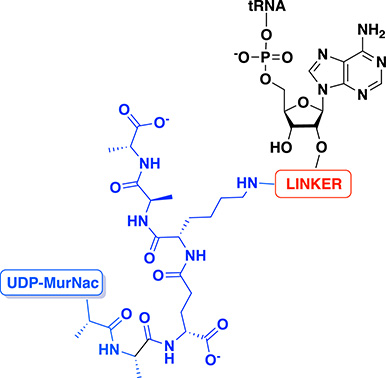
2.2. Synthesis of peptidyl–RNA conjugates
2.2.1. Peptidyl–RNA conjugates with a triazole linker
Synthesis of precursors
Peptidyl–RNA conjugates containing a triazole linker as the bisubstrate of Fem transferases were first synthesized by Fonvielle et al. in 2013 using the Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction [8]. The chemical strategy relies on the introduction of the azide function on the RNA moiety and the alkyne on the peptide mimicking the growing peptidoglycan.
The synthesis of 2′-azido-RNA helixes was achieved using two routes, either by solid phase synthesis (SPS) to obtain a series of short azido-RNAs (18, 12, 10, and 8 nucleotides) or by a chemoenzymatic approach to prepare azido-RNAs of different sizes simulating the acceptor arm of the tRNA [8]. In this study, 2′-azido-adenosine derivatives 7a and 7b were synthesized in a seven-step procedure starting from commercially available adenosine 1 (Scheme 2) [8, 9].
Synthesis of 2′-azido-2′-deoxyadenosine 7a and 7b.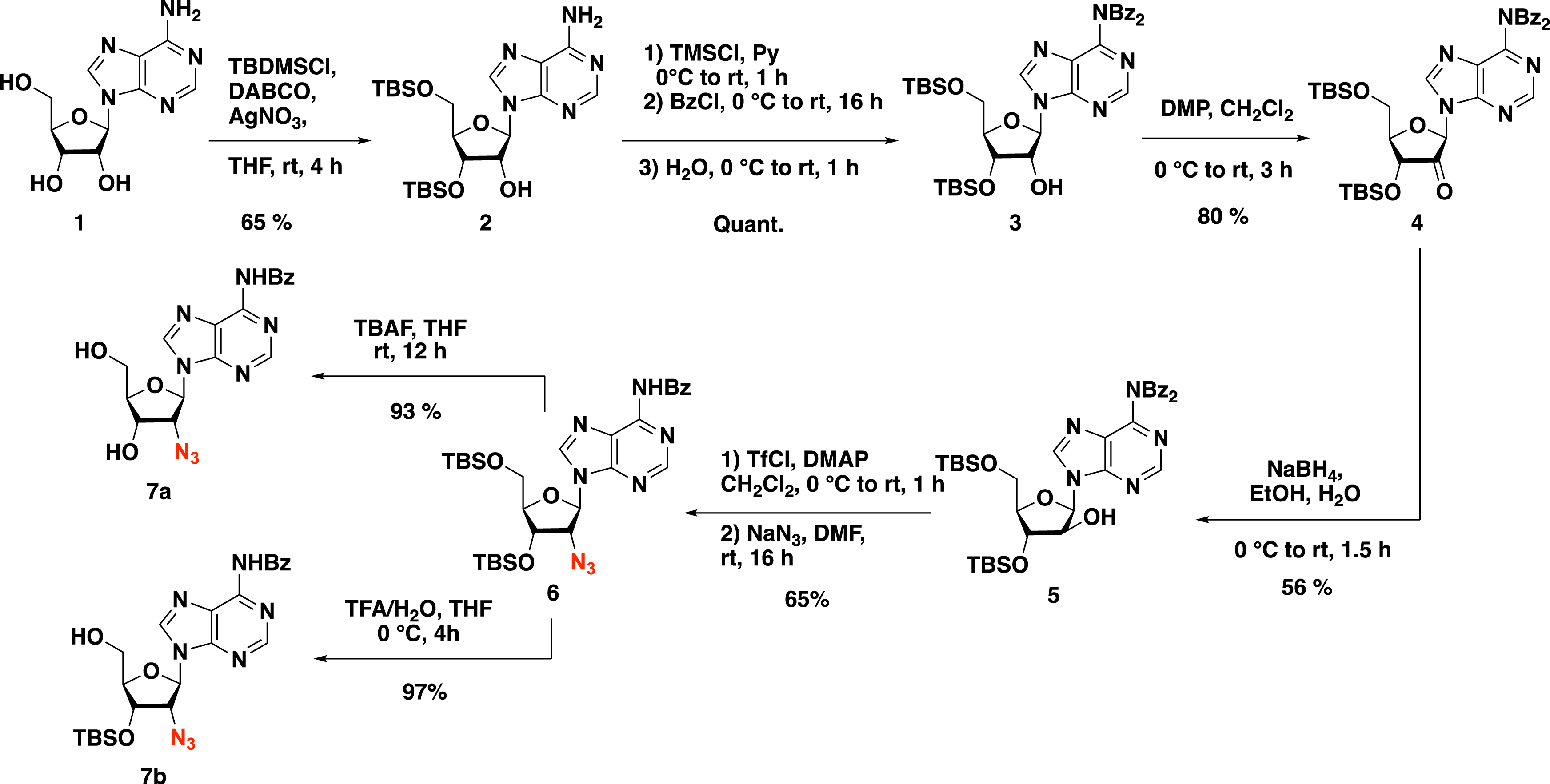
To achieve the synthesis of the short double-stranded azido-RNAs by SPS, 2′-azido-adenosine 7a was first grafted on the resin via a succinyl linker, the resulting adenosine 8 being then engaged in SPS (Scheme 3, Route A). The strategy allows the introduction of a hexaethylene glycol linker to obtain stabilized short double-stranded azido-RNAs [9].
The two routes for the synthesis of 2′-azido-RNAs. Route A: synthesis of short azido-RNAs by SPS. Route B: synthesis of azido-RNA by a chemoenzymatic approach. DMTr protecting group is removed from the SPS machine.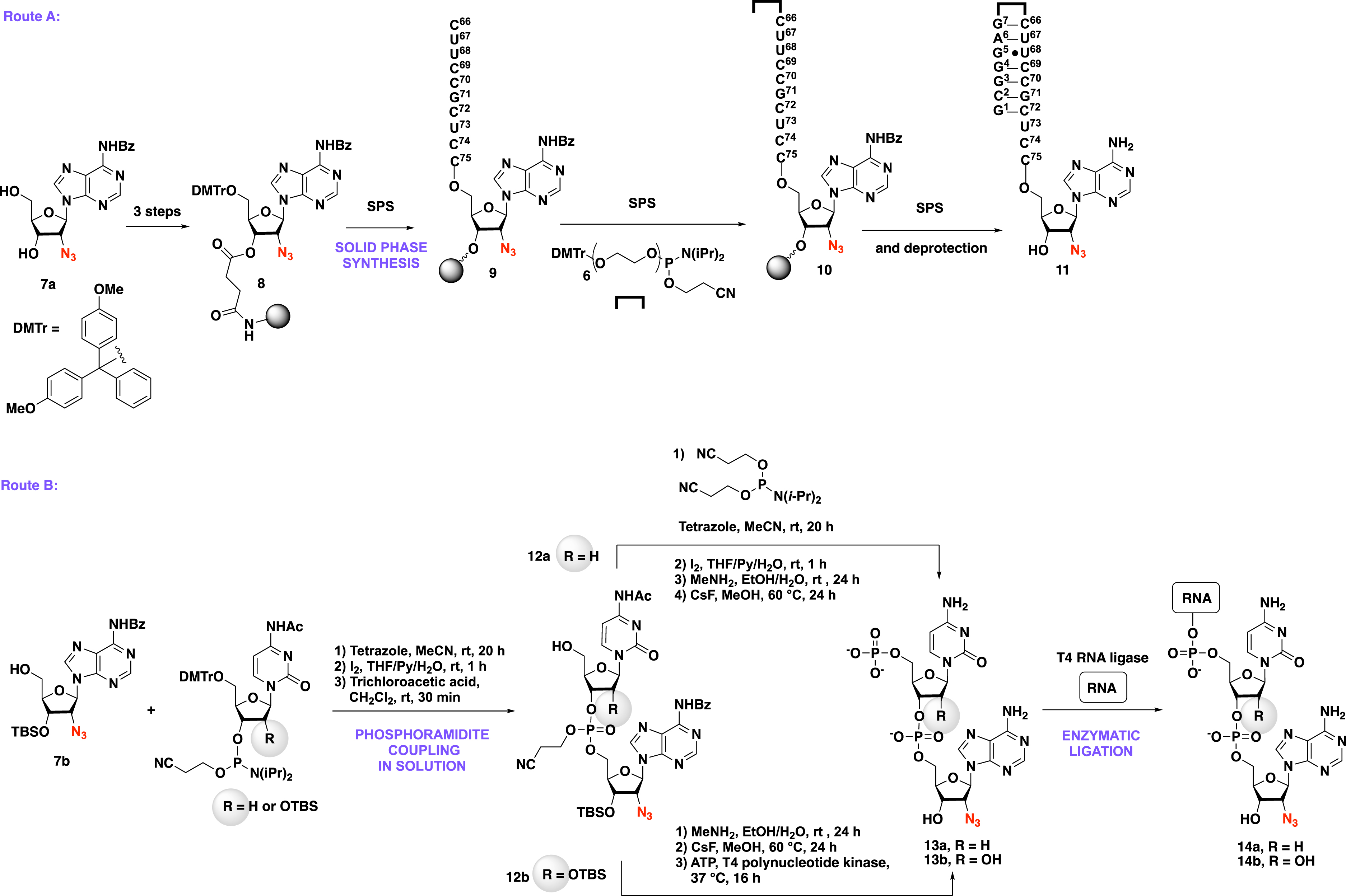
The use of a chemoenzymatic strategy was also investigated by Etheve-Quelquejeu and coworkers to provide azido-RNA with increased size (Scheme 3, route B). The key step of such an approach is based on the synthesis of dinucleotide derivatives 12a–b that were obtained by the phosphoramidite coupling between compound 7b and the commercially available Ac-dC-PCNE and Ac-C-PCNE, respectively. Since the T4 RNA ligase recognizes 5′-phosphorylated RNAs, a chemical [3] and an enzymatic phosphorylation [12] were used to access 5′-phosphorylated 2′-azido-dinucleotides 13a and 13b, respectively. Dinucleotides 13a–b were then ligated to a 22-nt RNA helix in the presence of the T4 RNA ligase to produce the 24-nt azido-RNA helixes 14a–b. This chemoenzymatic strategy was also used to access full tRNA analogues of 76 nucleotides with a sequence corresponding to tRNAAla [3], tRNAGly [10], and tRNAArg [13].
The second part of the bisubstrate structure, namely the peptide mimicking the growing peptidoglycan, was synthesized by semi-synthesis (Scheme 4). To access the peptidoglycan precursor containing an alkyne function 17, meso-cystine was enzymatically incorporated into the peptidoglycan precursor providing compound 15 (Scheme 4). A reduction step in the presence of dithiothreitol (DTT) followed by the conversion of the L-Cys into dehydroalanine in the presence of O-(mesitylenesulfonyl)hydroxylamine (MSH) led to the formation of UDP–MurNAc–pentapeptide 16 [8]. Finally, the addition of but-3-yne-1-thiol to the Michael acceptor produced alkyne-containing UDP–MurNAc–pentapeptide 17. The formation of the dehydroalanine into the peptide was optimized [5, 6, 7] by using 1,5-dibromohexanediamide instead of MSH in the presence of tris(2-carboxyethyl)phosphine (TCEP), providing a one-step process. Worthy of note, compound 17 is a mixture of diastereoisomers that were separated by HPLC purification.
Synthesis of a peptidoglycan fragment containing a propargyl group.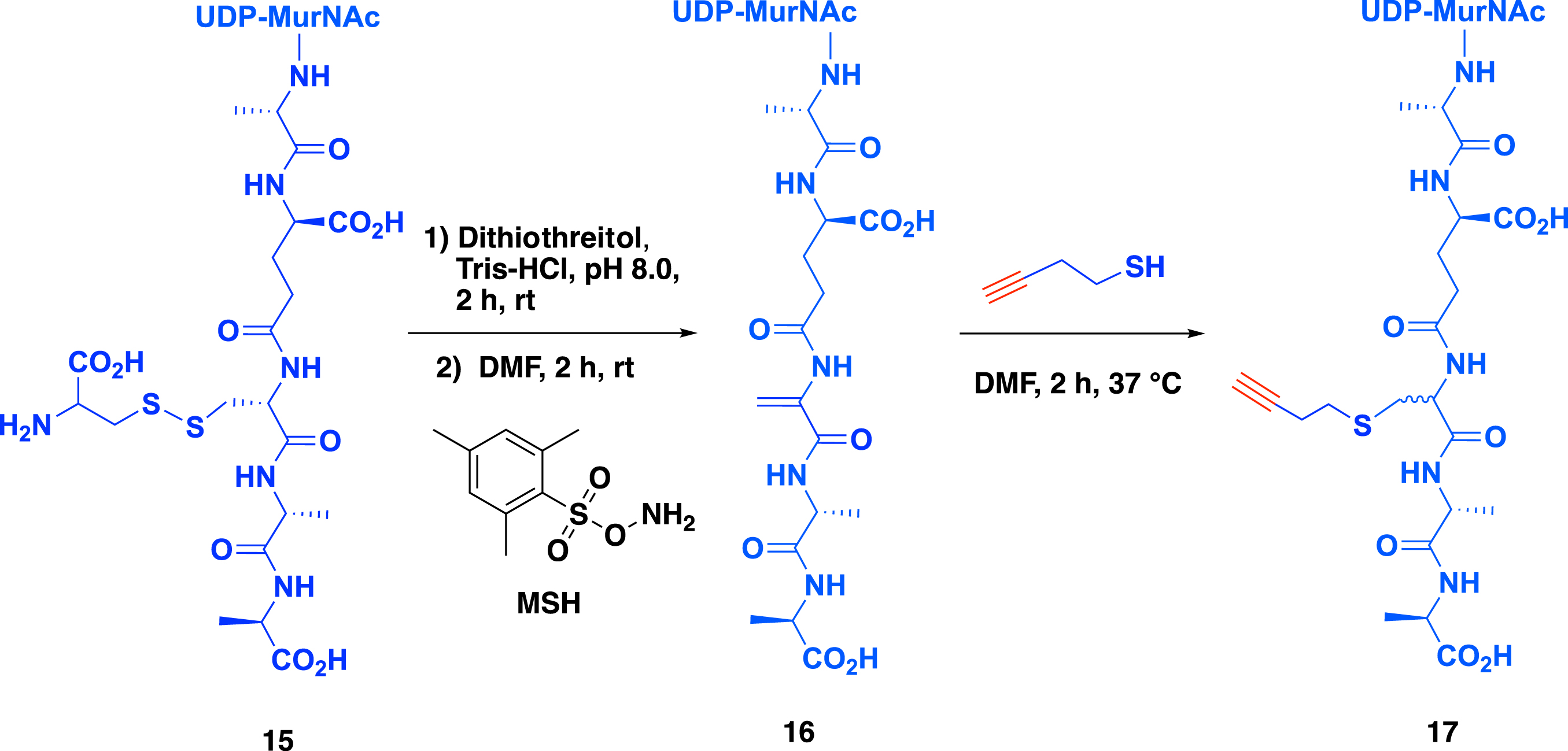
Synthesis of peptidyl–RNA conjugates by CuAAC
CuAAC is one of the most popular bioconjugation techniques reported in the past few decades for the late-stage modification of biomolecules. In 2013, the Etheve-Quelquejeu group reported the use of this reaction to access peptidyl–RNA conjugates as bisubstrates of Fem transferases.
The condition of the CuAAC reaction between 2′-azido-RNA 14a and alkyne UDP–MurNAc–pentapeptide 17 has to be optimized first. Using azido-RNA helix 14a (50 mM), alkyne peptide 17 (100 mM), copper sulfate (0.5 mM), sodium ascorbate (5 mM), and tris[(1-hydroxypropyl-1H-1,2,3-triazol-4-yl)methyl]amine (THPTA) (3.5 mM) in water for 24 h at 37 °C led to the expected peptidyl–RNA conjugate 18 in 36% yield after purification by denaturing polyacrylamide gel electrophoresis (Scheme 5A) [5]. The THPTA ligand was required to stabilize Cu(I) in aqueous buffer and avoid RNA degradation.
(A) The key CuAAC step to provide peptidyl–RNA conjugates to study the transferase FemXWv from Weissella viridescens. (B) Structure of the lipid–carbohydrate–peptidyl–RNA conjugates synthesized by the same strategy to study the transferase FmhB from Staphylococcus aureus.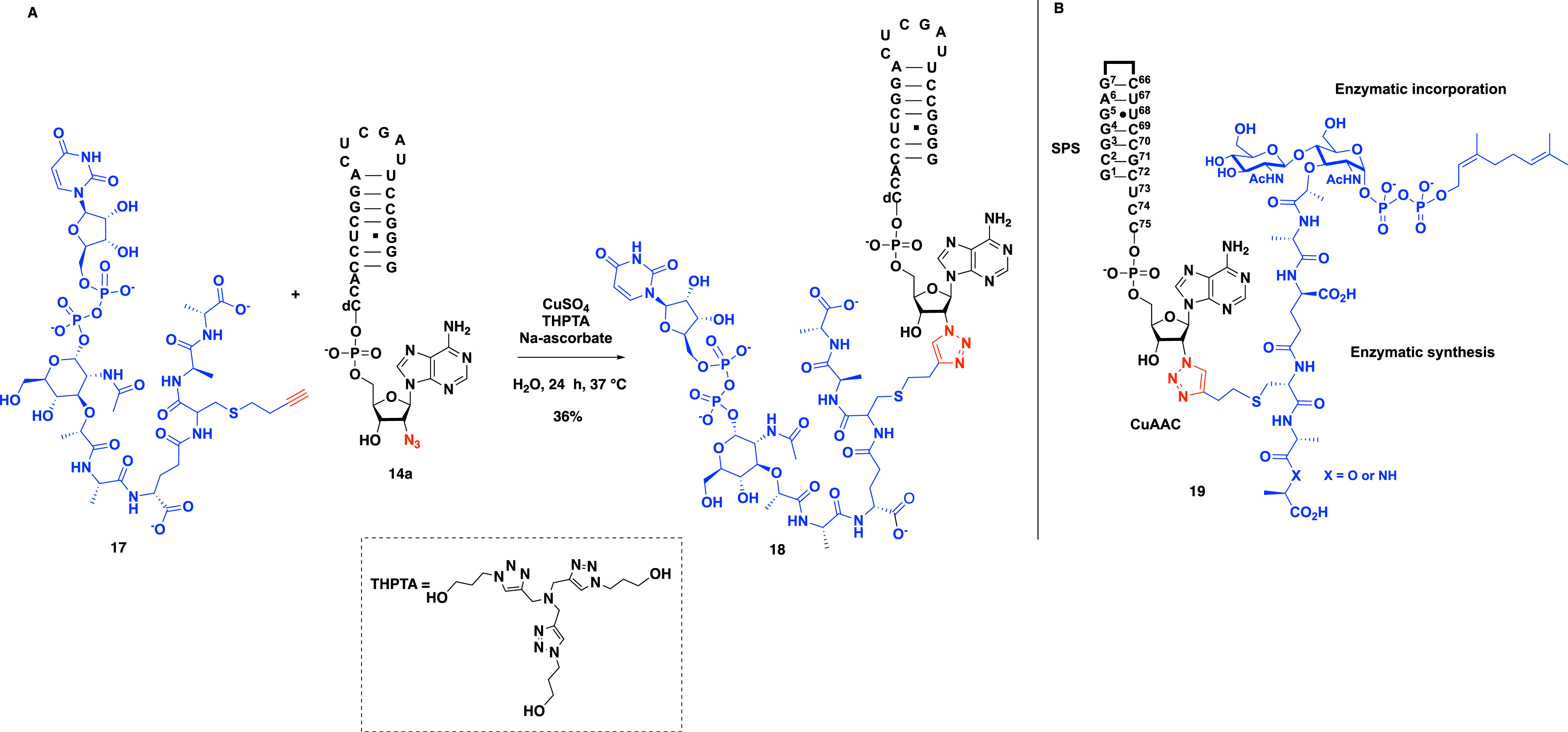
This versatile strategy was used in 2018 by Fonvielle et al. to access even more complex conjugates, namely lipid–carbohydrate–peptidyl–RNA conjugates 19 as bisubstrates of the transferase FmhB (Scheme 5B) [10].
2.2.2. Peptidyl–RNA conjugates with a squaramate linker
In 2016, Fonvielle et al. investigated the nature of the linker in bisubstrate compounds using a squarate motif to tether the RNA 3′-terminus and the peptide moiety. In this work, they described the introduction of a squaramide linkage by the reaction of 2′-amino-RNAs containing 4, 8, or 18 nucleotides with diethyl squarate diester followed by the reaction of the amino group of the lysine of the growing peptidoglycan peptide on the intermediate (Scheme 6) [14]. The synthesis starts with the reduction of the azido group of compounds 20a–c in the presence of TCEP forming 2′-amino-RNA 21a–c, which were then subjected to 1,4-addition with diethyl squarate diester to produce electrophilic RNA 22a–c with yields ranging from 75% to 79%. The reaction of RNAs 22a–c at pH 9.2 in the presence of UDP–MurNAc–pentapeptide 23 produced peptidyl–RNA conjugates with a squaramate linker in 23% to 45% yield. Importantly, increasing the size of the RNA moiety led to comparable reaction yields (Scheme 6). These results show that squarate-mediated ligation provides a versatile route to peptidyl–RNA conjugates. Significantly, this ligation is fully compatible with unprotected UDP–MurNAc–pentapeptide, which contains reactive functions such as phosphate, carbohydrate hydroxyl, and carboxyl groups.
Synthesis of peptidyl–RNA conjugates containing a squaramide linker.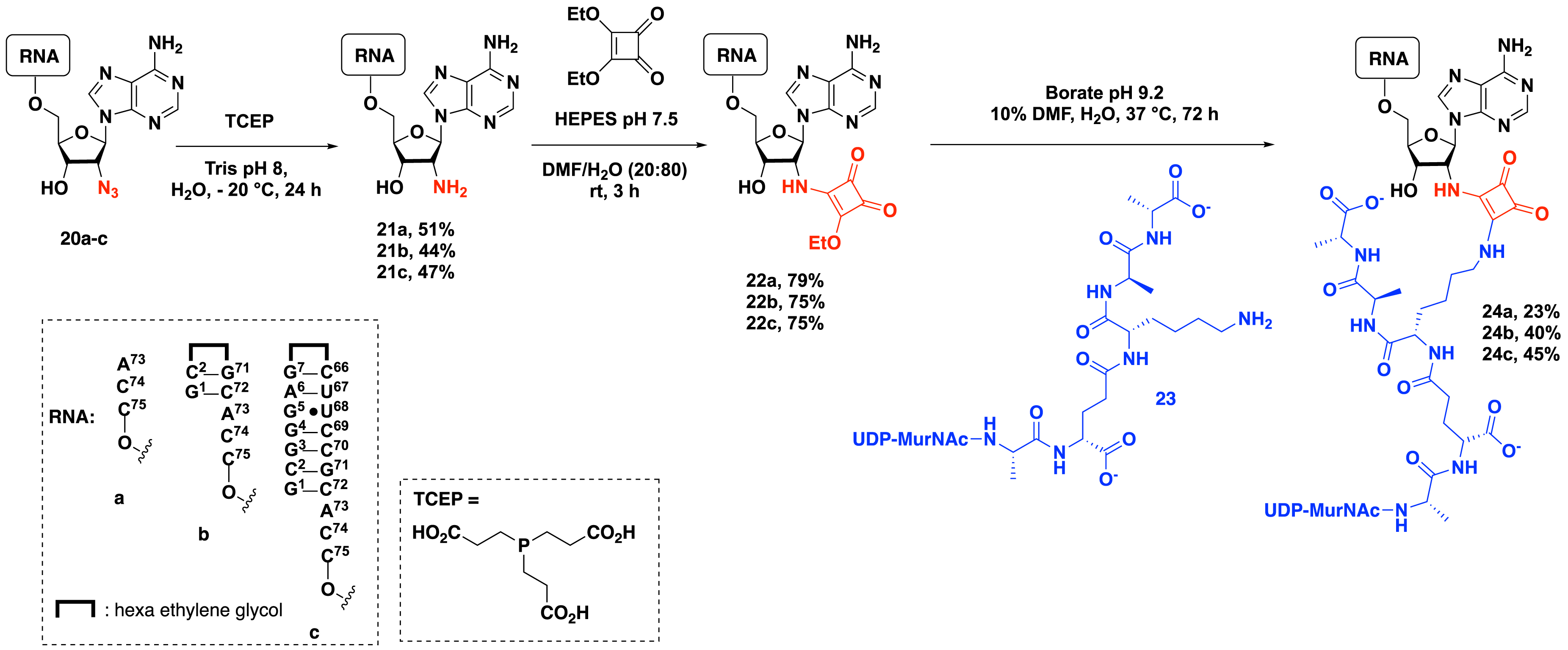
2.2.3. Synthesis of peptidyl–XNA conjugates
In 2022, Rietmeyer et al. explored the nature of the RNA by reporting the synthesis of peptidyl–xeno-nucleic acid (XNA) conjugates to evaluate the impact of the incorporation of XNA into the acceptor arm of tRNA (Scheme 7) [15]. The bisubstrate molecules described in this study were composed of the following: (1) a peptidyl part, mimicking the UDP–MurNAc–pentapeptide, one of the substrates of Fem transferases; (2) an XNA part in which the riboses of RNA were replaced by 2′-deoxy-2′-fluororibose, 1′,5′-anhydrohexitol, and 2′-deoxy-2′-fluoro-D-arabinose; and (3) a triazole linker. A series of 2′-azido-XNA 25a–j were prepared and then subjected to CuAAC in the presence of alkyne UDP–MurNAc–pentapeptide 17 using copper sulfate, sodium ascorbate, and THPTA. Ten peptidyl–XNA conjugates 26a–j were obtained under these reaction conditions (Scheme 7).
Synthesis of peptidyl–XNA conjugates.
2.3. Biological results
2.3.1. Evaluation of peptidyl–RNA conjugates as inhibitors
Triazole-containing peptidyl–RNA conjugates 12 and 13 were tested as inhibitors of the Fem family of enzymes, demonstrating picomolar inhibitory activity against the Fem transferases of Weissella viridescens and Staphylococcus aureus, with IC50 values of 89 ± 10 pM against FemXWv [8] and 56 ± 6 nM against FmhB [10], respectively. These results indicate that both components of the peptidyl–RNA conjugates are required for potent inhibitory activity since azido-RNA alone (compound 14a in Scheme 3) shows a 104 fold higher IC50 of 1.6 ± 0.3 μM [6] compared to 89 pM.
Squaramide-containing peptide–RNA conjugates were also potent inhibitors of FemXWv though the triazole linker was clearly preferred within the FemXWv active site (IC50 of 123 ± 6 nM for the squaramide-containing conjugate 24c compared to 0.15 ± 0.01 nM for the triazole-containing peptide–RNA conjugate 18). In addition, the squaramate unit also promoted specific cross-linking of RNA to the catalytic lysine in FemXWv but not to related transferases that recognize different aminoacyl-tRNAs [14].
The XNA conjugates were also evaluated as inhibitors [15]. The findings reveal that XNA residues located in the 3′-terminal single-stranded regions affect ligand binding within the enzyme catalytic site.
2.3.2. Crystallographic studies
The structure of FemXWv was previously elucidated for both the apo form and complexes containing UDP–MurNAc–pentapeptide or the reaction product UDP–MurNAc–pentapeptide (Ala) [16]. However, attempts to cocrystallize the enzyme with the RNA substrate were unsuccessful. In contrast, triazole-containing peptidyl–RNA or peptidyl–XNA conjugates enabled the successful crystallization of FemXWv in complex with these conjugates [9, 15].
In particular, the crystallographic structures of peptidyl–RNA conjugates in complex with the enzyme provide insights into the enzyme hot spots and catalytic mechanism. For instance, it has been demonstrated that the tetrahedral intermediate formed during catalysis is stabilized by Lys305 in FemXWv. This enzyme creates a unique environment for substrate-assisted catalysis by orienting both Ala-tRNAAla and UDP–MurNAc–pentapeptide into active conformations for aminoacyl transfer. This mechanism is unique to tRNA-dependent enzymes involved in protein-based peptide bond formation (Scheme 8) [9].
Mechanism of the aminoacyl transfer catalyzed by FemXWv [9].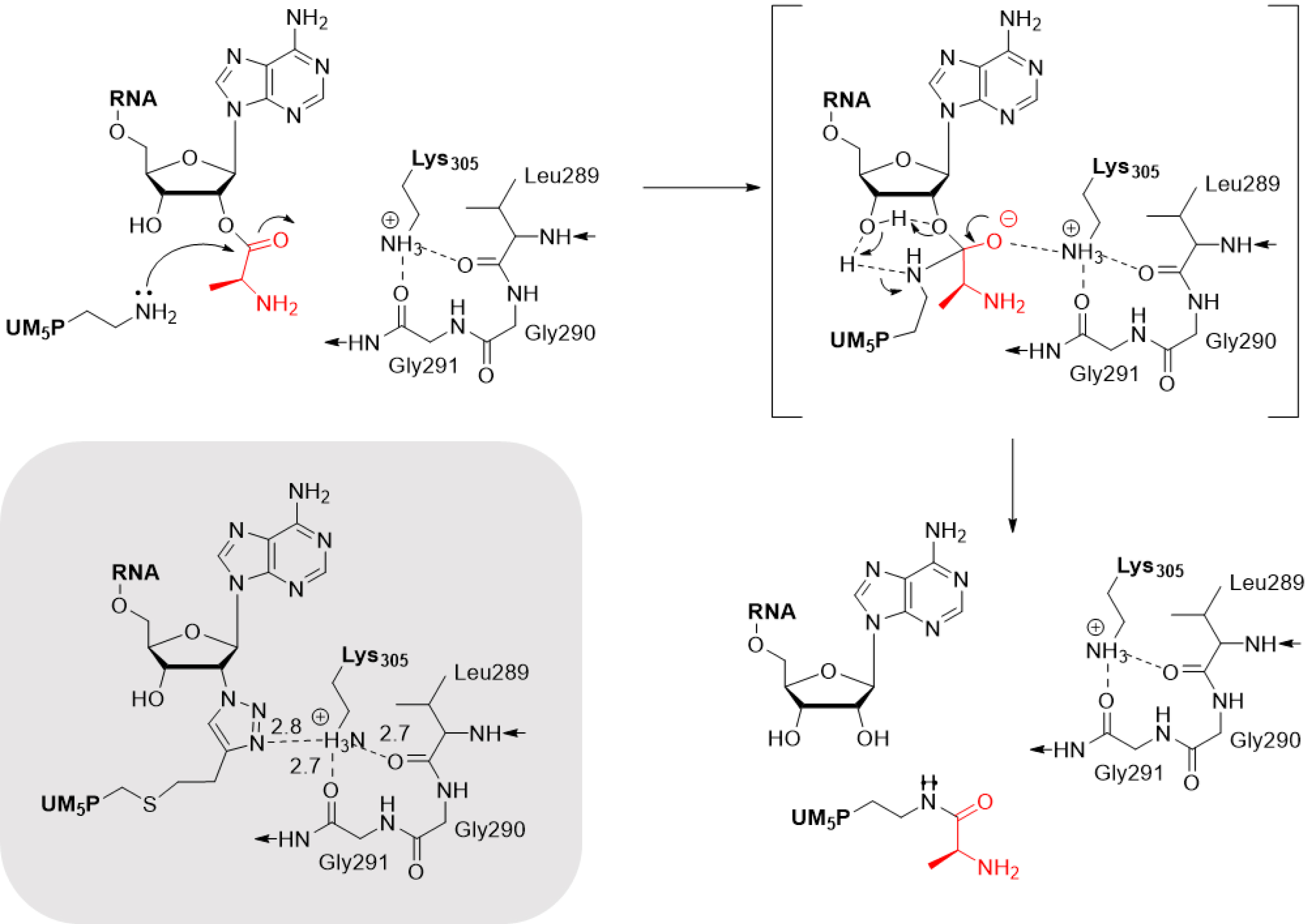
Structural data from peptidyl–XNA conjugates also revealed, for the first time, that the geometry of 1′,5′-anhydrohexitol nucleotides closely resembles that of ribonucleotides, with the 1′,5′-anhydrohexitol of hexitol nucleic acids mimicking ribose in its C-3′-endo sugar puckering. In contrast, the incorporation of 2′-deoxy-2′-fluoro-D-arabinose nucleic acids (2′F-ANA) and DNA residues into the single-stranded region resulted in the loss of a π-stacking interaction, leading to misalignment of the ACCA terminal moiety [15].
2.3.3. Conclusion
Collectively, these examples highlight the potential of peptidyl–RNA conjugates to explore non-ribosomal peptide synthesis, provide structural insights into RNA–enzyme interactions, and develop potent inhibitors.
3. Cofactor–RNA conjugates for the study of m6A methyltransferases
3.1. The m6A methyltransferases
The S-adenosyl-L-methionine (SAM) dependent methyltransferases (MTases) catalyze the transfer of a methyl group to their substrate using mainly the SAM cofactor as the methyl donor. This reaction leads to the release of the coproduct S-adenosyl-L-homocysteine (SAH) (Scheme 9). The MTases methylate several types of substrates including proteins, small molecules, and nucleic acids (DNA and RNA) on which they are able to deposit this mark on different positions.
Methylation catalyzed by MTases.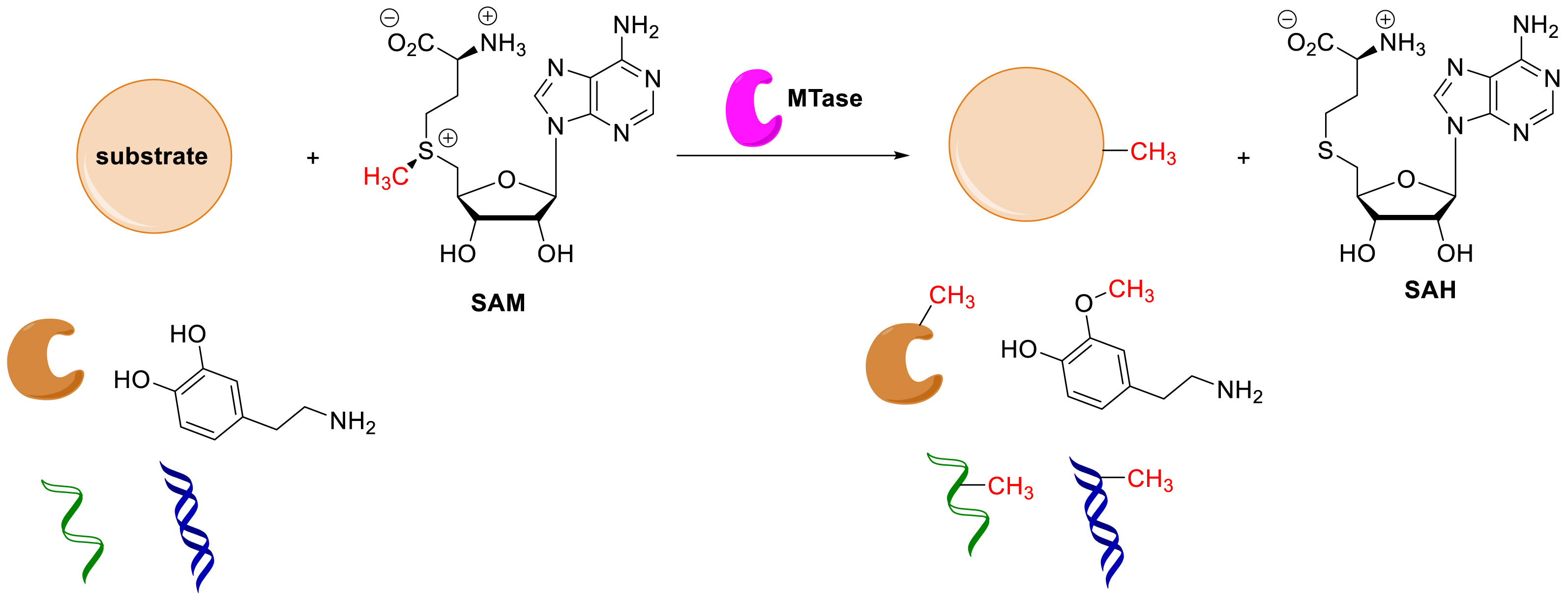
The installation of a methyl group is the most frequent modification found in RNA [17, 18]. In particular, the introduction of a methyl group on the exocyclic N-6 atom of adenosine (m6A) by m6A MTases is the most studied epitranscriptomic mark, identified and characterized across all domains of life and all types of RNA [19]. The m6A modification has been shown to be essential in stabilizing RNA/RNA and RNA/protein interactions [20, 21].
The m6A modification on mRNA is a dynamic and highly regulated process, involving three distinct families of proteins, that is, the writers (MTases) install the m6A mark, which is recognized by the reader proteins and removed by the erasers (Scheme 10). m6A is the most abundant internal modification in eukaryotic mRNA and long non-coding RNA [22, 23]. It regulates different aspects of RNA metabolism such as splicing [24, 25], stability [26], translation [27, 28, 29], and many more biological processes [30, 31].
Importantly, it has been shown that dysregulation of MTase expression is correlated to human diseases including cancers [32, 33, 34], type 2 diabetes [35], neurological disorders [36], cardiovascular diseases [37], and viral infections [38, 39].
The m6A modification process.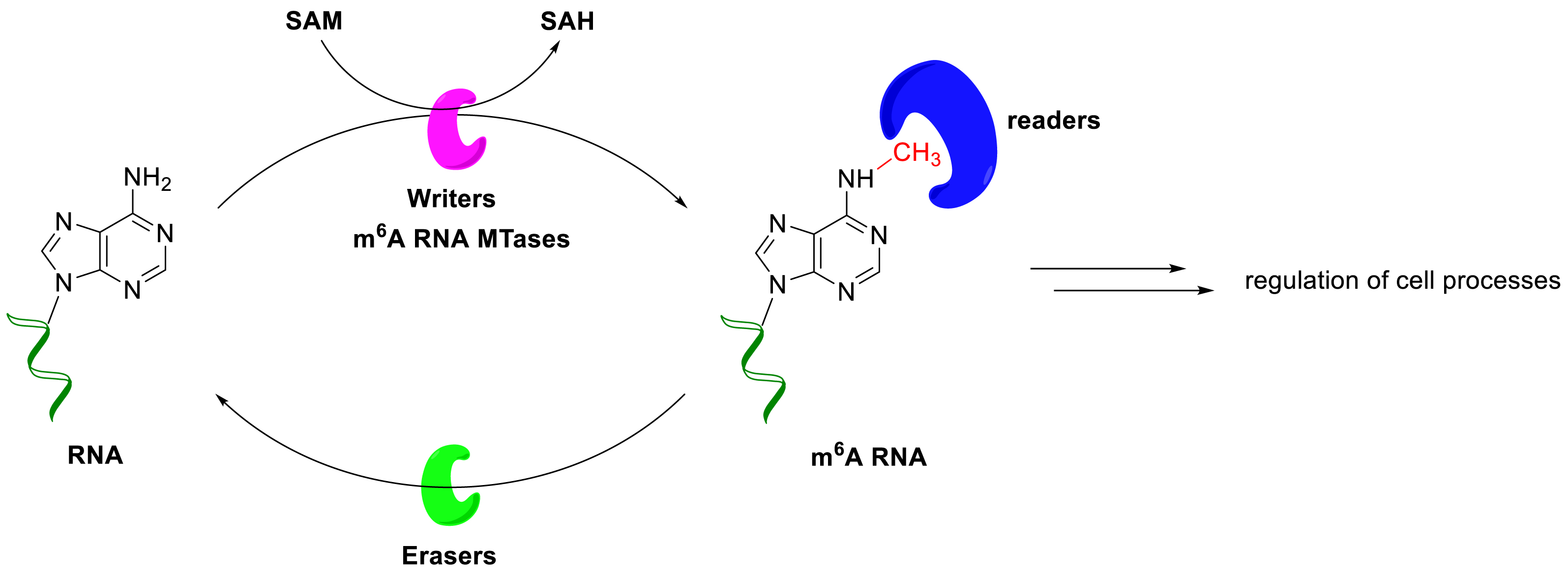
Accumulating evidence indicates that m6A MTases are promising drug targets, and several pharmaceutical companies have already developed inhibitors of the METTL3/METTL14 complex. For example, STORM Therapeutics has announced the entry into Phase I of the selective METTL3 inhibitor STC-15 for the treatment of acute myeloid leukemia and solid tumors. An inhibitor of this enzyme complex identified by a fragment-based approach has also been reported by Gotham Therapeutics [40]. Very recently, the first inhibitors of METTL16 have been also described [41].
However, still little is known about the molecular mechanism of m6A modification, including the recognition of the RNA, which requires a sequence motif (DRACH, where A is the methylated adenosine; D=A, G, or U; R=A or G; and H=A, C, or U) and/or RNA structure, the binding in the active site of the MTase, and finally the release of the m6A substrate. This can be explained by difficulties encountered in obtaining RNA–protein crystallographic structures, leading to a limited number of structurally characterized m6A MTases. To date, only the catalytic domain of METTL16 has been crystallized with a substrate RNA [42], and no structure of the ternary complex has been reported. The structural studies carried out on human RNA MTases show that the substrate-binding site is largely open on the SAM-binding pocket, favoring the design of bisubstrate compounds [43]. In this context, bisubstrate analogues of MTases designed to accommodate the SAM-binding domain and the substrate-binding site appear as important chemical tools to access crystallographic structures that would help in understanding better the enzymatic reaction. These compounds are SAM–adenosine and SAM–RNA conjugates, which contain an analogue of the cofactor SAM covalently attached to a substrate surrogate via an appropriate linker to mimic the transition state of the SN2 mechanism of the methylation reaction (Figure 3).
General structure of SAM–RNA conjugates.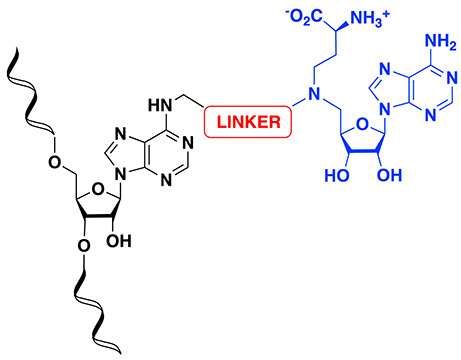
3.2. Synthesis of SAM–adenosine and SAM–RNA conjugates
In respect of the bisubstrate strategy, Atdjian et al. reported in 2018 [44] a synthetic route to access SAM–adenosine conjugates. The structures of these compounds contain a SAM analogue covalently attached to a substrate surrogate via an alkyl linker that mimics the transition state of the SN2 mechanism of the methylation. The key step of the synthesis is based on the concept of the convertible nucleoside using the O6-(benzotriazol-1-yl)inosine [45, 46, 47, 48] derivative as the electrophilic nucleoside to ensure the crucial connection between the SAM moiety bearing an alkyl linker and the N-6 position of the adenosine.
Bisubstrate molecules containing a SAM analogue with an alkyl linker 31a–b were first synthesized (Scheme 11) by Atdjian et al. [44]. Protected inosine 27 was activated in situ in the presence of PyBOP and DBU in DMF to generate the corresponding activated inosine intermediate, which was directly subjected to a nucleophilic aromatic substitution (SNAr) with 5′-aminoadenosine derivatives 28a–b, leading to the formation of SAM–adenosine conjugates 29a and 29b in 85% yield. The removal of the protecting groups was then achieved using ZnBr2 salt, yielding 30a and 30b in 71% and 46%, respectively. Treatment with ammonium fluoride gave access to the fully deprotected bisubstrates 31a and 31b.
Synthesis of adenosine–SAM bisubstrates containing an alkyl linker by a convertible nucleoside approach.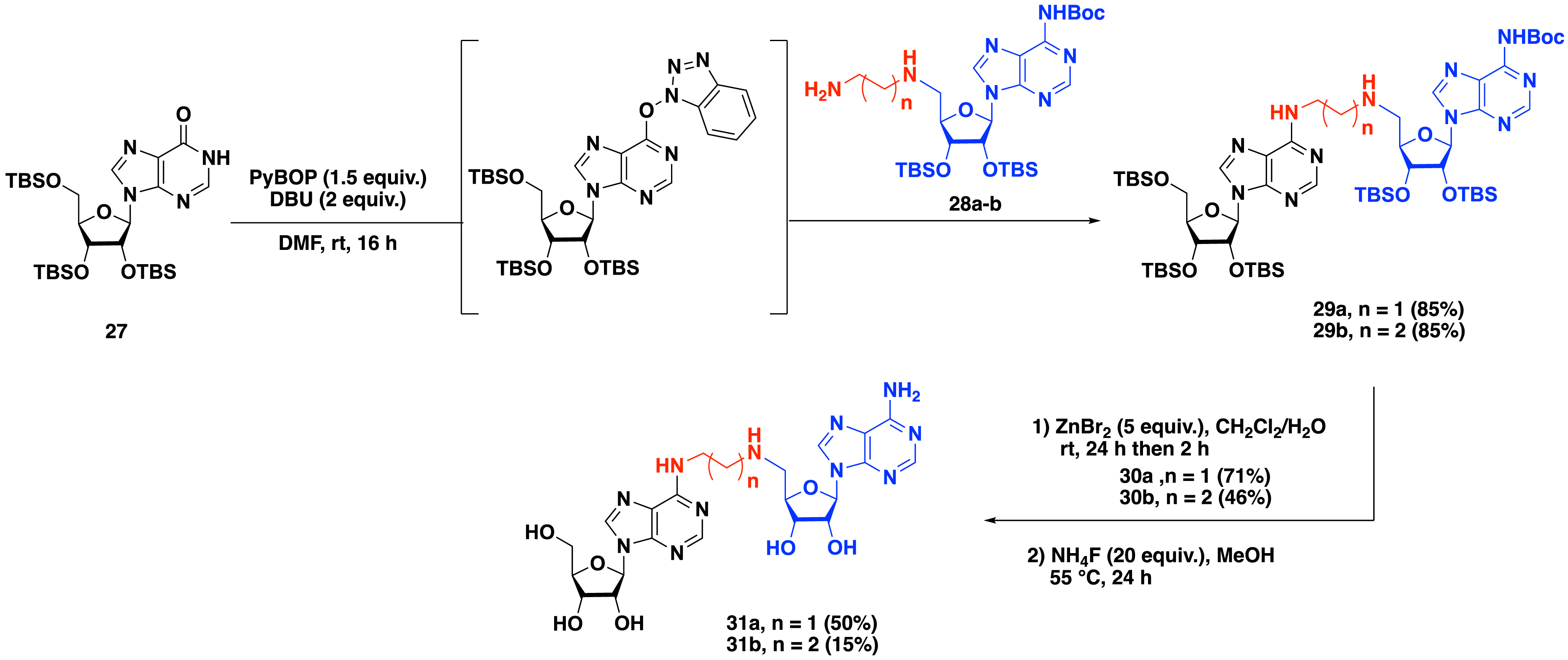
To ensure better affinity with methyltransferase, a second generation of SAM–adenosine conjugates with an alkyl linker was prepared in which an α-amino-acid motif mimicking the α-amino-acid side chain of the cofactor SAM was introduced [44]. The convertible nucleoside approach was used in the presence of adenosine derivatives 32a–b substituted at the C-5′ position by an α-amino-acid residue and an alkyl linker containing two to three carbon atoms to access compounds 33a and 33b in 80% and 81% yield, respectively. A two-step procedure was applied to remove the protecting groups of the adenosine analogues and the α-amino-acid side chain, leading to the formation of bisubstrate molecules 35a and 35b (Scheme 12).
Synthesis of SAM–adenosine bisubstrates containing an alkyl linker and α-amino-acid side chain.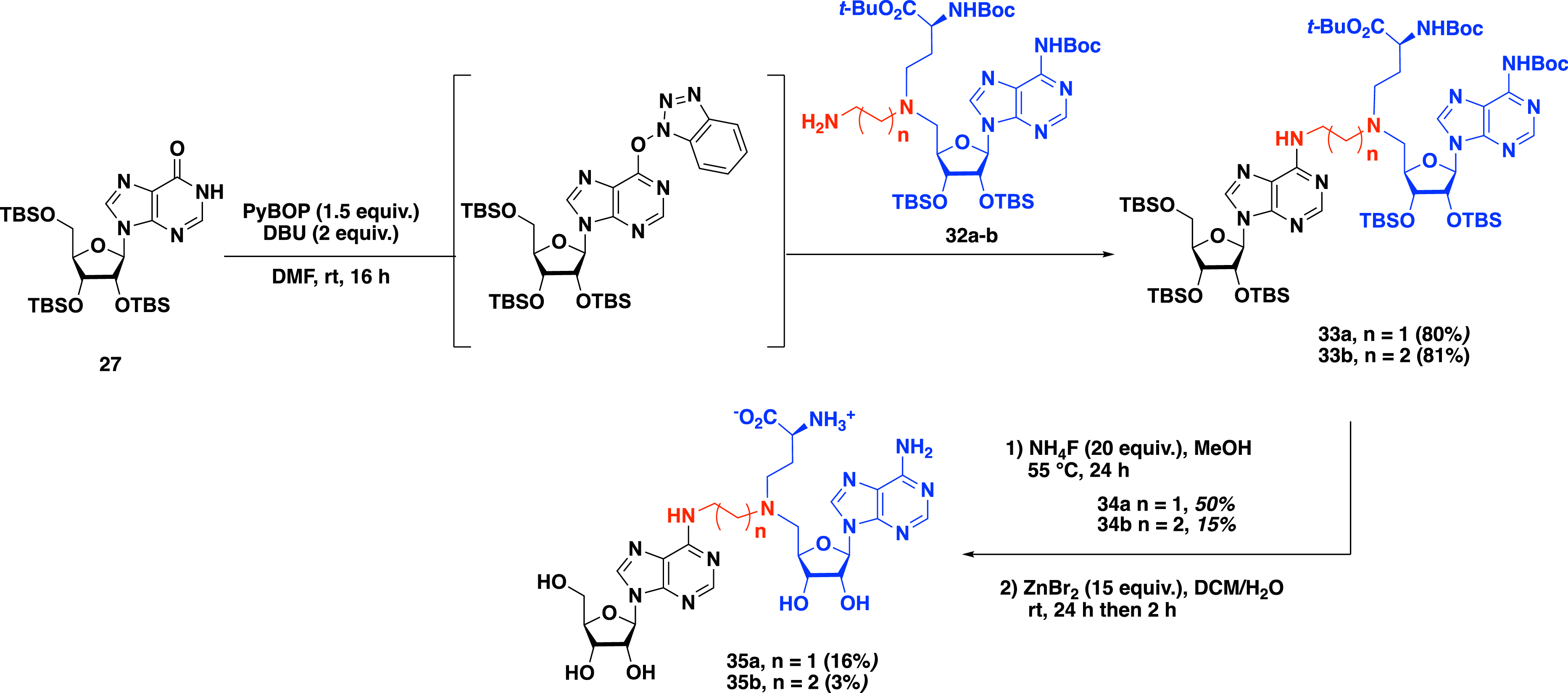
To increase the size of the part mimicking the RNA substrate, Etheve-Quelquejeu and coworkers applied the convertible nucleoside strategy to short RNA strands [49]. Activated inosine 36 was subjected to a phosphoramidite coupling in the presence of the commercially available guanosine phosphoramidite and tetrazole in acetonitrile. The triester phosphite intermediate was then oxidized with a solution of diiodine in a water/THF/pyridine mixture to access the corresponding phosphotriester. Deprotection of the C-5′ position was carried out by treatment with trichloroacetic acid in order to remove the 4,4′-dimethoxytrityl group. Dinucleotide 37 was thus obtained in three steps in 56% yield (Scheme 13). The SNAr reaction with SAM analogue 32b followed by three deprotection steps finally yielded the dinucleotide GA*.
Synthesis of GA* dinucleotide–SAM conjugate.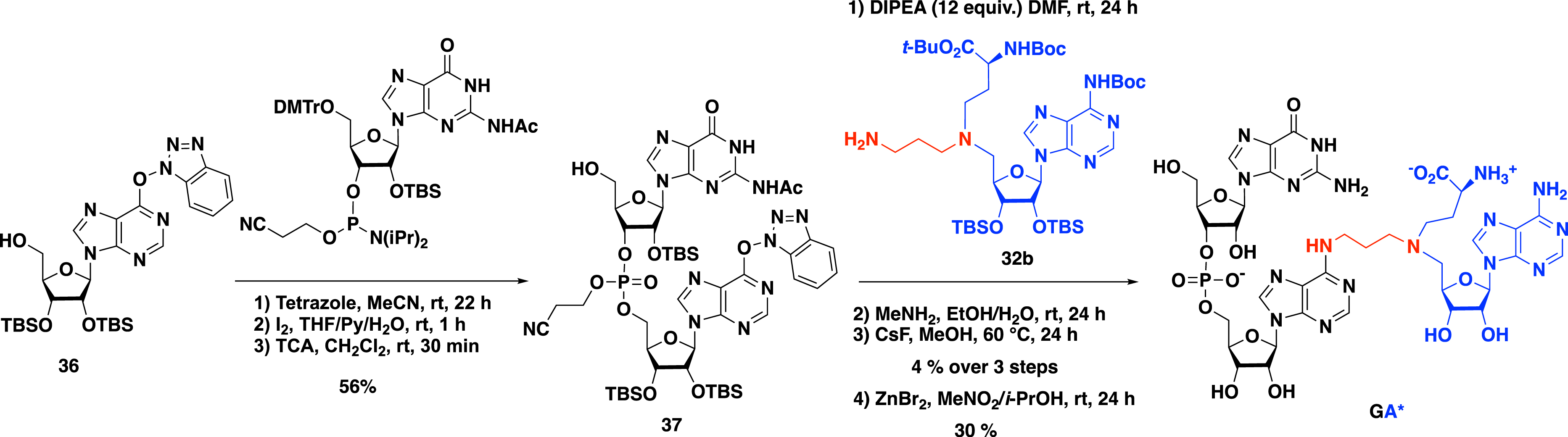
The introduction of the SAM analogue in internal position requires the preparation of the convertible phosphoramidite derivative 33, in which the N-6 position is activated with an O6-(benzotriazol-1-yl) group in six steps starting from inosine (Scheme 14). The A*A dinucleotide– and the GA*A trinucleotide–SAM conjugates were obtained following the phosphoramidite coupling strategy described above [49].
Synthesis of convertible phosphoramidite 33 to access SAM–A*A and SAM–GA*A conjugates.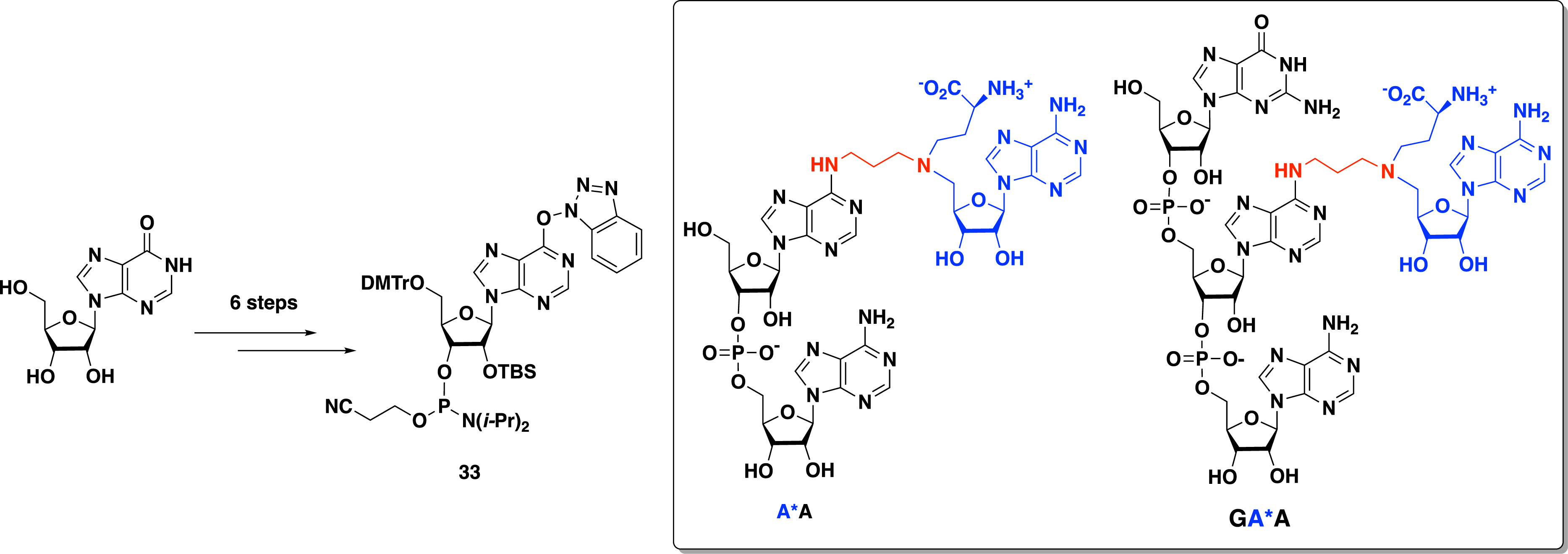
The synthesis of a SAM–RNA conjugate with a longer RNA strand was then performed by a chemoenzymatic strategy [49]. An enzymatic ligation catalyzed by the T4 RNA ligase was used to link a dinucleotide covalently tethered to the SAM analogue (pdGA*) to a synthetic 11-nt RNA molecule to produce the corresponding 13-nt SAM–RNA conjugate (Scheme 15).
Enzymatic ligation with T4 RNA ligase to access 13-nt SAM–RNA conjugate.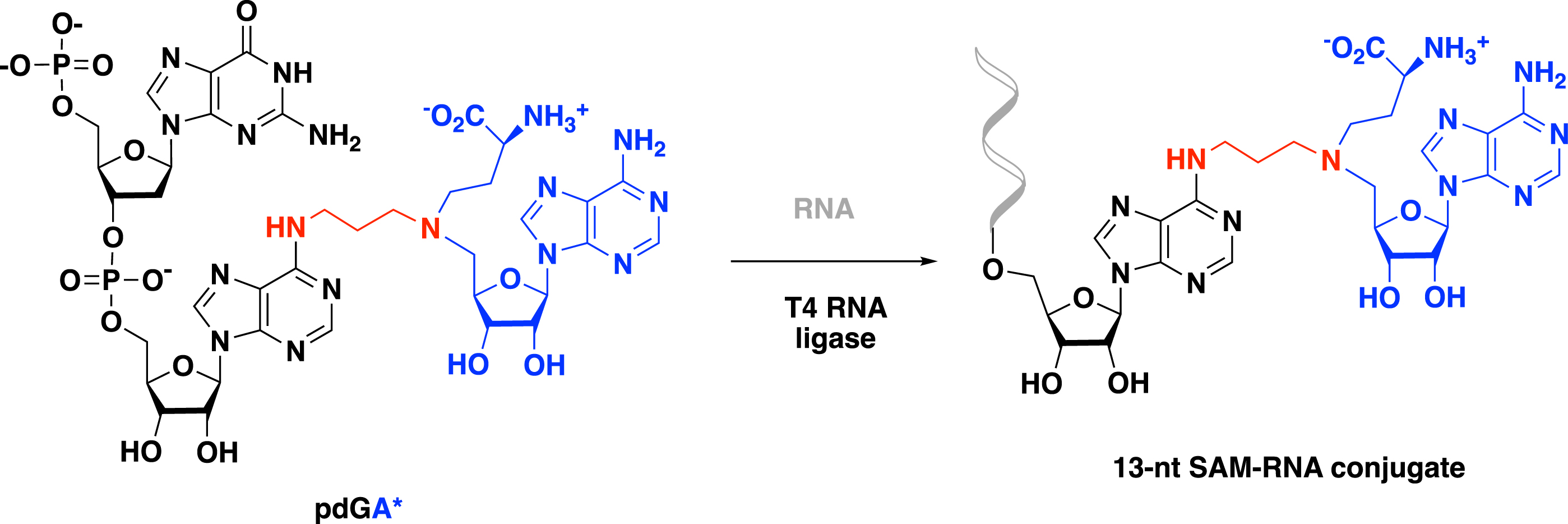
Urea was also chosen to provide bisubstrates with a linker containing only one carbon atom as in the SN2 mechanism of the methyl transfer catalyzed by the MTases [44]. To synthesize such molecules, the exocyclic amine at the N-6 position of adenosine 34 was first activated with isopropenyl chloroformate to access the corresponding isopropenyl carbamate 35 in 75% yield (Scheme 16). Then, the condensation with secondary amine 36 in pyridine produced urea 37 in 85% yield. The deprotection of the silyl groups with NH4F followed by the removal of the Boc groups and tert-butyl ester allowed the formation of urea bisubstrate 38 in 47% yield.
Synthesis of a SAM–adenosine conjugate with a urea linker.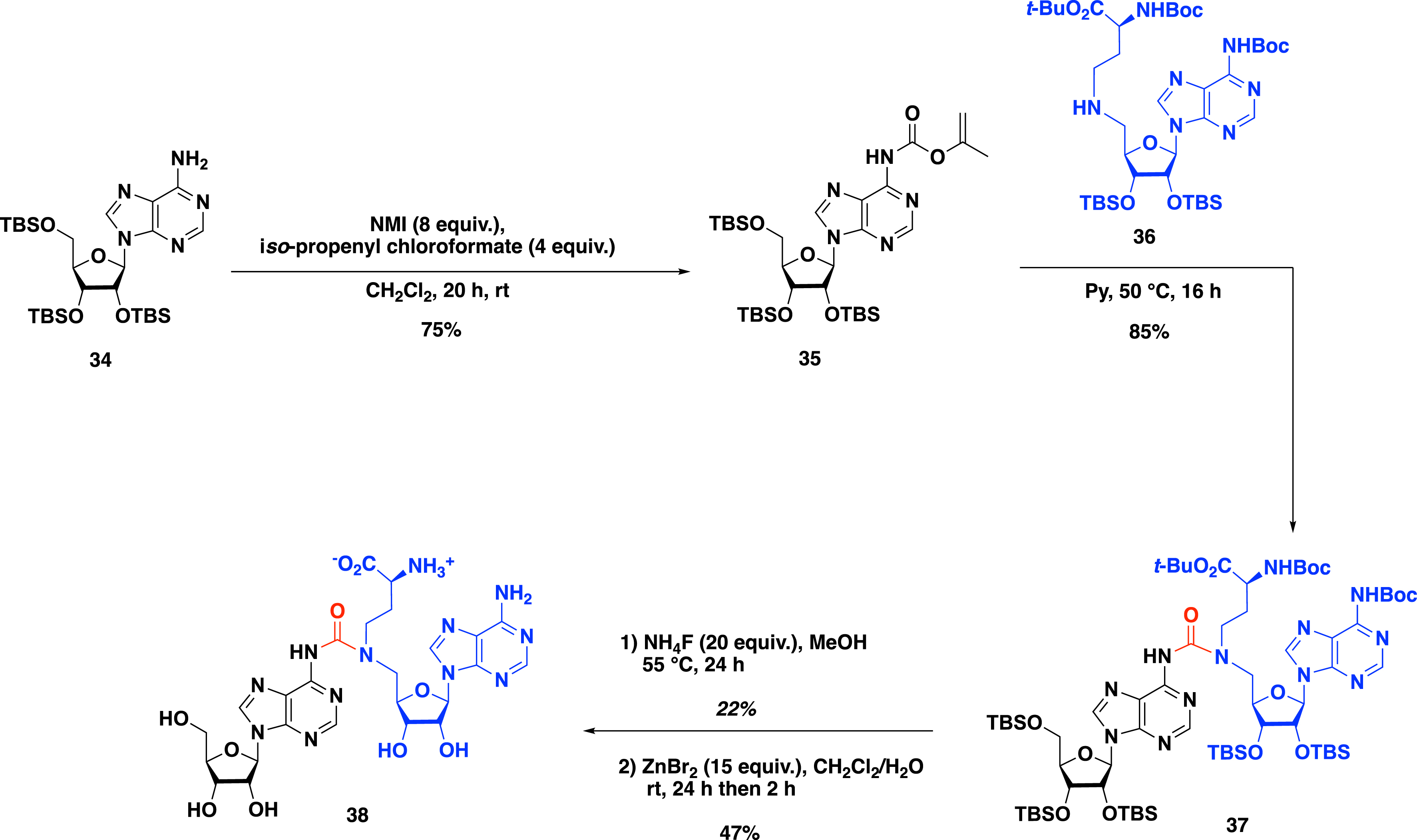
Moving forward to explore the impact of the linker of the SAM–adenosine conjugates, Atdjian et al. [50] examined in 2020 the CuAAC reaction to introduce 1,4-disubstituted-triazole ring as a linker (Scheme 17). The CuAAC reaction between N-6-propargyl adenosine 39 and 5′-azido-adenosine 40 was conducted in the presence of sodium ascorbate and copper sulfate in DMF/H2O to obtain triazole bisubstrate 41 in 36% yield (Scheme 17). Interestingly, this approach, allowing the formation of a 1,2,3-triazole link between the adenosine and the SAM analogue, does not require any protecting group on the reactive functions of the nucleosides.
Synthesis of N-6 and N-1 SAM–adenosine conjugates by CuAAC.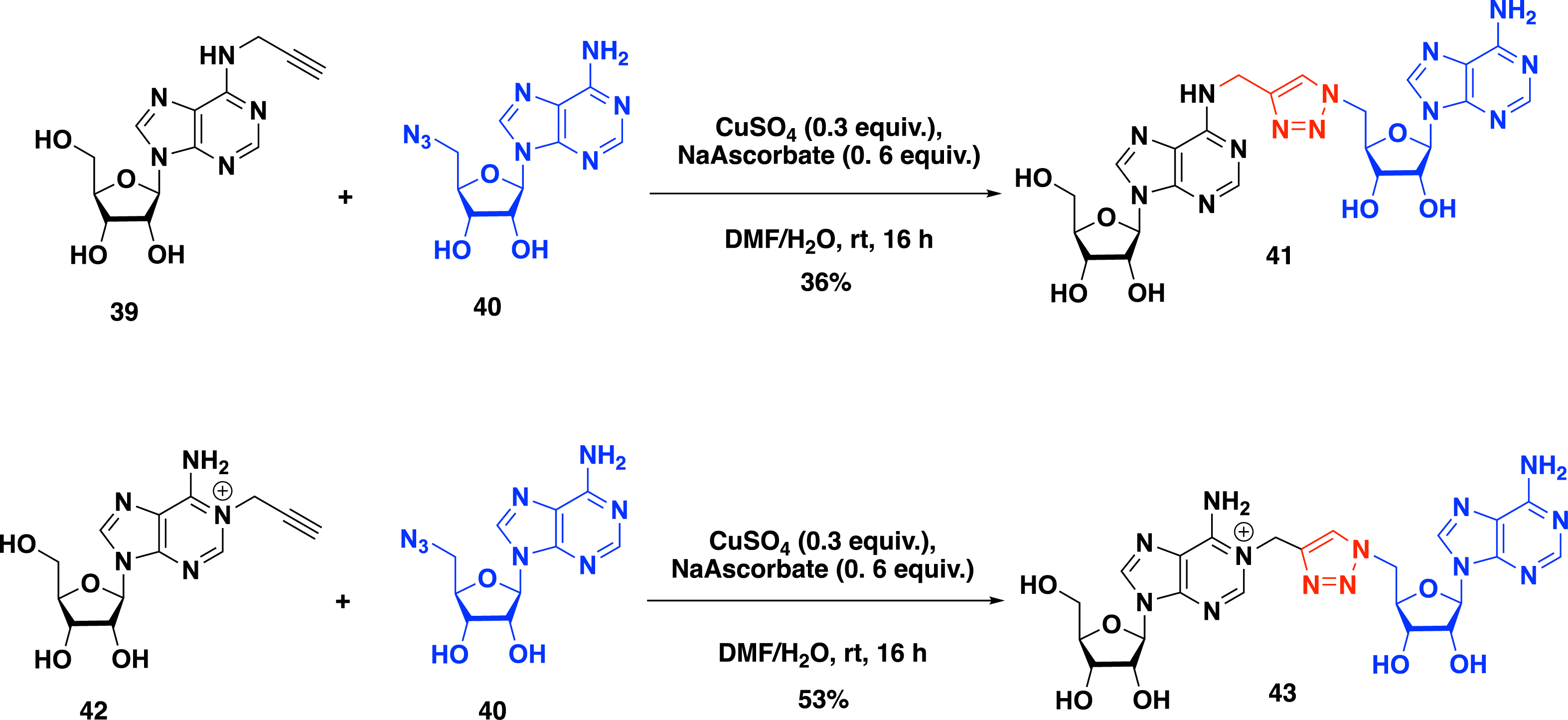
In addition to their effort to access the SAM–adenosine conjugate connected at the N-6 position, Etheve-Quelquejeu and coworkers explored in the same year [50] the reactivity of the N-1 position of adenosine to prepare bisubstrate molecules to study m1A MTase. The synthesis of SAM–adenosine conjugate 43 with a triazole linker connected at the N-1 position was achieved by the CuAAC between N-1 propargyl adenosine derivative 42 and 5′-azido-adenosine 40. In the presence of sodium ascorbate and copper sulfate, the expected positively charged conjugate 43 was isolated in 53% yield (Scheme 17).
As seen above, CuAAC efficiently provides bisubstrate analogues with a 1,2,3-triazole linker. However, this strategy does not allow the introduction of the α-amino-acid motif of the SAM cofactor. To overcome this issue, Coelho et al. developed in 2023 a two-step metal-catalyzed procedure to access bisubstrate analogues of RNA MTase [51]. In this work, 5-iodotriazole was modified by metal-catalyzed reactions. The reactivity of adenosine derivatives in iodo copper(I) azide–alkyne cycloaddition (iCuAAC) reaction was achieved between adenosine 39 substituted at the N-6 position with an alkyne function and 5′-azido-adenosine 40. The 5-iodotriazole compound 44 was obtained in 38% yield in the presence of a substoichiometric amount of Cu(ClO4)2, an excess of NaI as the iodinating source, N,N-diisopropylethylamine (DIPEA), and tris(benzyltriazolylmethyl)amine as the ligand in DMF (Scheme 18).
Synthesis of 5-iodotriazole compound 44 by iCuAAC.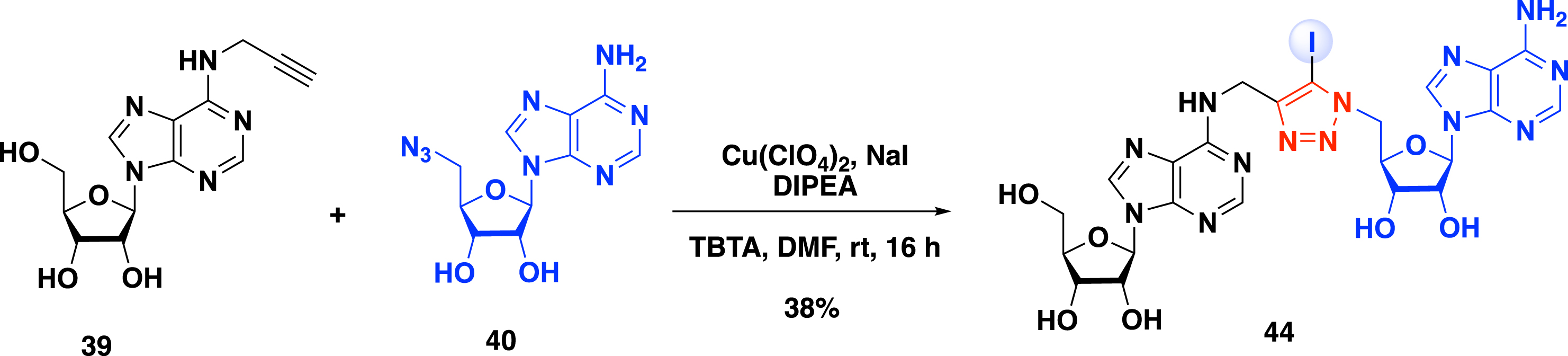
A second metal-catalyzed promoted reaction was optimized to introduce the α-amino-acid motif mimicking the methionine side chain of the SAM cofactor [51]. First, a Sonogashira cross-coupling reaction was examined between compound 44 and alkyne 45 using Pd(OAc)2, CuI, triphenylphosphine trisulfonate, and DIPEA in DMF to obtain the corresponding coupling product in 13% yield (Scheme 19). The tert-butyl and the Boc protecting groups were then removed using ZnBr2 salt in a mixture of isopropanol and nitromethane, producing compound 46 in 41% yield. The 5-iodotriazole 44 is also a suitable substrate for the Suzuki–Miyaura cross-coupling reaction (Scheme 19). In the presence of pinacolboronic ester 47 with a water-soluble palladium species in tris buffer, the corresponding cross-coupling product was isolated in 57% yield. A similar approach using the Stille cross-coupling reaction in the presence of vinylstannane 48 was reported to obtain the same analogue. A second step of deprotection led to the formation of bisubstrate analogue 49 containing a vinyl side chain in 47% yield.
Functionalization of 5-iodotriazole 44 by metal-catalyzed cross-coupling.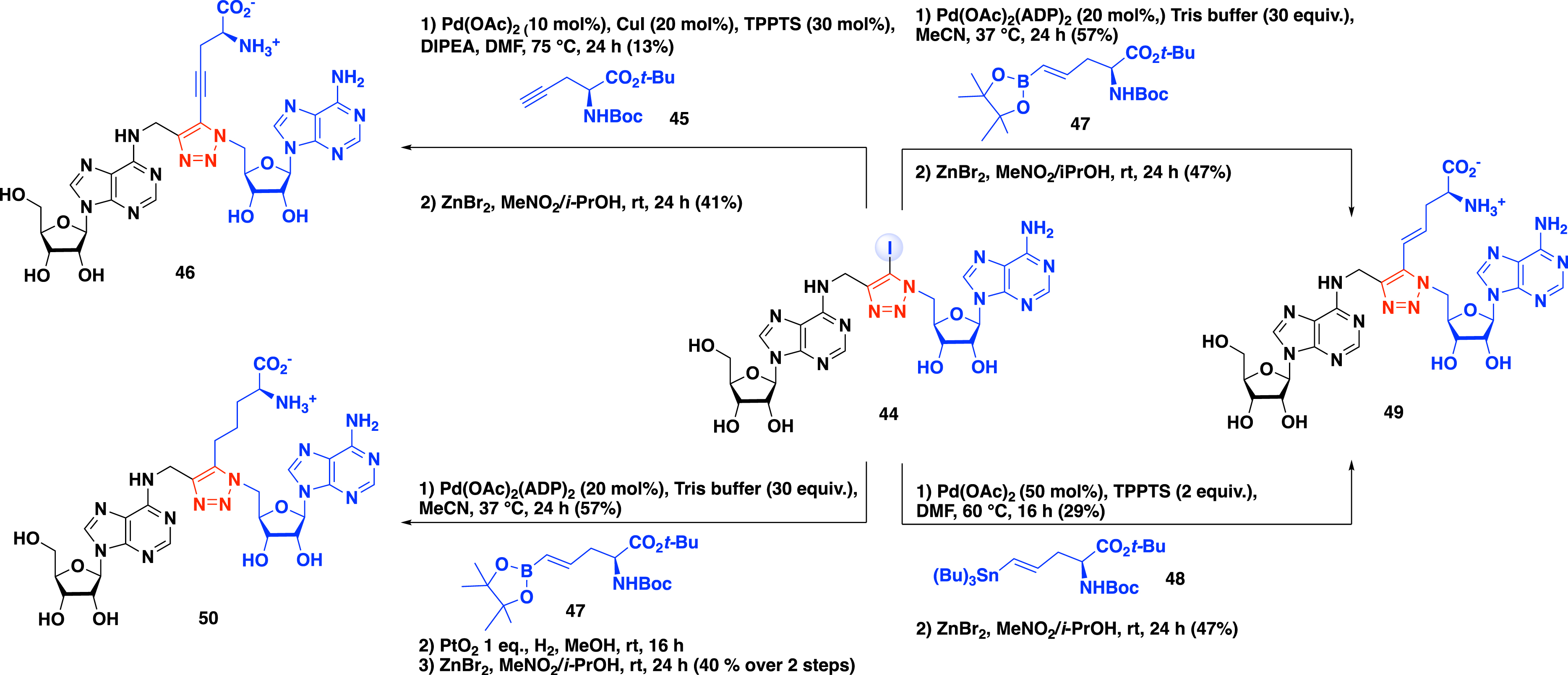
The procedure developed by Coelho et al. additionally allows access to bisubstrate analogue 50 with an alkyl chain [51]. A three-step procedure beginning with a Suzuki–Miyaura cross-coupling reaction in the presence of alkyne 47, followed by the reduction of the double bond under hydrogen atmosphere with PtO2, and a final deprotection step of the α-amino-acid protecting groups provided compound 50 in 40% yield over two steps (Scheme 19).
The two-step metal-catalyzed procedure developed by the group of Etheve-Quelquejeu was next considered to synthesize bisubstrate molecules with a longer RNA strand mimicking the substrate of the methyltransferase (Scheme 20) [51]. This approach requires the presence of an alkyne function at the internal position of the oligonucleotide. This was achieved by the incorporation of a phosphoramidite nucleoside containing an alkyne function at the N-6 position of the adenosine during oligonucleotide synthesis. The resulting propargyl-N-6-A-RNA 51 was then subjected to iCuAAC in the presence of 5′-azido-adenosine 52 to form 5-iodotriazole derivative 53. A second step of late-stage modification of the SAM-RNA conjugate, in the presence of pinacolboronic ester 47, followed by the removal of tert-butyl and Boc protecting groups led to the formation of bisubstrate analogue 54 containing a SAM analogue covalently attached to the RNA substrate via a 1,2,3-triazole linker substituted at the C-5 position with an α-amino-acid motif (Scheme 20).
Synthesis of SAM–RNA conjugate 54 with a triazole linker substituted by an analogue of the methionine side chain.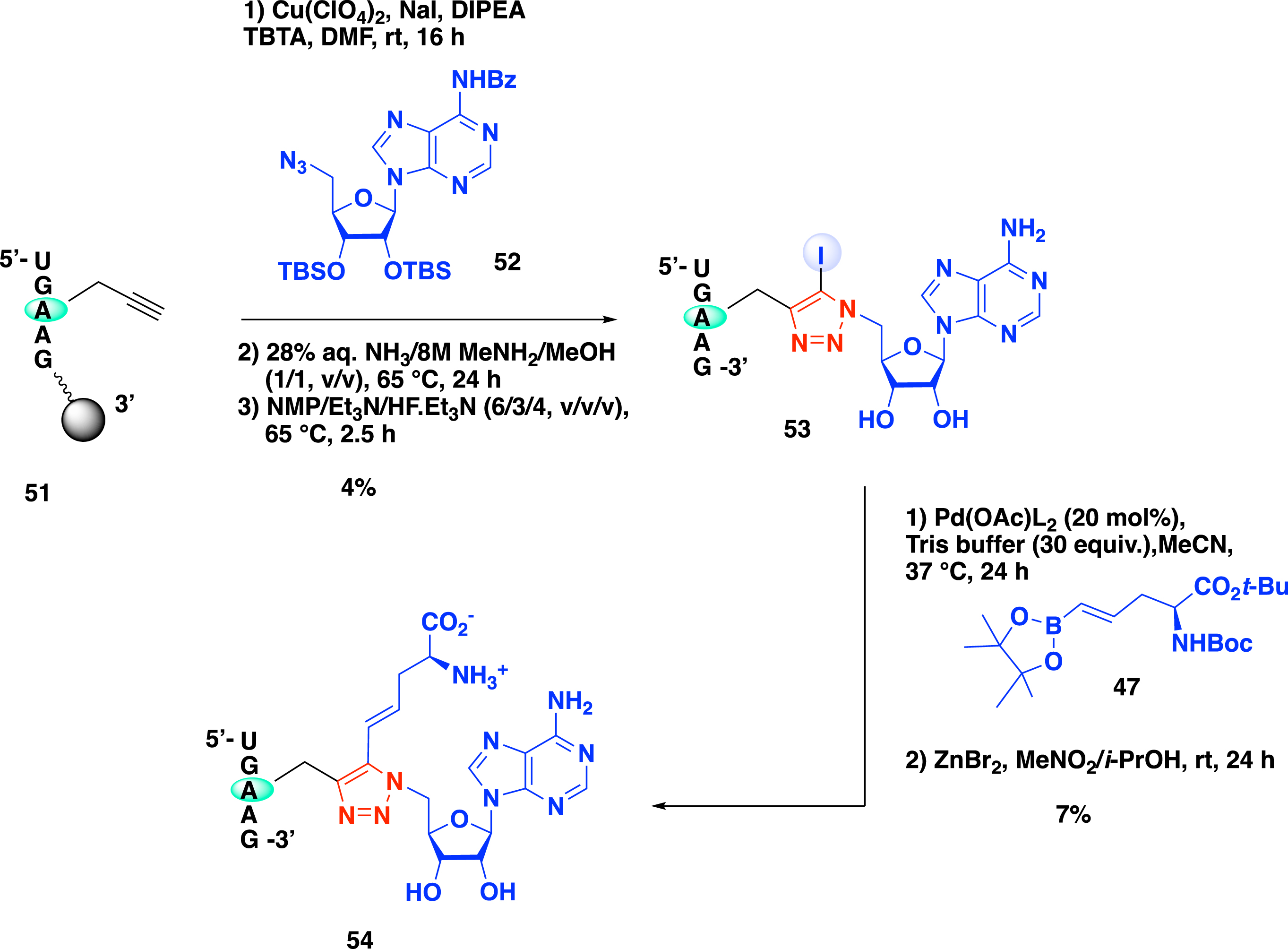
3.3. Biological results
The SAM–adenosine conjugates were evaluated on the bacterial rRNA MTase RlmJ, which methylates the 23S rRNA at position A2030 and on METTL3–14, which deposits the m6A mark on human mRNA. This enzymatic complex is involved in human diseases and considered a promising drug target [32, 35, 38].
The binding of SAM–adenosine conjugates 31a–b, 35a–b, and 38 to RlmJ was first assayed using differential scanning fluorimetry [52]. Compounds 31a–b show an increase in the Tm value, indicating that they establish stabilizing interactions with the protein compared to other potential bisubstrates. This result also underlines the importance of the methionine part of the SAM analogue likely to be involved in the interaction and an aliphatic linker. In addition, compounds 35a and 35b display KD values of 25 and 30 μM, respectively. X-ray structures were solved for the complexes 35a/RlmJ and 35b/RlmJ. In the complex 35b/RlmJ, one structure shows that the RNA moiety of the conjugate binds the presumed substrate pocket of the enzyme while the cofactor analogue occupies partly the SAM-binding site, showing a rotation of 120° of the adenosine out of the pocket (Figure 4A). Importantly, compound 35b interacts with key residues involved in the catalytic mechanism, and the crystallographic structure confirms the interactions of the methionine part with the active site (Figure 4B). All together, these results indicate that the positioning of conjugate 35b in RlmJ resembles the transition state of the SN2 mechanism of the methyl transfer.
(A) Structure of 35b/RlmJ (blue) aligned with SAH/RlmJ (yellow) [52]. (B) Interaction map of bisubstrate 35b in the active site of RlmJ [52].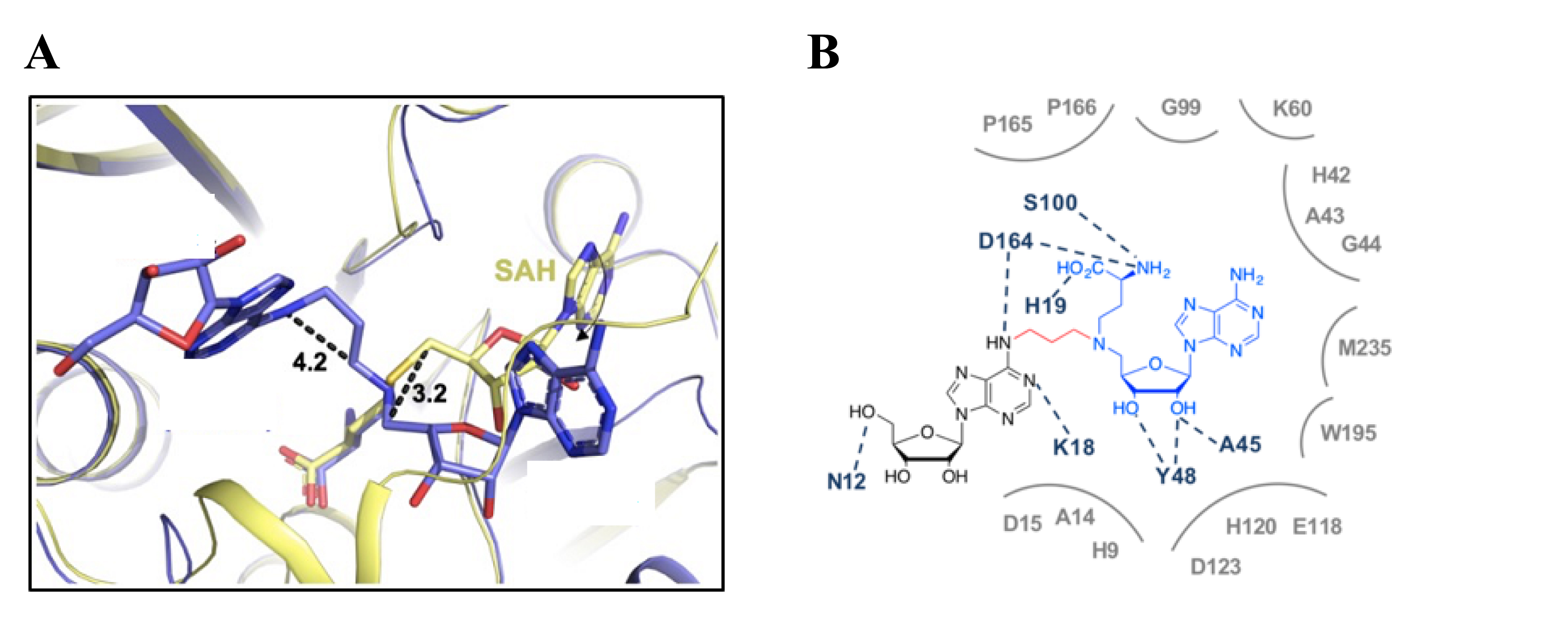
In order to fully decipher the mechanism of methylation catalyzed by METTL3–14, conjugates 35a–b were also used as tools in a combined experimental and computational approach [53]. First, the inhibitory potency of the compounds was determined using a reader-based TR-FRET assay. Under these conditions, the conjugates exhibit IC50 values in the low micromolar range. Several crystallographic structures of METTL3–14 in complex with compounds 35a–b were solved. Analysis of the interaction networks between the active site and the conjugates validated their use as tools to decipher the catalytic mechanism. In particular, compound 35a was identified as the transition state analogue of the methyl transfer catalyzed by METTL3. Using the crystal structure of the complex 35a–METTL3–14 (Figure 5), QM/MM simulations allowed for the proposition of the mechanism catalyzed by METTL3–14.
(A) Superposition of the crystal structure of METTL3–14 bound to 35a (magenta) and SAM (blue) [53]. (B) Interaction map of bisubstrate 35a in the active site of METTL3–14 [53].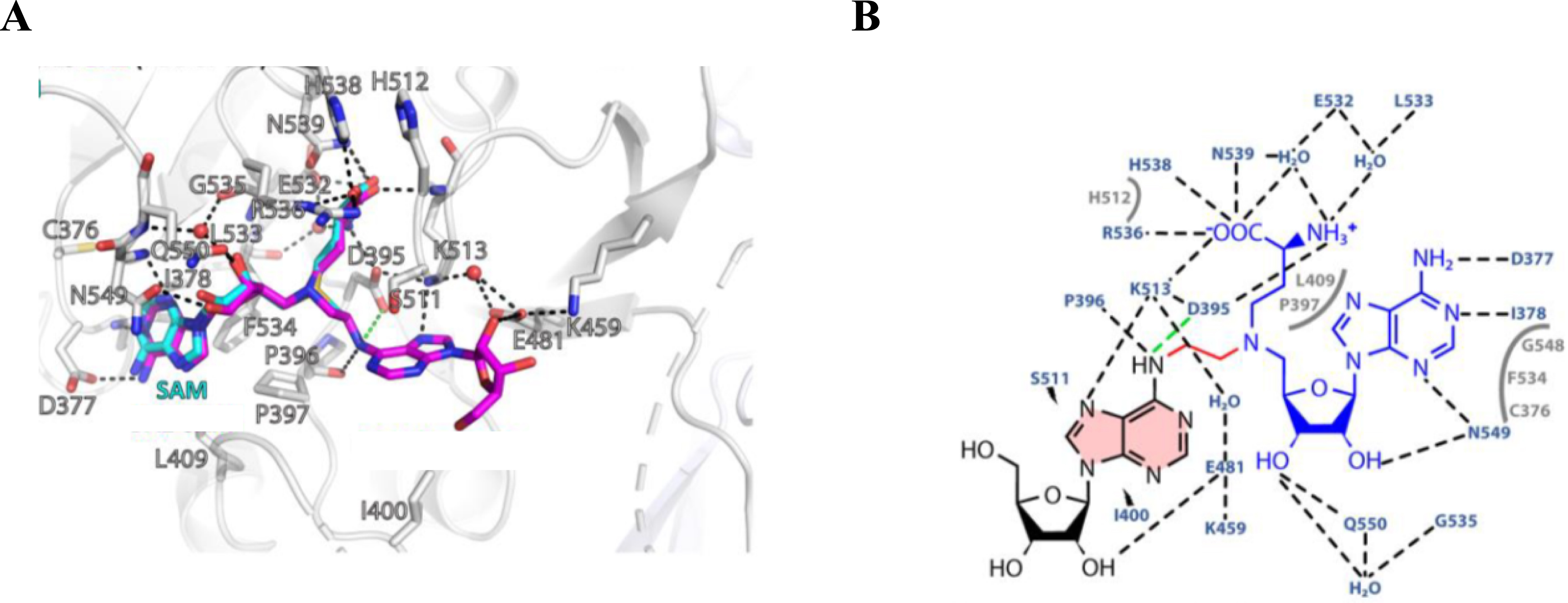
To go further, SAM-RNA conjugates with increased size of the RNA part were also used in crystallization assays with RlmJ to gain insights into the m6A methylation process [49]. Two crystallographic structures were obtained with SAM-RNA conjugates GA* and GA*A containing a dinucleotide and a trinucleotide as the substrate, respectively. In the case of the dinucleotide GA*, the adenosine of the SAM analogue shows a correct orientation in the SAM pocket. All together, these two RX structures allowed for the construction of a model showing the SAM cofactor and a trinucleotide with the sequence GA2030A of E. coli 23S rRNA bound to RlmJ. Thanks to this model, we were able to propose a mechanism for the methylation reaction catalyzed by RlmJ (Scheme 21) [49].
Proposed mechanism for the methylation reaction catalyzed by RlmJ [49].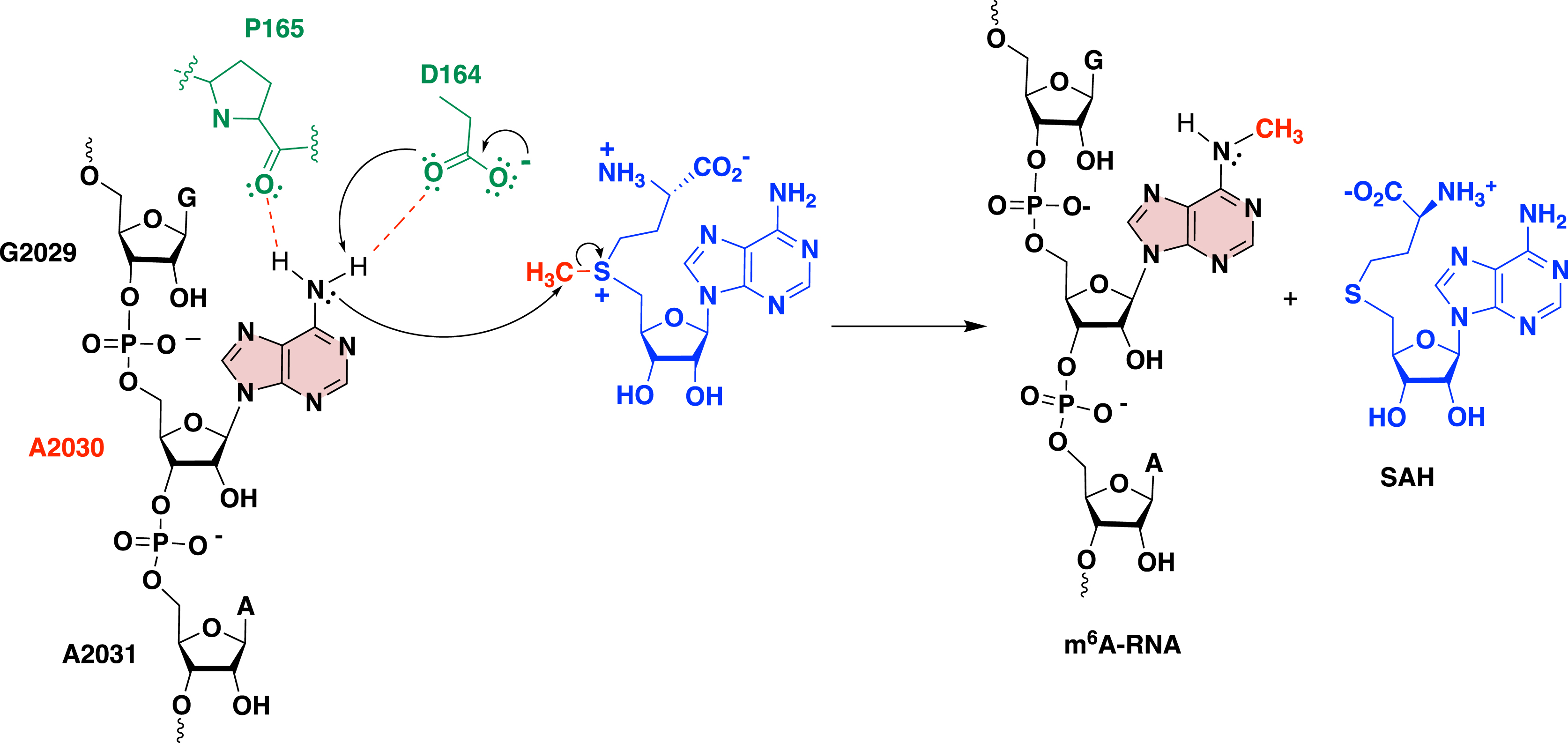
In summary, the SAM–adenosine conjugates are valuable tools to decipher the full mechanism of other RNA MTases and also for the design of potent inhibitors.
4. Conclusion and outlook
The chemical approaches discussed in this review highlight the versatility and efficiency of nucleoside and nucleotide chemistry in addition to SPS and the chemoenzymatic approach in the design of RNA conjugates for the study of RNA transferases. These methods allow for significant flexibility in modifying the bisubstrate compounds, including the RNA sequence and size, the nature of the linkers, and the peptide or cofactor components. Such adaptability is crucial for tailoring these chemical tools to explore a wide range of transferases beyond the examples provided in this work, extending their utility to new classes of RNA-dependent enzymes.
The insights they provide into enzyme–substrate interactions are particularly valuable not only for fundamental research in RNA biology but also for drug discovery. Indeed, since new transferases continue to be discovered and considered as promising new drug targets [54], these chemical tools will be instrumental in the development of new therapeutic strategies.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Funding
The authors thank the CNRS and Paris Cité University for their support and the Agence Nationale de la Recherche (ANR: ARNTools, SyntRNA) and La Ligue contre le Cancer for funding.